

Abstract
Background:
Oxygen-glucose deprivation-nutrition resumption (OGD-NR) models on H9c2 cells are commonly used in vitro models of simulated myocardial ischemia-reperfusion injury (MIRI), but no study has assessed whether these methods for establishing in vitro models can effectively imitate the characteristics of MIRI in vivo. This experiment was designed to analyze the feasibility of six OGD-NR models of MIRI.
Methods:
By searching the PubMed database using the keywords “myocardial reperfusion injury H9c2 cells,” we obtained six commonly used OGD-NR in vitro models of MIRI performed on H9c2 cells from more than 400 published papers before January 30, 2017. For each model, control (C), simulated ischemia (SI), and simulated ischemia-reperfusion (SIR) groups were assigned, and cell morphology, lactate dehydrogenase (LDH) release, adenosine triphosphate (ATP) levels, reactive oxygen species (ROS), mitochondrial membrane potential (MMP), and inflammatory cytokines were examined to evaluate the characteristics of cell injury. Subsequently, a coculture system of cardiomyocyte-endothelial-macrophage was constructed. The coculture system was dealt with SI and SIR treatments to test the effect on cardiomyocytes survival.
Results:
For models 1, 2, 3, 4, 5, and 6, SI treatment caused morphological damage to cells, and subsequent SIR treatment did not cause further morphological damage. In the models 1, 2, 3, 4, 5 and 6, LDH release was significantly higher in the SI groups than that in the C group (P < 0.05), and was significantly lower in the SIR groups than that in the SI groups (P < 0.05), except for no significant differences in the LDH release between C, SI and SIR groups in model 6 receiving a 3-h SI treatment. In models 1, 2, 3, 4, 5, and 6, compared with the C group, ATP levels of the SI groups significantly decreased (P < 0.05), ROS levels increased (P < 0.05), and MMP levels decreased (P < 0.05). Compared with the SI group, ATP level of the SIR groups was significantly increased (P < 0.05), and there was no significant ROS production, MMP collapse, and over inflammatory response in the SIR groups. In a coculture system of H9c2 cells-endothelial cells-macrophages, the proportion of viable H9c2 cells in the SIR groups was not reduced compared with the SI groups.
Conclusion:
All the six OGD-NR models on H9c2 cells in this experiment can not imitate the characteristics of MIRI in vivo and are not suitable for MIRI-related study.
Keywords: Feasibility, In vitro Models, Myocardial Ischemia-Reperfusion Injury
摘要
背景:
H9c2细胞糖氧剥夺-营养恢复(OGD-NR)模型是最常用的模拟在体心肌缺血-再灌注损伤(myocardial ischemiareperfusion injury (MIRI) 的离体建模方法。但是,至今尚无研究对该模型的可行性进行评估。该实验旨在评价常用的6种 OGD-NR模型模拟在体MIRI的可行性。
方法:
通过Pubmed检索,从既往发表的400余篇文献中获得6种常用的模拟在体MIRI的H9c2细胞OGD-NR模型。对每种模型 设置对照(C)组、模拟缺血(SI)组和模拟缺血再灌注(SIR)组。实验结束检测细胞形态学、乳酸脱氢酶(LDH)释放情况、三磷酸腺苷(ATP)水平、活性氧(ROS)水平、线粒体膜电位(MMP)水平以及炎症因子水平。随后构建H9c2细胞-内皮细胞-巨噬细 胞共培养系统,对共培养系统进行实验,以观察SI处理和SIR处理对心肌细胞存活情况的影响。
结果:
在模型1、2、3、4、5和6,SI处理可导致细胞形态学损伤,随后实施SIR处理未造成进一步的细胞形态学损伤。除外接 受3h SI处理模型6的LDH释放在C、SI和SIR组之间无明显差异之外,模型1, 2, 3, 4, 5和6的SI组LDH释放明显高于C组(P < 0.05) ,SIR组LDH释放明显低于SI组(P < 0.05)。在模型1、2、3、4、5和6,与C组相比,SI组的ATP明显降低(P<0.05)、ROS生成 明显增加(P<0.05)、MMP水平明显降低(P<0.05)。与SI组相比,SIR组的LDH释放明显降低(P<0.05)、ATP水平明显升 高(P<0.05)、无大量ROS生成、无MMP坍塌以及无过度的炎症反应。在H9c2细胞-内皮细胞-巨噬细胞共培养系统实验中, 与SI组相比,SIR组的活细胞比例无明显降低。
结论:
在H9c2细胞建立的6种常用OGD-NR模型均不能成功模拟在体MIRI的特征,它们不适用于MIRI的相关研究。
INTRODUCTION
Ischemic heart disease is a major cause of morbidity and mortality worldwide. Reperfusion is the only method to rescue salvageable myocardium, but reperfusion itself may induce further myocardial injury, a phenomenon has been termed as myocardial ischemia-reperfusion injury (MIRI).[1] Since the discovery of the MIRI phenomenon in dogs by Jennings et al.[2] in 1960, numerous studies have been conducted to reveal the underlying mechanisms of MIRI and to explore the effective treatments for MIRI.[3] Experimental studies in this field have used in vivo, ex vivo, and in vitro models. It is generally believed that in vivo and ex vivo models of MIRI have a proven success rate by occluding and releasing vessels in specific animals.[4] Furthermore, there have been diverse methods for generating in vitro models of MIRI. Cells used for in vitro MIRI models include H9c2 cells, neonatal rat cardiomyocytes (NRCs), adult rat cardiomyocytes, and neonatal mouse cardiomyocytes, HL-1 cells. Establishment methods of in vitro MIRI models include oxygen-glucose deprivation-nutrition resumption (OGD-NR) and specific ischemic buffer-reperfusion buffer.[5,6,7,8] H9c2 cells are immortalized cells with a cardiac phenotype, which are widely used for the study of cardiac disease. Although H9c2 cells demonstrate some similar characteristics as primary cardiomyocytes including high ATP levels, mitochondrial mass, and respiratory activity, they still possess a number of differences such as the inability of pulse and the ability of infinite proliferation.[9] In addition, H9c2 cells were the most used cell type for the convenient access and cultivation, and usually, the cells were performed with OGD-NR for constructing in vitro model. Although hundreds of MIRI studies have used OGD-NR models on H9c2 cells for the mechanism exploration, no study has assessed whether these methods for establishing in vitro MIRI models can effectively imitate the characteristics of MIRI in vivo.
The most obvious and classical method of discerning reperfusion injury is comparing cells that have been reperfused for a few minutes to cells that have not been reperfused.[2] This method was selected to compare cells subjected to simulated ischemia (SI) treatment and those subjected to simulated ischemia-reperfusion (SIR) treatment for a short period (mainly compared SI group vs. SIR-0.5 h group). Furthermore, it is well known that pathophysiological features of lethal reperfusion injury include oncosis, hypercontracture, large generation of reactive oxygen species (ROS), over inflammatory response, and mitochondrial permeability transition pore (MPTP) opening.[1,10,11] Previous in vivo studies considered these pathophysiological characteristics as the most important determinants for the occurrence of MIRI. In this study, thus, these variables were measured to evaluate whether in vitro SIR injury can resemble the characteristics of MIRI in vivo. By analyzing the characteristics of six commonly used OGD-NR models on H9c2 cells, the main aims of this experiment were to determine whether they could imitate some aspects of in vivo MIRI and be reliably used for the related studies in the field of MIRI.
METHODS
Cell culture and grouping
Cell culture
H9c2 cells were purchased from a cell bank of the Chinese Academy of Medical Sciences and were cultured with a complete medium (Dulbecco's modified Eagle media with 10% fetal bovine serum) in a humidified incubator at 37°C in 5% CO2 and 21% O2. The culture medium was changed every other day, and cells were passaged when the cell density reached 80–90%.
Macrophages
Macrophages (raw 264.7) were a kind gift from professor Zhao in our laboratory. Cells were cultured in a complete medium and passaged when the cell density reached 80–90%. A cell scraper was used to detach the cells from the culture dish.
Endothelial cells
Human umbilical vein endothelial cells were a kind gift from professor Zhao in our laboratory. The cells were cultured with complete medium in a humidified incubator at 37°C in 5% CO2 and 21% O2. Culture medium was changed every other day, and the cells were passaged when the cell density reached 80–90%.
Coculture
To determine the roles of macrophages and endothelial cells in generating an in vitro MIRI model, a transwell plate was used to construct the coculture system. Cardiomyocytes were seeded at a density of 1 × 106 cells/well in the lower chamber, while macrophages and endothelial cells were seeded in the upper chamber.
Cell groups and experimental protocol
By searching the PubMed database using the keywords “myocardial reperfusion injury H9c2 cells,” we obtained six commonly used OGD-NR in vitro models of MIRI performed on H9c2 cells from more than 400 published papers before January 30, 2017. The six OGD-NR in vitro models assessed in this experiment are shown in Table 1. In each model, H9c2 cells were randomly divided into three groups: (1) C group: cells were cultured as normal; (2) SI group: cells were subjected to SI; and (3) SIR group: cells were subjected to SI and then simulated reperfusion for given period according to the different experimental designs. A detailed flowchart of the experiment is shown in the Supplementary Figure 1 (275.6KB, tif) .
Table 1.
The six in vitro OGD-NR models of MIRI used in previous literatures
| Models | SI | SIR |
|---|---|---|
| 1 | No glucose, no FBS DMEM; anoxia | DMEM + 10% FBS; normoxia |
| 2 | FBS-hungry overnight; no glucose, no FBS DMEM; anoxia | DMEM + 10% FBS; normoxia |
| 3 | No glucose, no FBS DMEM; anoxia | DMEM; normoxia |
| 4 | Low-glucose DMEM, no FBS DMEM; anoxia | DMEM + 10% FBS; normoxia |
| 5 | High-glucose DMEM, no FBS DMEM; anoxia | DMEM + 10% FBS; normoxia |
| 6 | DMEM + 10%FBS; anoxia | DMEM + 10% FBS; normoxia |
OGD-NR: Oxygen glucose deprivation-nutrition resumption; MIRI: Myocardial ischemia reperfusion injury; SI: Simulated ischemia; SIR: Simulated-ischemia reperfusion; FBS: Fetal bovine serum; DMEM: Dulbecco’s modified Eagle media.
Experimental grouping and flow chart. (1) To measure cell morphology, LDH release and ATP levels, for the models 1-6, 5 different SI durations (3 h, 6 h, 9 h, 12 h and 15 h) were designed, and SI, SIR-0.5 h, SIR-2 h and SIR-12 h groups were included. The reasons for choice of different SI durations are that the injury by SI in the models 1-6 is slight by SI-3 h treatment, and then aggravate with the prolongation of SI duration, and becomes severe at SI-15 h. (2) To measure the levels of ROS, MMP and inflammatory cytokines, a 6 h duration of SI treatment was chosen for the models 1-6. For each model, C, SI and SIR-0.5 h groups were included. The injury by SI-6 h treatment in the models 1–6 is moderate. Reperfusion injury seldom occurs when ischemia injury is too slight or severe in vivo, 6 h of SI duration was chosen. SI: Simulated ischemia; SIR: Simulated-ischemia reperfusion; LDH: Lactate dehydrogenase; ATP: Adenosine triphosphate; ROS: Reactive oxygen species; MMP: Mitochondrial membrane potential.
Simulated ischemia and simulated ischemia-reperfusion treatments
Cell culture media was replaced with SI buffer and SIR buffer as shown in Table 1 according to the different models. Anoxia was achieved by placing the H9c2 cells in Tri-gas incubator (Huaxi electricity, China) to maintain an atmosphere of 0.5% O2, 5% CO2, and 94.5%N2. Nomoxia was accomplished by placing the H9c2 cells in an atmosphere of 5% CO2, 21% O2, and balance N2 at 37°C (Thermo, USA).
Measurements
Morphology
Morphological changes of cardiomyocytes were observed by inverted phase-contrast microscopy and recorded by imaging software (Leica, Germany).
Lactate dehydrogenase release
The LDH release was measured according to the manufacturer's protocol. The cell culture supernatant was collected, and 100 μl was used to analyze the LDH activity using diagnostic kits (Sigma, USA). Results are represented as a percentage of LDH activity in the supernatant medium over the total enzyme activity.
Adenosine triphosphate
The ATP Level was analyzed using an ATP detection kit (Beyotime, China) according to the manufacturer's instructions. Briefly, the supernatant medium was removed, and the cells were harvested using lysis buffer and centrifuged at 12,000×g for 5 min. Next, the mixture of lysis and reagent was analyzed by a multimode reader (EnSpire, USA). The ATP level was normalized by protein level measured using a BCA assay (Beyotime, China) and was expressed in nanomoles per milligram protein.
Reactive oxygen species
At the end of the experiment, the cell culture supernatant was removed, and cardiomyocytes were incubated with 10 μmol/L dichloro-dihydrofluorescein diacetate (Beyotime, China) in a serum-free medium at 37°C for 10 min in the dark. Then, cells were washed twice and collected for the measurement by flow cytometry (BD, USA). The results of ROS levels are presented as the FITC-positive cells/total cells (corresponding FITC geometric mean). The FITC-positive cells/total cells present the percentage of cells with ROS generation, and the corresponding FITC geometric mean presents the mean fluorescence intensity, indicating the low or high degree of ROS generation.
Mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured using a kit according to the manufacturer's protocol. At the end of the experiment, the cell culture supernatant was removed, and cells were incubated with the Ro123 probe kit (Beyotime, China) for 10 min at 37°C in the dark. Cardiomyocytes were washed twice and then analyzed by flow cytometry (BD, USA). The results of MMP levels are presented as the FITC-positive cells/total cells (corresponding FITC geometric mean).
Inflammatory cytokines
Levels of inflammatory cytokines in the cell culture supernatant were measured using an ELISA kit (R and D, USA) according to the manufacturer's instructions. Briefly, the cell culture supernatant was collected and added to an antibody-coated plate. After the supernatant was incubated with detection antibody for 1 h at 37°C, enzyme antibody conjugate, substrate solution, and stop solution were added. The optical density of each well was measured using a microplate reader at 450 nm.
Live H9c2 cells percentage
Percentage of live H9c2 cells was measured using a Muse Cell Analyzer with a live cell counting kit (Merck and Millipore). Cells were collected through trypsin digestion and resuspended in 200 μl of DPBS. Next, 50 μl of cells was added to 450 μl of reaction mixture. The mixture was incubated for 5 min and measured using the Muse Cell Analyzer.
Statistical methods
All data with a normal distribution are presented as the means ± standard deviation (SD). Differences between groups were analyzed using a one-way analysis of variance or Student's t-test. A P < 0.05 indicated a statistically significant difference. Data were analyzed with SPSS.19 software (San Diego, CA, USA).
RESULTS
Simulated ischemia-reperfusion did not cause further injury to cardiomyocyte morphology
For H9c2 cells, the SI-3 h treatment caused mild cardiomyocyte injury in models 1, 2, and 3, as demonstrated by more granule generation in cytoplasm [Supplemental Figure 2a (1.4MB, tif) ]. With the progression of SI treatments, SI-6 h treatment resulted in severe cardiomyocyte injury in models 1, 2, and 3, with an obvious decrease in cell number [Figure 1], and SI-9 h treatment produced more severe injury and a further decrease in cell number [Supplemental Figure 2b (1.4MB, tif) ]. The SI-12 h treatment produced morphology changes in models 4, 5, and 6, which appeared as slight shape changes but without severe deformation or membrane rupture [Supplemental Figure 2c (1.4MB, tif) ]. After SIR treatment, no further injury was observed in the SIR-0.5 h groups, and even cellular morphology improvements were noticed in the SIR-12 h groups [Figure 1 and Supplemental Figure 2 (1.4MB, tif) ].
Figure 1.
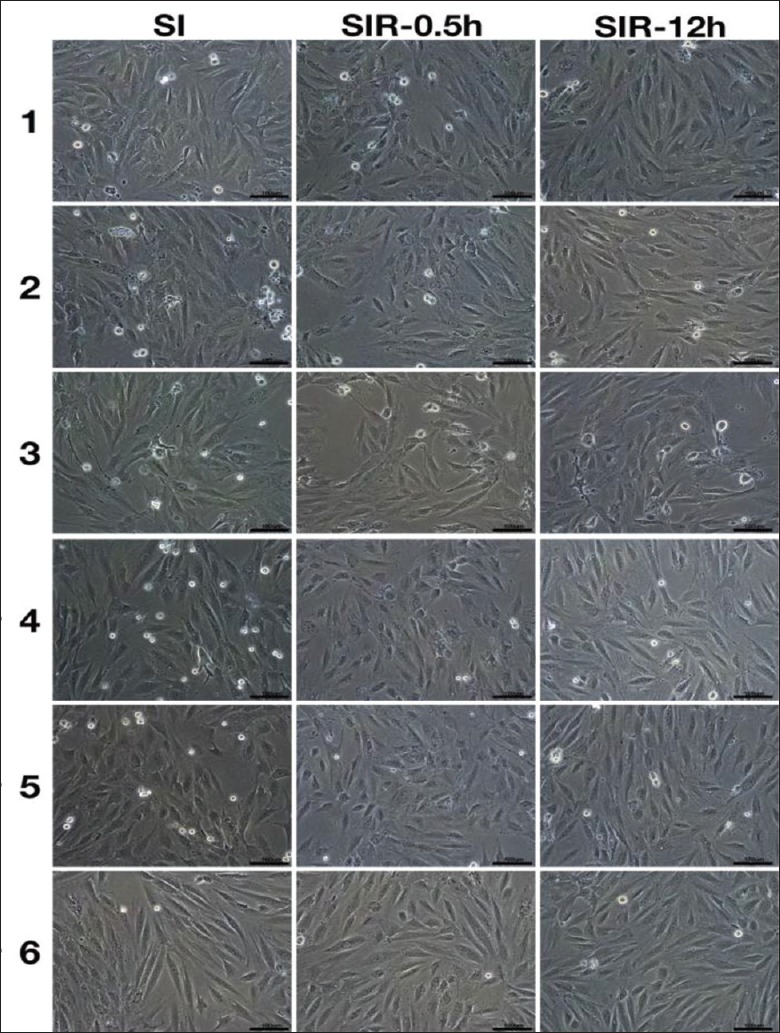
SIR did not cause further injury to cell morphology. When duration of SI treatment was 6 h, cell morphology of SI, SIR-0.5 h and SIR-12 h groups in the models 1–6 using H9c2 cells. Scale bars, 100 μm. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion.
The cell morphology of SI, SIR-0.5 h and SIR-12 h groups with different durations of SI treatment when using the H9c2 cells. Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 3 h in the models 1–6 (a); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 9 h in the models 1–6 (b); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 12 h in the models 1-6 (c); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 15 h in the models 1-6 (d). Scale bars, 100 μm. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion.
Simulated ischemia-reperfusion reduced lactate dehydrogenase release after simulated ischemia treatment
After SI treatment, LDH release increased significantly in models 1, 2, 3, 4, and 5 compared with the C group (P < 0.05), indicating that SI treatment produces severe cardiomyocyte injury [Figure 2]. After SIR treatment, compared with the corresponding SI groups, LDH release decreased sharply in models 1, 2, 3, 4, and 5 (P < 0.05) [Figure 2]. In model 6, when the duration of SI treatment was 3h, LDH release was not significantly different among C, SI and SIR groups; when the durations of SI treatment were 6h, 9h, 12h, and 15h, LDH release significantly increased in the SI groups compared with the C group (P < 0.05) and significantly decreased in the SIR groups compared with corresponding SI groups (P < 0.05), even though degrees of LDH release changes were relatively small.
Figure 2.

SIR reduced LDH release after SI treatment. When durations of SI treatment were 3, 6, 9, 12, and 15 h, LDH release of SI, SIR-0.5 h, SI-2 h, and SIR-12 h groups in the models 1–6 using H9c2 cells. -: Indicates SI; -: Indicates SIR. Data are presented as mean ± SD (n = 6). *P < 0.05 compared to control group; †P < 0.05 compared to SI group with the same model. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion; LDH: Lactate dehydrogenase; SD: Standard deviation.
Simulated ischemia-reperfusion increased adenosine triphosphate levels after simulated ischemia treatment
ATP level of C group was 12.88 ± 0.63 nmol/mg. When the duration of SI treatment was 3 h, ATP levels in the models 1–6 decreased to 4.05 ± 0.63, 4.31 ± 0.28, 4.01 ± 0.63, 4.38 ± 0.36, 3.87 ± 0.42, 4.52 ± 1.28 nmol/mg, respectively (P < 0.0001, t = 17.18; P < 0.0001, t = 21.59; P < 0.0001, t = 17.18; P < 0.0001, t = 20.34; P < 0.0001, t = 20.58; and P = 0.0005, t = 10.17, respectively, compared with the C group); after SIR treatment, ATP levels of SIR-0.5 h groups in the models 1–6 increased to 5.41 ± 0.48, 5.62 ± 0.24, 5.32 ± 0.41, 5.55 ± 0.45, 5.61 ± 0.48,7.23 ± 0.43 nmol/mg (P = 0.041, t = 2.980; P = 0.0035, t = 6.179; P = 0.040, t = 2.993; P = 0.025, t = 3.500; P = 0.009, t = 4.720; and P = 0.025, t = 3.476, respectively, compared with the corresponding SI groups); ATP levels of SIR-2 h and SIR-12 h groups in the models 1–6 increased further. No matter how long the duration of SI treatments was in our experiment, ATP levels all showed the similar trends as above. Compared with the C group, H9c2 cells exposed to SI treatment displayed a marked decrease in the ATP levels. However, ATP levels significantly recovered after SIR-0.5 h treatment and further recovered after SIR-2 h and SIR-12 h treatments [Figure 3].
Figure 3.

SIR increased ATP levels after SI treatment. When durations of SI treatment were 3, 6, 9, 12, and 15 h, ATP levels of SI, SIR-0.5 h, SI-2 h, and SIR-12 h groups in the models 1–6 using H9c2 cells. Data are presented as mean ± SD (n = 3–4). *P < 0.05 compared to control group; †P < 0.05 compared to SI group with the same model. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion; ATP: Adenosine triphosphate; SD: Standard deviation.
Simulated ischemia-reperfusion did not result in large generation of reactive oxygen species after simulated ischemia treatment
ROS level of C group was 69.9% ± 9.7% (32.6 ± 5.6) (data are presented as the FITC-positive cells/total cells [corresponding FITC geometric mean]). After SI treatment, ROS levels in the models 1–6 changed to 91.5 ± 3.9 (47.0 ± 1.6), 87.6 ± 1.6 (40.1 ± 3.7), 91.8 ± 4.6 (52.8 ± 1.7), 42.6 ± 2.5 (24.5 ± 3.2), 50.8 ± 2.0 (25.3 ± 3.7), and 58.8 ± 2.4 (22.0 ± 1.5), respectively. After SI treatment, compared with the C group, ROS levels significantly increased in the models 1, 2, and 3 (P = 0.017, t = 3.503; P = 0.031, t = 2.966; P = 0.017, t = 3.482, respectively), but significantly decreased in the models 4, 5, and 6 (P = 0.005, t = 4.739; P = 0.019, t = 3.372; P = 0.034, t = 3.171, respectively). After SIR treatment, compared with the corresponding SI group, ROS levels of SIR-0.5 h groups decreased in the model 3 (P = 0.011, t = 4.508, compared with the corresponding SI group), and increased in models 4, 5, and 6 (P = 0.049, t = 2.785; P = 0.022, t = 3.650; P = 0.031, t = 3.263, respectively). The ROS level was not significantly different between the SI and SIR groups of models 1 and 2 [Figure 4].
Figure 4.

SIR did not result in large generation of ROS after SI treatment. ROS levels of SI and SIR-0.5 h groups in the models 1–6 using H9c2 cells. FITC-positive percentage of cells refers to the FITC-positive cells/total cells; geometric mean refers to the geometric mean of FITC fluorescence intensity. Data are presented as mean ± SD (n = 3–4). *P < 0.05, †P < 0.01 compared to control group; ‡P < 0.05 compared to SI group with the same model. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion; ROS: Reactive oxygen species; FITC: Fluorescein isothiocyanate; NS: No significance.
Simulated ischemia-reperfusion did not lead to dissipation of mitochondrial membrane potential after simulated ischemia treatment
MMP level of C group was 96.5% ± 2.9% (2032.0 ± 92.9) (data are presented as the FITC-positive cells/total cells [corresponding FITC geometric mean]). After SI treatment, MMP levels decreased to 96.7 ± 3.8 (1743.3 ± 50.5), 96.4 ± 4.6 (1663.7 ± 36.1), 97.2 ± 2.8 (1686.3 ± 95.7) in the models 1, 2, and 3, respectively (P = 0.0091, t = 4.732; P = 0.0031, t = 6.405; P = 0.0109, t = 4.492, respectively, compared with the C group), and increased to 96.8 ± 4.0 (2260.7 ± 74.7), 96.6 ± 2.8 (2368.0 ± 44.0), 96.6 ± 3.7 (2346.0 ± 60.9) in models 4, 5, and 6 (P = 0.0295, t = 3.317; P = 0.0048, t = 5.654; P = 0.0081, t = 4.889, respectively, compared with the C group). After SIR treatment, MMP levels were not significantly different between the SI and SIR-0.5 h groups of the models 1–6 [Figure 5].
Figure 5.

SIR did not lead to MMP dissipation after SI treatment. MMP levels of SI and SIR-0.5 h groups in the models 1-6 when using H9c2 cells. FITC-positive percentage of cells refers to the FITC-positive cells/total cells; geometric mean refers to the geometric mean of FITC fluorescence intensity. Data are presented as mean ± SD (n = 3–4). *P < 0.05, †P < 0.01 compared to control group. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion; MMP: Mitochondrial membrane potential. NS: No significance; SD: Standard deviation; FITC: Fluorescein isothiocyanate.
Inflammatory cytokines in the supernatant of cardiomyocytes
When only cardiomyocytes were treated with SI and SIR, relatively low levels of inflammatory cytokines in the supernatant was observed in few groups, and no any inflammatory cytokine in the supernatant was detected in many groups [Table 2].
Table 2.
The concentrations of inflammatory cytokines (pg/ml) after SI and SIR - 0.5 h treatments in the models 1-6 using H9c2 cells
| Groups | H9c2 cells | |
|---|---|---|
| IL-6 | TNF-α | |
| 1-SI | 0.44 ± 0.02 | 0.00 |
| 1-SIR | 1.69 ± 0.76 | 0.00 |
| 2-SI | 0.07 ± 0.01 | 0.00 |
| 2-SIR | 0.06 ± 0.01 | 0.00 |
| 3-SI | 0.07 ± 0.02 | 0.00 |
| 3-SIR | 0.07 ± 0.01 | 0.00 |
| 4-SI | 0.06 ± 0.01 | 0.00 |
| 4-SIR | 0.01 ± 0.02 | 0.06 ± 0.01 |
| 5-SI | 0.07 ± 0.01 | 0.00 |
| 5-SIR | 0.06 ± 0.01 | 0.00 |
| 6-SI | 0.07 ± 0.02 | 0.00 |
| 6-SIR | 0.07 ± 0.01 | 0.00 |
SI: Simulated ischemia; SIR: Simulated ischemia reperfusion; IL-6: Interleukin 6; TNF-α: Tumor necrosis factor α.
Coculturing endothelial cells, macrophages, and cardiomyocytes did not result in a successful model
After the endothelial cells, macrophages and H9c2 cells were cocultured, the live H9c2 cells% of C group was 93.2% ± 0.7%. After SI treatment, live H9c2 cells% in the models 1–6 were 82.7 ± 2.8, 79.8 ± 3.6, 83.5 ± 3.2, 84.9 ± 1.9, 85.3 ± 1.3, and 91.7 ± 1.2, respectively. The live H9c2 cells% of SI groups in the models 1–5 decreased significantly compared with the C group (P = 0.0016, t = 6.154; P = 0.0031, t = 6.392; P = 0.0037, t = 5.114; P = 0.0022, t = 6.984; P = 0.0008, t = 9.149, respectively). After SIR treatment, live H9c2 cells% in the models 1–6 were 86.47 ± 1.28, 88.17 ± 3.59, 84.01 ± 3.32, 85.73 ± 1.65, 86.60 ± 0.92, 93.03 ± 0.45, respectively. The live H9c2 cells% of SIR groups in the model 2 increased significantly (P = 0.00452, t = 2.876, compared with the corresponding SI groups), and there was no significant difference between SI and SIR groups in the models 1, 3, 4, 5, and 6. The live H9c2 cells% of SIR groups were not decreased compared with the SI groups [Figure 6].
Figure 6.

Coculturing endothelial cells, macrophages, and cardiomyocytes did not produce a successful model. The percentage of live H9c2 cells of SI and SIR-0.5 h groups in the models 1-6 when using H9c2 cells. The percentage of live H9c2 cells of SIR-0.5 h groups were not decreased compared with corresponding SI groups. Data are presented as mean ± SD (n = 3–4). *P < 0.05 compared to control group; †P < 0.05 compared to SI group with the same model. SI: Simulated ischemia; SIR: Simulated-ischemia reperfusion; NS: No significance; SD: Standard deviation.
DISCUSSION
Simulated ischemia-reperfusion cannot imitate myocardial ischemia-reperfusion injury with H9c2 cells
Using six OGD-NR in vitro models on H9c2 cells, our results showed that morphology of cells experienced SI treatment was not further deteriorated by SIR treatment. LDH is an intracytoplasmic enzyme, and LDH release can be induced by exocytosis and shedding of membrane blebs when cells suffer from injury.[12] The percent of LDH release can indicate the degree of cell injury.[12,13] Our results showed that after SI treatment, LDH levels were significantly increased in the models 1, 2, 3, 4, and 5, indicating that SI treatment led to severe cardiomyocytes injury. After SIR treatment, however, LDH levels sharply decreased. In the model 6, when the duration of SI treatment was 3 h, LDH release was not significantly different between C, SI and SIR groups; when the durations of SI treatment were 6 h or longer, LDH release significantly increased in the SI groups compared with the C group (P < 0.05) and significantly decreased in the SIR groups compared with corresponding SI groups (P < 0.05), degrees of LDH release changes were relatively small. These results indicate that oxygen deprivation-reoxygenation alone does not cause severe cardiomyocytes injury. In addition, our experiment showed that ATP levels recovered along with SIR, which was significantly different from MIRI in vivo.[14] The ATP recovery reflects the improvement of mitochondrial oxidative respiration. Furthermore, our results showed no large generation of ROS, occurrence of MMP dissipation and over inflammatory response during SIR, which were also significantly different from the features of MIRI in vivo.[1,10,11] An interesting phenomenon obtained in the H9c2 cells was that ROS level of C group was relatively high, and ROS levels of SI groups in the models 4, 5, and 6 were significantly lower than that of C group. Afterward, ROS levels of SIR-0.5 h groups in the models 4, 5, and 6 were increased than that of SI group. This may result from the complete medium, which is the culture medium for the C group and SIR medium for the models 4, 5 and 6. We assumed that the complete medium, to some extent, would cause injury to H9c2 cells and result in an increased ROS level. When cell injury caused by SI-6 h treatment was not severe (in the models 4, 5, and 6), ROS levels decreased compared with that of the C group, which would then increase after SIR treatment in a complete medium. Corresponding to the ROS changes, MMP levels of SI groups in the models 4, 5, and 6 increased compared with the C group.
Previous experiments of myocardial ischemia-reperfusion injury in the isolated cardiomyocytes
According to our review of original MIRI in vitro studies over the past several decades, many have used several kinds of cardiomyocytes, including the embryonic chick cardiomyocytes, primary adult rat cardiomyocytes, primary adult rat cardiomyocytes, NRCs, HL-1 cells, H9c2 cells, and so on. At first, Altschuld et al.[15] observed that isolated rat cardiomyocytes were round-shaped and hypercontracture in response to SIR treatment; however, the membrane integrity of a sarcolemma was preserved without extensive releases of enzymes and cytosolic components, which was different from the “oxygen paradox” in situ.[16] Siegmund et al.[17,18] found a similar phenomenon in adult rat cardiomyocytes. All these initial experiments on the isolated rat cardiomyocytes are the sources and references of the OGD-NR models in subsequent studies. The H9c2 cells are different from these primary cells, with a strong ability of beating and hypercontracture. Hypercontracture in the first minutes of reperfusion really plays a significant role in the pathogenesis of MIRI in vivo and it can result in mechanical rupture and the following necrosis.[11] The inability of hypercontracture of H9c2 cells may be a crucial reason that no further injury could be caused by SIR treatment.
The underlying reasons for model failure
The mechanisms underlying lethal reperfusion injury mainly include: (a) oncosis, (b) hypercontracture, (c) large generation of ROS, (d) over inflammatory response, and (e) opening of MPTP.[1,10,11]
During myocardial ischemia, ATP level quickly declines, resulting in an energy deficiency, dysfunction of sodium pump and the following intracellular Na+ accumulation.[19] Aerobic metabolism causes intracellular accumulation of H+ and metabolites, and increases of osmotic stress.[20] Interestingly, the interstitial compartment in the myocardium is small to selectively enable small amounts of fluid to traverse into cells during ischemia. However, reperfusion could rapidly wash out H+, resulting in the Na+/H+ exchanger activity and exacerbating intracellular Na+ accumulation. During myocardial reperfusion, a continuous supply of isotonic fluid enters cells and eventually causes oncosis.[19,20] Furthermore, reperfusion can lead to hypercontracture in the myocardium, which causes mechanical stress to sarcolemma and exacerbates cell injury in the first minutes of reperfusion.[11]
For the isolated cardiomyocytes in our experiment, however, the morphological phenotype of oncosis was not observed after SIR treatment. This may be because the cell culture supernatants during SI and SIR treatments were the same, and the increase in osmotic pressure had reached equilibrium during SI treatment. Due to the lack of continuous supply of isotonic fluid during SIR treatment, a large influx of fluid into the cardiomyocytes did not occur. Furthermore, hypercontracture has been observed in the intact heart, isolated heart, and isolated cardiomyocytes; however, this phenomenon in the isolated cardiomyocytes is different from that in the other two models. The hypercontracture observed in isolated cardiomyocytes was incomplete, as demonstrated by pulsating at an increased rhythm, with membrane integrity, significantly decreased LDH release and recovery of ATP level. Hypercontracture is not equal to irreversible injury, and it can only result in mechanical rupture of the membrane based on enough muscle mass of the heart.[16] For the H9c2 cells, they do not have the ability to pulsate or hypercontract; therefore, the absence of development of cardiomyocyte hypercontracture cannot initiate several injury processes during the first minutes of reperfusion.
After myocardial reperfusion, large generation of ROS and extensive infiltration of proinflammatory neutrophils into ischemic tissues can exacerbate the myocardial injury.[1] These severe pathological events and following injury caused by these, can lead to constitutive MPTP opening, which may indicate a common end-effector of pathological events initiated by ischemia/reperfusion.[1,21] In MIRI, main sources of ROS production include xanthine oxidase (present in the endothelium), neutrophils, mitochondria, and autoxidation of catecholamines.[22] Overproduction of ROS through the electron transport chain and other sources may produce a robust positive feedback circuit that induces MPTP opening and ROS-induced ROS production (RIRP).[23] For the H9c2 cells used in our experiment, however, production of ROS was limited due to the lack of all the sources except mitochondria, which are unable to cause such a robust surge in ROS production and RIRP. Furthermore, we noticed that the levels of inflammatory cytokines in the culture supernatant of isolated cardiomyocytes was very low, even could not be detected in many groups. Unlike inflammatory cells, H9c2 cells have an inability to produce large amounts of inflammatory cytokines. For the H9c2 cells, the lack of large ROS production, pronounced inflammatory response, cell membrane rupture caused by oncosis and hypercontracture and related injury effects would not result in constitutive MPTP opening. Just as our results had shown, thus, no MMP reduction occurred following SIR-0.5 h after SI treatment.
In our experiment, co-cultured H9c2 cells with macrophages and endothelial cells also did not inflict further injury caused by SIR after SI treatment. In fact, other than macrophages and endothelial cells, multiple other cells are also involved in the pathophysiology of MIRI, including neutrophils, lymphocytes, platelets, mast cells, monocytes, and Kupffer cells.[24] The deficiency of other cells and cell-to-cell interactions in the coculturing system may explain the model failure.[25,26]
Perspectives: Translational outlook implications
The OGD-NR models on H9c2 cells are widely used in vitro modelsof MIRI for the convenience. These models have extensively been using to explore fundamental mechanisms excluding the influence of neurohumoral factors. However, no study has assessed whether all these methods used for generatingmodels can effectively imitate the characteristics of MIRI in vivo. By assessing the six OGD-NR in vitro models commonly used in previous works, we found that the cardiomyocyte injury did not meet the characteristics of MIRI in vivo, neither further injury caused by SIR treatment nor pathophysiological characteristics produced by SIR treatment. In the future, these six OGD-NR in vitro models on H9c2 cells may no longer be suitable for simulating in vivo MIRI research.
Limitations
There are some limitations in our design, such as a small experimental size of five-time points. As the duration of the six models assessed in our experiment from more than 400 published papers range from 2 to 24 h, which is really a wide range, we only included five different durations of SI treatments and the corresponding SIR groups for each assessed model. It would have excluded all the possible effects between these time points. In addition, we could not cover all cell types used in the past experiments of MIRI, just studied the most commonly used, which also lead to limitations.
In conclusion, our results confirm that these 6 in vitro OGD-NR models using H9c2 cells assessed in this experiment can not cause or imitate the morphological and pathophysiological characteristics of MIRI in vivo. They maybe not suitable for MIRI-related study.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
The work was supported by grants from the major projects of Scientific and Research Found of Plastic Surgery Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College (No. Z2017001) and the National Natural Science Foundation of China (No. 81470019).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors would like to thank the assistance of Professor Zhao for the kind gift of macrophages.
Footnotes
Edited by: Li-Shao Guo
REFERENCES
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol. 1960;70:68–78. [PubMed] [Google Scholar]
- 3.Xia Z, Li H, Irwin MG. Myocardial ischaemia reperfusion injury: The challenge of translating ischaemic and anaesthetic protection from animal models to humans. Br J Anaesth. 2016;117(Suppl 2):ii44–62. doi: 10.1093/bja/aew267. doi: 10.1093/bja/aew267. [DOI] [PubMed] [Google Scholar]
- 4.Vidavalur R, Swarnakar S, Thirunavukkarasu M, Samuel SM, Maulik N. Ex vivo and in vivo approaches to study mechanisms of cardioprotection targeting ischemia/reperfusion (i/r) injury: Useful techniques for cardiovascular drug discovery. Curr Drug Discov Technol. 2008;5:269–78. doi: 10.2174/157016308786733555. doi: 10.2174/157016308786733555. [DOI] [PubMed] [Google Scholar]
- 5.Almohammedi A, Kapetanaki SM, Wood BR, Raven EL, Storey NM, Hudson AJ. Spectroscopic analysis of myoglobin and cytochrome c dynamics in isolated cardiomyocytes during hypoxia and reoxygenation. J R Soc Interface. 2015;12 doi: 10.1098/rsif.2014.1339. pii: 20141339 doi: 10.1098/rsif.2014.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boccalini G, Sassoli C, Formigli L, Bani D, Nistri S. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: Involvement of the Notch-1 pathway. FASEB J. 2015;29:239–49. doi: 10.1096/fj.14-254854. doi: 10.1096/fj.14-254854. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Jia KY, Zhang HL, Fu J. MiR-195 enhances cardiomyocyte apoptosis induced by hypoxia/reoxygenation injury via downregulating c-myb. Eur Rev Med Pharmacol Sci. 2016;20:3410–6. [PubMed] [Google Scholar]
- 8.Habener A, Chowdhury A, Echtermeyer F, Lichtinghagen R, Theilmeier G, Herzog C. MitoNEET protects HL-1 cardiomyocytes from oxidative stress mediated apoptosis in an in vitro model of hypoxia and reoxygenation. PLoS One. 2016;11:e0156054. doi: 10.1371/journal.pone.0156054. doi: 10.1371/journal.pone.0156054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muscari C, Gamberini C, Bonafe’ F, Giordano E, Bianchi C, Lenaz G, et al. Evaluation of cellular energetics by the pasteur effect in intact cardiomyoblasts and isolated perfused hearts. Mol Cell Biochem. 2004;258:91–7. doi: 10.1023/b:mcbi.0000012839.79429.42. [DOI] [PubMed] [Google Scholar]
- 10.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piper HM, Abdallah Y, Schäfer C. The first minutes of reperfusion: A window of opportunity for cardioprotection. Cardiovasc Res. 2004;61:365–71. doi: 10.1016/j.cardiores.2003.12.012. doi: 10.1016/j.cardiores.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Hüser M, Stegemann E, Kammermeier H. Is enzyme release a sign of irreversible injury of cardiomyocytes? Life Sci. 1996;58:545–50. doi: 10.1016/0024-3205(95)02262-7. doi: 10.1016/0024-3205(95)02262-7. [DOI] [PubMed] [Google Scholar]
- 13.Gaucher S, Jarraya M. Technical note: Comparison of the PrestoBlue and LDH release assays with the MTT assay for skin viability assessment. Cell Tissue Bank. 2015;16:325–9. doi: 10.1007/s10561-014-9478-1. doi: 10.1007/s10561-014-9478-1. [DOI] [PubMed] [Google Scholar]
- 14.Pasque MK, Wechsler AS. Metabolic intervention to affect myocardial recovery following ischemia. Ann Surg. 1984;200:1–2. doi: 10.1097/00000658-198407000-00001. doi: 10.1097/00000658-198407000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschuld RA, Hostetler JR, Brierley GP. Response of isolated rat heart cells to hypoxia, re-oxygenation, and acidosis. Circ Res. 1981;49:307–16. doi: 10.1161/01.res.49.2.307. doi: 10.1161/01.res.49.2.307. [DOI] [PubMed] [Google Scholar]
- 16.Hearse DJ, Humphrey SM, Chain EB. Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: A study of myocardial enzyme release. J Mol Cell Cardiol. 1973;5:395–407. doi: 10.1016/0022-2828(73)90030-8. doi: 10.1016/0022-2828(73)90030-8. [DOI] [PubMed] [Google Scholar]
- 17.Siegmund B, Koop A, Klietz T, Schwartz P, Piper HM. Sarcolemmal integrity and metabolic competence of cardiomyocytes under anoxia-reoxygenation. Am J Physiol. 1990;258:H285–91. doi: 10.1152/ajpheart.1990.258.2.H285. doi: 10.1152/ajpheart.1990.258.2.H285. [DOI] [PubMed] [Google Scholar]
- 18.Siegmund B, Klietz T, Schwartz P, Piper HM. Temporary contractile blockade prevents hypercontracture in anoxic-reoxygenated cardiomyocytes. Am J Physiol. 1991;260:H426–35. doi: 10.1152/ajpheart.1991.260.2.H426. doi: 10.1152/ajpheart.1991.260.2.H426. [DOI] [PubMed] [Google Scholar]
- 19.Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–41. doi: 10.1016/s0022-2828(03)00043-9. doi: 10.1016/s0022-2828(03)00043-9. [DOI] [PubMed] [Google Scholar]
- 20.Raedschelders K, Ansley DM, Chen DD. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Ther. 2012;133:230–55. doi: 10.1016/j.pharmthera.2011.11.004. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Ibáñez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–71. doi: 10.1016/j.jacc.2015.02.032. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 22.Bagheri F, Khori V, Alizadeh AM, Khalighfard S, Khodayari S, Khodayari H. Reactive oxygen species-mediated cardiac-reperfusion injury: Mechanisms and therapies. Life Sci. 2016;165:43–55. doi: 10.1016/j.lfs.2016.09.013. doi: 10.1016/j.lfs.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Moris D, Spartalis M, Tzatzaki E, Spartalis E, Karachaliou GS, Triantafyllis AS, et al. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann Transl Med. 2017;5:324. doi: 10.21037/atm.2017.06.17. doi: 10.21037/atm.2017.06.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonaventura A, Montecucco F, Dallegri F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur J Clin Invest. 2016;46:590–601. doi: 10.1111/eci.12633. doi: 10.1111/eci.12633. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Dorado D, Inserte J, Ruiz-Meana M, González MA, Solares J, Juliá M, et al. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation. 1997;96:3579–86. doi: 10.1161/01.cir.96.10.3579. doi: 10.1161/01.cir.96.10.3579. [DOI] [PubMed] [Google Scholar]
- 26.García-Dorado D, Théroux P, Desco M, Solares J, Elizaga J, Fernandez-Avilés F, et al. Cell-to-cell interaction: A mechanism to explain wave-front progression of myocardial necrosis. Am J Physiol. 1989;256:H1266–73. doi: 10.1152/ajpheart.1989.256.5.H1266. doi: 10.1152/ajpheart.1989.256.5.H1266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental grouping and flow chart. (1) To measure cell morphology, LDH release and ATP levels, for the models 1-6, 5 different SI durations (3 h, 6 h, 9 h, 12 h and 15 h) were designed, and SI, SIR-0.5 h, SIR-2 h and SIR-12 h groups were included. The reasons for choice of different SI durations are that the injury by SI in the models 1-6 is slight by SI-3 h treatment, and then aggravate with the prolongation of SI duration, and becomes severe at SI-15 h. (2) To measure the levels of ROS, MMP and inflammatory cytokines, a 6 h duration of SI treatment was chosen for the models 1-6. For each model, C, SI and SIR-0.5 h groups were included. The injury by SI-6 h treatment in the models 1–6 is moderate. Reperfusion injury seldom occurs when ischemia injury is too slight or severe in vivo, 6 h of SI duration was chosen. SI: Simulated ischemia; SIR: Simulated-ischemia reperfusion; LDH: Lactate dehydrogenase; ATP: Adenosine triphosphate; ROS: Reactive oxygen species; MMP: Mitochondrial membrane potential.
The cell morphology of SI, SIR-0.5 h and SIR-12 h groups with different durations of SI treatment when using the H9c2 cells. Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 3 h in the models 1–6 (a); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 9 h in the models 1–6 (b); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 12 h in the models 1-6 (c); Cell morphology of SI, SIR-0.5 h and SIR-12 h groups when duration of SI treatment was 15 h in the models 1-6 (d). Scale bars, 100 μm. SI: Simulated ischemia; SIR: Simulated ischemia-reperfusion.