

Abstract
Efficient intracellular delivery of target macromolecules remains a major obstacle in cell engineering and other biomedical applications. We discovered a unique cell biophysical phenomenon of transient cell volume exchange by using microfluidics to rapidly and repeatedly compress cells. This behavior consists of brief, mechanically induced cell volume loss followed by rapid volume recovery. We harness this behavior for high-throughput, convective intracellular delivery of large polysaccharides (2000 kDa), particles (100 nm), and plasmids while maintaining high cell viability. Successful proof of concept experiments in transfection and intracellular labeling demonstrated potential to overcome the most prohibitive challenges in intracellular delivery for cell engineering.
Subject terms: Microfluidics, Cell deformation, Delivery, Cell engineering
Graphical abstract
This microfluidic device implements the innovative mechanism of cell volume exchange for convective transfer (VECT) of large macromolecules to the cell interior.
Introduction
Studies of the physical response of cells to deformation using micropipettes, microcantilevers, and microfluidic manipulations have shown substantial change in cell shape but not cell volume. Cell manipulations due to significant deformations of up to 85% strain applied across a range of timescales from ~10 μs to >1 s [1–7] have described cell deformation and shape change but have not described cell volume change [4,7–11].
In this study, we have discovered a new cell behavior of transient and significant (up to 30%) cell volume change in response to large magnitude deformations with ultrafast timescales (~10 μs) without impairing cell viability. We attained fast deformations by rapidly flowing cells through microfluidic constrictions with an abrupt, stepwise compression profile. To characterize this new behavior, we employed high resolution video microscopy and quantitative fluorescent marker delivery. We found that volume change increased with smaller cell compression gap, and that cells were partially able to recover their volume on the time scale of ~1 ms after each compressive event and fully recover shape and volume within 100 ms. Characterization of cell integrity, viability, and related gene expression demonstrated no detrimental effects even for volume change up to 30%.
This surprising ability of cells to rapidly exchange fluid with their surroundings in response to ultrafast mechanical compressions opens a potent new way to deliver large extracellular molecules and particles into cells. We utilized this method of cell volume exchange for convective transfer (VECT) to intracellularly deliver molecules and particles suspended in extracellular fluid. We demonstrated rapid delivery into human cells of a variety of molecule types and sizes, including dextran (4–2000 kDa), plasmids, mRNA, nanoparticles, and even 100 nm beads. The ability to efficiently deliver large molecules contrasts with currently described delivery methods that rely on diffusion for transmembrane transfer of molecules, which is inefficient for large macromolecules [6,12–17]. Thus, this new phenomenon of cell volume exchange under ultrafast mechanical deformation enables a multitude of highly valuable cell engineering processes.
Results
Microfluidic cell deformation
Cell deformation was caused by microfluidic flow through ridges with rectangular cross-section that were repeated within a microchannel to precisely exert abrupt and brief compressions upon cells. Hydrodynamic forces maintained high cell velocity throughout multiple constrictions, while the angled ridges cleared dead cells and clusters of cells which could cause occlusions [18–20]. As cells encountered the rectangular ridges, abrupt shape change was observed as cells compress under the ridges to conform to a gap that is smaller than their diameter (Fig. 1a). Cell compression time was determined by convolution of the cell with a steep edge of the ridge (<1 μm as determined by optical profilometry) at the measured cell velocity (~100 mm/s). During this time, cells were observed to compress vertically up to 50%, for a vertical compression velocity on the order of 1 m/s. The sudden shape change caused by the abrupt deformation structure of the rectangular ridge was quantitatively analyzed by high speed video analysis (Fig. 1b, Supplementary Video 1).
Figure 1.
Overview of device and cell volume measurement. (a) Profilometric image of the microfluidic channel layout with diagonal ridges. The arrow indicates cell flow direction. (b) Overlay of a single cell (outlined) at multiple positions passing through the ridges. (c) Image analysis of a single cell inside the device. The schematic diagram of a cell at (i) the captured top view at each respective position; (ii) three dimensional schematic representation of the cell at positions before and during compression under first two ridges (iii) approximation of the side view of the cell based on channel height at the corresponding positions; overlaid view of the cell at different positions (iv) top view; (v) spherical projection of cells with same volume as uncompressed and compressed conditions and (vi) side view. (d) Percent of cell volume that was lost under the first ridge increased with smaller device ridge gap, n>250, bars are interquartile range. (e) Normalized volume of cells at different ridge positions in the channel, n≥45, bars are standard deviation. (f) Measured cell size showed minimal impact by device, viability stain showed device processing caused <5% cell death, n = 2.
Measurement of cell volume change
Using a computational cell deformation model [19] combined with area analysis of high speed videos of individual cells in the microfluidic channel, we evaluated the change in cell volume at several points in the channel (Fig. 1c). Measurements were taken of K562 myelogenous leukemia cell area before compression, and then when entirely constrained under each ridge (Fig. 1ci). Before compression, each cell was approximated as an ellipsoid, while the cell shape under each ridge was approximated to a truncated ellipsoid, as determined by a cell deformation model [19] (Fig. 1cii,iii). The compressed cell height was equal to the ridge gap, which was independently measured by profilometry. Due to the uncertainty of cell shape and orientation between ridges, the cell volume between ridges cannot be deduced from its area measurement.
From the known gap and modeled cell shape, we determined the cell volume before and during compressions. An overlay of cell area measurements at the various positions shows subtle area change, suggesting that the vertical constraint from the ridge mainly accounts for the volume change (Fig. 1civ). A view of spherical cells with the same volume as the compressed cells visualizes the volume change when projected on the pre-compression cell (Fig. 1cv). Cells exhibited the most significant volume decrease at the first ridge due to the sudden change in shape from ellipsoid to truncated ellipsoid (Fig. 1cvi). Decreasing the gap size of the microfluidic device led to a greater volume decrease between the pre-compression cell and the cell compressed under the first ridge (Fig. 1d). The cell volume proceeded to slightly decrease with each subsequent compression to a plateau volume after approximately 8 ridges (Fig. 1e).
While the volume was observed to decrease by up to 30% during compressions, cells were quickly restored to their initial size with little impact on cell integrity, viability, and related gene expression. After microfluidic processing, cell culture and expansion was successfully conducted with no change in cell growth rate. Analysis of still images of >800 cells immediately after microfluidic processing shows <3% change in mean cell size compared to cells without microfluidic processing (Fig. 1f). Similarly, ethidium homodimer-1 (EthD-1) staining of processed cells showed <3% cell death compared to the No Device group (Fig. 1f). We used RT PCR immediately after microfluidics to further quantify that the compressions in the microchannel did not impact the expression of apoptotic, cytoskeletal, and other signaling genes (Supplementary Fig. 1). A separate, detailed study on cell viability after rapid compressions, including expression of apoptotic genes, was consistent with this observation [21]. These results suggested that cells recovered normal volume and function after the brief volume loss.
Characterizing volume exchange through molecular delivery
The volume reduction of compressed cells indicated that a portion of cytosol was expelled from the cell interior through a mechanically compromised cell membrane. Cell volume recovery, on the other hand, requires extracellular fluid to enter the cell. Since the video analysis does not allow us to evaluate cell volume in between the ridges, we further characterized the dynamics of volume exchange and fluid transfer through the compromised cell membrane using fluorescently labeled dextran (Sigma-Aldrich) as a tracker molecule. Dextran of various sizes was added to the cell suspension immediately before compression experiments. We hypothesized that cell relaxations after each compression will cause the extracellular fluid to enter the cell interior transporting dispersed fluorescent molecules, and that the molecules will partially remain in the cell interior after consecutive compressions serving as an indicator of volume exchange (Fig. 2a).
Figure 2.
Characterizing molecule delivery. (a) Cross-sectional view of a cell undergoing compression under the ridges and relaxation, illustrating volume exchange and molecule uptake. (b) Confocal microscopy images of a live single cell delivered with 2000 kDa TRITC-dextran with diffuse fluorescence profile throughout the cell interior. Scale bar 5 μm. (c) Molecule delivery increased with smaller size of ridge gap through which cells pass. (d) Molecule delivery was greater with increased volume change. K562 cells were processed in 7-ridge devices with 2000 kDa FITC-dextran. (e) Molecule delivery decreased with faster flow rate. However, 200 μm spacing between ridges consistently demonstrated higher delivery than 100 μm spacing across several flow rates. (f) Overall trend indicates delivery increased with greater cell relaxation time between the ridges until a plateau was observed.
Confocal imaging determined that molecular delivery by cell VECT was dispersed throughout the cell interior, suggesting non-endocytic delivery (Fig. 2b) [22]. We experimentally observed that greater compressions from smaller ridge gaps resulted in higher delivery of fluorescent molecules (Fig. 2c, Supplementary Fig. 2). The fluorescent signal showed a positive correlation with the measured volume loss associated with the gap size (Fig. 2d). The measured delivery to cells with smaller gap dimensions (5.6 μm) was confounded at the conditions tested due to cells flowing around the ridges rather than passing through the smaller gap underneath the ridges. Ridges with gaps larger than the K562 cell diameter (14.5 ± 1.5 μm) did not cause volume change, and showed lower delivery of 2000 kDa dextran macromolecules (Fig. 2c) in a manner consistent with existing studies that used fluid shear mechanoporation to induce membrane pores, allowing diffusive delivery of molecules [13,19]. Increasing gap size from 16 μm to 20 μm does not change delivery, but gaps smaller than the cell diameter significantly increase delivery with smaller gap. Therefore, a gap smaller than the relaxed cell diameter can be identified as a threshold at which delivery behavior changes.
Based on the correlation between volume loss and molecule delivery, we hypothesized that altering the time that the cell relaxes as it moves between consecutive constrictions can affect the volume uptake and, therefore, molecular delivery. The relaxation time between ridges was controlled either by varying the ridge spacing or the flow rate. We observed that increased flow rate resulted in decreased delivery, while the 200 μm spacing between ridges consistently resulted in higher delivery than the 100 μm spacing (Fig. 2e, Supplementary Fig. 3). Therefore, the increased relaxation time between ridges led to greater delivery (Fig. 2f), despite differences in flow speed and ridge spacing. We also observed that molecular delivery showed diminishing returns past a certain duration of cell relaxation between ridges (~1 ms), suggesting a saturation point of relaxation (Fig. 2f). This result is in contrast with diffusive delivery, which increases with faster flow rates [6,23].
Characterization of convective molecular delivery
To further confirm that intracellular delivery occurs due to cell volume change, we tested whether cell VECT is affected by the size of the molecule. Since diffusion rate is inversely proportional to molecule size, diffusive delivery typically shows lower efficiency for larger macromolecules [12–17]. In contrast, cell VECT demonstrated intracellular delivery with high efficiency (~90% of cells uptake molecules) regardless of molecule size for the range tested (Fig. 3a,b). This study used equal mass per volume of molecules ranging from 4 kDa, roughly the molecular weight (MW) of a small molecule drug, to 2000 kDa. This size-independent delivery supported our hypothesis that molecule uptake was achieved predominantly by advection of material from outside the cell due to cell volume recovery, rather than molecular diffusion through membrane pores.
Figure 3.
Investigating properties of cell VECT. (a) Delivery was independent of molecule size for the range tested (4–2000 kDa FITC-dextran). 10.2 μm gap device with 30 ridges used. (b) ~90% of cells showed positive delivery regardless of molecule size. (c, d) Molecule delivery was greater with increasing number of constrictions. The trend plateaued after 14 ridges. 9 μm gap devices were used. (e, f) Cells processed with 1–300 μg/mL of 2000 kDa FITC-dextran showed a linear trend for median cell fluorescence intensity. 10.2 μm gap device with 30 ridges used. (g) FITC-dextran delivered to K562 cells was removed by processing the cells in the device with a FITC-free buffer, n=2. 7 μm gap device with 22 ridges used. (h) Isolation of delivery inside the channel demonstrated that >80% of cells successfully uptake molecules during the brief time inside the channel. Only ~33% of cells showed delivery after incubating in a molecule-rich bath upon leaving the device, n = 3. 9 μm gap device with 14 ridges used.
The use of multiple ridges greatly increased volume exchange and molecular delivery to the cells. We observed a positive and non-linear correlation between the number of ridges and molecule delivery, which saturated at 14 ridges for these experimental conditions (Fig. 3c,d). The final molecular delivery was also found to be linearly dependent on the extracellular concentration (Fig. 3e,f), indicating that saturation of the intracellular and extracellular molecule concentration was reached.
To further explore the hypothesis that cell VECT causes the cytosol to reach equilibrium with extracellular molecule concentration, we processed previously dextran-positive cells with dextran-free buffer to remove the dextran from within the cells. We first delivered 2000 kDa FITC-dextran to K562 cells using VECT, then resuspended these delivered cells in FITC-free buffer and processed them in the device again for the Removal group. We found that the Removal group has a mean fluorescence intensity that matches the No Device group, indicating that this method is highly effective in removing previously delivered molecules (Fig. 3g). These results support our assertion that cell VECT achieved molecule concentration equilibrium and can remove unbound molecules from the cell interior, a capability not demonstrated with diffusive delivery [23].
To determine the time scale at which delivery occurs during cell VECT, we designed an experiment to analyze the relative amount of delivery that occurs during the brief time (<0.1 s) of cell compressions inside the device channel and immediately after leaving the device. Delivery inside the channel was determined by flowing K562 cells through the channel with the target delivery molecules, 2000 kDa FITC-dextran, and then inhibiting delivery after the channel by immediately diluting the outlet sample into a molecule-free bath. Delivery after the channel was isolated by flowing cells through the channel in the absence of target molecules, then exposing the cells to a molecule-rich bath immediately after leaving the channel. Molecules were delivered to over 80% of cells during their <0.1 s transit through the channel, while only ~33% of cells exhibited delivery when provided dextran immediately after transit through the compressions, even after incubation in the outlet well for >10 minutes. A threshold set at the brightest 10% of the No Device control was used to define the lower bound of fluorescence for positive delivery (Fig. 3h). The high delivery obtained primarily during compressions inside the channel supports that cell VECT delivers large macromolecules by fluid exchange during compression and relaxation.
Modeling volume exchange and molecular delivery
To better understand the relationship between volume change and intracellular delivery, we constructed a simple mathematical model of molecular delivery due to repeated volume exchange events. The model assumes the cell interior and exterior are well mixed and incompressible liquids. Therefore, as cells are compressed to volume VC under the ridge, a corresponding amount of intracellular volume is driven out of the cell carrying out a mass of target molecules dictated by the intracellular concentration of that species. Conversely, as a cell recovers lost volume, any species outside the cell are drawn in at a rate set by their external concentration and the time-dependent cell volume recovery V(t) (Fig. 4a). The cell fluorescence intensity is used to represent the amount of intracellular molecule delivery for the model and experimental results, with the assumption that cell fluorescence intensity is proportional to intracellular molecule concentration.
Figure 4.
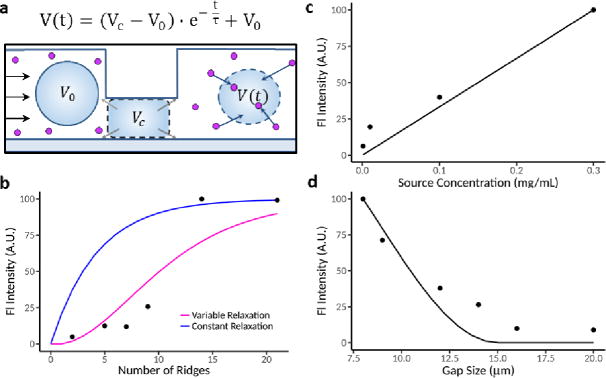
Development of mechanistic model to incorporate cell volume exchange. (a) An illustration showing the assumptions made in our model. Before encountering any ridges, cells have initial volume V0. Under each ridge, we assumed cell volume decreased to some constant VC which was dependent on the ridge gap. After clearing the ridge, we assumed that the cell volume increases with time t after ridge compression and approaches V0 asymptotically with timescale τ. Cell relaxation was assumed to either be constant or to occur more rapidly with more compressions. Total volume exchange is therefore dictated by the amount that the cell relaxes between ridges. (b-d) Comparisons between the median fluorescence intensity observed in our experiments (dots) to the predictions of our model (solid lines).
We considered a model in which cells behave as a Kelvin-Voit viscoelastic material and expand after compression to exponentially to approach their original volume V0. We expressed this asymptotic recovery using an exponential function where t is the time elapsed after the most recent compression (Fig. 4a, Supplementary Note 1). We first assumed constant volume exchange per ridge, where the factor τ, the time for cells to recover 66.7% of lost volume, is independent from the number of compressions. However, we determined that the results from our molecule delivery experiments are inconsistent with constant volume exchange per ridge (Fig. 4b).
We next considered a model in which volume exchange increases with consecutive compressions (Fig. 4b). We assume that relaxation time τ decreases with repeated compressions, asymptotically approaching some final value (Supplementary Note 1). Using atomic force microscope (AFM) cell relaxation measurements, we observed that cell shape recovery can indeed occur more rapidly after several compressions (Supplementary Fig. 4). We then fit this model to our experimental data, which yielded τ that decreased from an initial value of ~1 s to <1 ms after many ridges (Supplementary Fig. 5). Prior experiments suggest that relaxation of cells can indeed occur at time scales as slow as >10 s and as fast as ~10 μs with different compression conditions [2,4]. The experimental results by cell VECT are consistent with the model of molecule delivery in which a nonlinear positive dependence is observed with increasing number of ridges, a linear dependence occurs with the source concentration, and a threshold gap size smaller than the cell diameter is needed for delivery (Fig. 4b–d). Based on this result and existing studies, we hypothesize that repeated compressions by multiple ridges can lead to cell biophysical changes that result in faster cell deformation and recovery [4,24].
Applications of VECT to intracellular delivery
The application of cell VECT can address important limitations of microfluidic delivery platforms, particularly those that primarily use diffusive transport. To demonstrate the capabilities of the use of VECT as a highly efficient delivery platform for transfection agents, we successfully delivered Cy5-mRNA (TriLink) into K562 cells. The cells were stained with Hoechst nucleus stain to visualize the intracellular localization of the Cy5-mRNA (Fig. 5a). Using confocal microscopy, the mRNA was shown to permeate the cell interior. A No Device control of K562 cells exposed to Cy5-mRNA without device processing was imaged for comparison. A proof of concept transfection experiment successfully induced EGFP expression after delivery of EGFP mRNA (TriLink) and EGFP plasmid (OZ Biosciences) to K562 cells (Fig. 5b, Supplementary Fig. 6).
Figure 5.
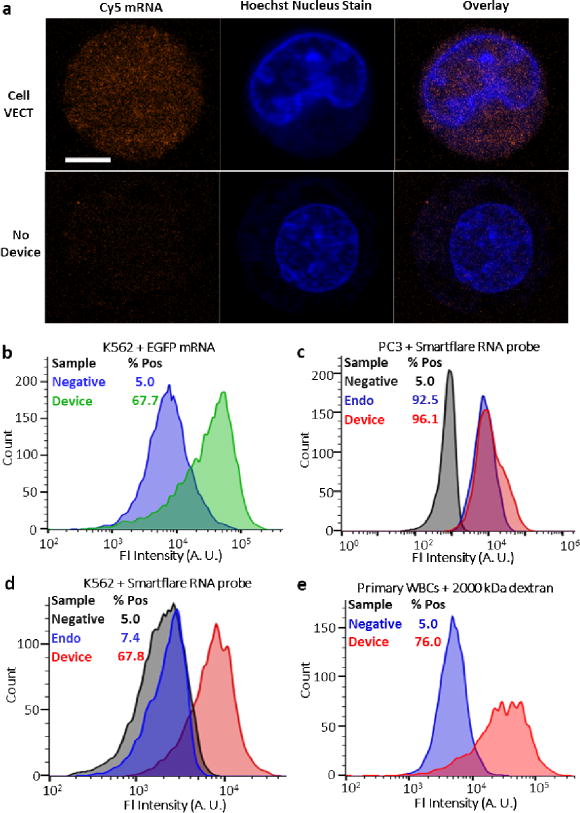
Using cell VECT to deliver a variety of molecules to cells. (a) Confocal microscopy showed diffuse delivery of Cy5-labeled mRNA throughout the interior of a fixed cell with nucleus staining. No Device control showed no such delivery. Scale bar 5 μm, n = 2. Images shown are representative of ~50 cell images. (b) Transfection of EGFP mRNA in K562 cells with substantial efficiency. (c) Device delivery of SmartFlare RNA microparticle probes to PC3 cells was competitive with the established method of 24 hr endocytosis. (d) Device successfully delivered SmartFlare to K562 cells which do not endocytose SmartFlares. (e) Successful delivery of 2000 kDa FITC-dextran to primary leukocytes isolated from discarded donor blood.
We also tested this platform’s potential applications for intracellular labeling and analysis by delivering SmartFlare Live Cell RNA probes (Millipore) to detect GAPDH RNA in K562 cells and adherent PC3 prostate cancer cells. Delivery to PC3 cells was competitive with the established method of 24 hr endocytosis, and was completed in less than 30 mins (Fig. 5c). Importantly, K562 cells, which do not uptake SmartFlare particles through endocytosis, showed successful delivery using cell VECT (Fig 5d). Our success in delivering to PC3 and K562 cells demonstrated this method’s robustness for delivery to both adherent and nonadherent cells.
We also successfully delivered 100 nm diameter fluorescent beads to K562 cells as a demonstration of this method’s ability to deliver extremely large particles (Supplementary Fig. 7). To address applications in cell engineering, we also used cell VECT to transfect and deliver large macromolecules to primary peripheral blood mononuclear cells (PBMCs) isolated from whole blood (Fig. 5e, Supplementary Fig. 8). Furthermore, because the design of the angled ridges can avoid cell clogging, the cell VECT processing was easily scaled up to multichannels to successfully process 50 million cells in 10 minutes without clogging. The demonstrated success in transfection and intracellular labeling for multiple cell types revealed the potential of this platform to compete with established delivery techniques for an array of cell engineering applications (Supplement Table S1).
Discussion
By using microfluidics to precisely induce rapid, brief, large strain compressions, we elucidated the surprising phenomenon of temporary cell volume exchange that maintains cell integrity, viability, and function. We discovered a behavior wherein cells initially undergo sudden volume loss followed by fast volume recovery. We found that induced volume change is greater for larger strains imposed through smaller constrictions. We also found that increased volume exchange required multiple ridges spaced such that there was sufficient time for cells to recover lost volume between each ridge. We used this effect of volume change and relaxation as a new approach to deliver molecules to cells. Specifically, rapid compression-driven volume loss worked in conjunction with cell relaxation to convectively drive volume and molecules into the cell interior.
The physical cause of this surprising cell behavior can be explained by considering the relevant forces imposed on the cell by the ridges. The sudden inertial compression under a ridge with stepwise profile is equivalent to a high velocity (~1 m/s) vertical impact on the cell to disrupt the membrane in a manner akin to a droplet splatter upon a surface. The subsequent physical constriction of the cell under the ridge results in rapid transfer of momentum to the liquid of the cell interior to drive fluid volume out of the cell. The brief nature of this compression causes cells to relax on a rapid time scale to uptake volume after compression. The observed rapid recovery is consistent with rapid, poroelastic recovery behavior of the cytoplasm at short time scales (<0.5 s) after brief compression [7,25]. The ability of the cytoskeleton to regulate cell volume and retain solutes could explain the minimal impact of cell VECT on cell viability despite the initial volume loss [26].
Cell VECT was shown to be distinct from current diffusive mechanoporation platforms, both in mechanism and capability. Diffusive microfluidic mechanoporation methods used gradual constrictions to impart shear stress on cells in a manner that facilitates smooth cell flow and thus slower deformation [4,7–11]. The compression creates a shear force on the cell membrane leading to membrane poration and extracellular molecular diffusion into the cell interior. While diffusion is a universal transport mechanism, it imposes constraints on delivery due to the inverse relationship between diffusivity and molecule size. Indeed, diffusive approaches to microfluidic mechanoporation have shown limited efficiency in the delivery of large macromolecules [12–17].
In our studies, we found cell VECT utilizes an advection-dominated molecular driving mechanism to efficiently deliver molecules of a wide range of sizes and structures for many cell types, while maintaining high viability. The microfluidic approach avoids many of the prohibitive drawbacks of detrimental changes to cell state associated with using chemical, viral, or electrical processing [27–33]. The simplicity of use and successful delivery of an array of biologically relevant macromolecules to various cell types demonstrated great potential for a wide range of highly valuable biomedical applications.
Conclusions
In this study, we discovered a new cell behavior wherein multiple, rapid, high strain compressions caused cell volume change and relaxation without impacting cell viability. We found that this volume exchange caused extracellular molecules to be convected into the cell interior. We refer to this process as cell VECT, a powerful driving mechanism for intracellular delivery, especially in comparison to diffusion alone. Cell VECT enables new applications for microfluidic molecular delivery, including high-throughput delivery of large macromolecules and particles. We have determined the device and experiment parameters that govern cell VECT and provide basic understanding to mechanically induced cell volume exchange. This work has elucidated a new cell phenomenon with great potential to serve as a nearly universal intracellular delivery platform for a variety of biotechnology applications.
Methods
Device design
The microfluidic device design used constrictions in the form of angled ridges in a single large channel for the rapid processing of high numbers of cells. The large channel allowed a multitude of cells to pass simultaneously under each ridge while hydrodynamic drag forces maintained cell velocity through the constrictions, allowing cell processing to continue rapidly even after many constrictions [18–20]. The angled ridges also served as an escape mechanism for nonviable cells and cell aggregates that would otherwise clog the device or dilute the processed cell population [21]. Therefore, this design functioned effectively even with localized clogs, and rapidly self-cleared. A multi-channel design of this device successfully processed 50 million cells in 10 minutes without clogging.
Fabrication of microfluidic channels
The microfluidic features of this device were molded onto polydimethylsiloxane (PDMS) and plasma bonded to a glass slide. A reusable SU-8 mold was made using standard two-step photolithography on a silicon wafer. The processes of fabricating the molds and characterizing the dimensions of their features were detailed in previous publications[18–20]. Constriction gaps of 50–60% of the average relaxed cell diameter (14.5 ± 1.5 μm) were used for optimal delivery, but gaps of 40–130% of the average cell diameter were also studied [19]. The cell inlet flow directed cells through the constrictions, preventing the cells from preferentially flowing around the ridges without compressing. To fabricate the devices, a 10:1 ratio of PDMS and crosslinking agent was mixed and poured onto the SU-8 mold to form the microfluidic channel features by replica molding. The PDMS was then degassed in a vacuum chamber and cured for 6 hrs at 60°C. The cooled PDMS was then removed from the molds and outlets and inlets were punched using biopsy punches. The PDMS was then bonded to sonicated glass slides using a plasma bonder (PDC-32G Harrick) followed by 1 hr in a 60°C oven. After cooling, the channels were passivated using 1% bovine serum albumin (BSA) for an overnight incubation at 4°C.
Cell culture
K562 cells (CCL-243) from ATCC were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. PC3 prostate cancer cells (CRL-1435), a gift from BD Biosciences, were cultured in F-12K with 10% FBS and 1% penicillin-streptomycin. The cells were incubated at 37°C with 5% CO2. The PC3 cells were passaged using 0.25% Trypsin-EDTA. Deidentified and discarded blood sample was collected from Lam Lab, Georgia Institute of Technology, under an institutional review board (IRB) approved study for laboratory research on discarded clinical samples and all methods were performed in accordance with the relevant guidelines and regulations. Primary leukocytes were isolated from whole donor blood by density gradient centrifugation. Whole donor blood was centrifuged at 700 RCF for 10 mins with Ficoll density centrifugation media and the concentrated leukocyte band (buffy coat) was collected.
Microfluidic experimental setup
A cell flow buffer consisting of DPBS (−/−) with 0.1% BSA, 0.04% EDTA, and trace Tween 20 was used to maintain a single-cell suspension throughout the experiment. Experiments performed using pure DPBS (−/−) and serum-free RPMI-1640 without BSA, EDTA, or Tween determined that these agents had no observable effect on molecular delivery. Transfection and RNA probe delivery experiments were done using Opti-MEM and serum-free RPMI-1640, respectively. The cells were isolated from culture media and resuspended in buffer at ~5×106 cells/mL with the desired concentration of target molecules. Multiple sizes of FITC-dextran were purchased from the same maker (Sigma-Aldrich) with very little variation in labeling fraction. The dextran molecules had an average FITC/dextran molar ratio of 0.00525 ± 0.0017. Our largest molecule, 2000 kDa, had a labeling fraction of 0.006, almost identical with the smallest molecule, 3–5 kDa, labeling fraction of 0.007. We also used the same mass per volume, so the mass of fluorophore in solution is the same across all molecules sizes. The cell-buffer suspension was infused into the microfluidic device at a controlled rate using syringe pumps (PHD 2000, Harvard Apparatus). A cell flow rate of ~100 mm/s through the channel was used unless the flow rate was the independent variable. Following collection from the outlets, the cells were washed 2X with DPBS (−/−) to remove residual molecules external to the cells.
High-speed video microscopy
The experiments were carried out on the stage of an inverted bright-field microscope (Eclipse Ti, Nikon), with a high-speed camera attachment (Phantom v7.3, Vision Research). High speed (~5,000 fps) videos were taken of cells during processing at various segments of the device.
Video analysis for cell volume change
To measure the cell volume inside the device, we took measurements of the cell area from video data and applied volume assumptions based on a cell deformation model. A custom cell tracking algorithm was used to automatically track the trajectory and area of cells in the video, with manual measurements used to verify. For each tracked cell, the algorithm identified all video frames where the cell was visible, and extracted the position and number of pixels it occupied (area). For each manual measurement, we took the ellipse that fit to the pixels of the sharpest gray scale intensity gradient to represent the maximum projected cell boundary. We calibrated the length scales of each image based on known ridge dimensions, which enabled us to translate the number of pixels into an area measure. For each cell, we measured the area before it entered the ridge region of the device to determine its uncompressed volume and the area when completely under each ridge to determine the compressed volumes. The volume of the unperturbed cell was taken as an ellipsoid where the projected area of the ellipse was revolved about the major axis, resulting in the minimum reasonable volume for the unperturbed cell. To take the compressed cell measurements, we applied the same revolved ellipsoid procedure to the compressed cell area and cut equal caps that represent the volume of the ellipsoid that intersected with the constraints of the ridge and channel bottom. This was considered the maximum reasonable volume for the compressed cell as it approached the cylindrical case for smaller gap sizes and collapsed back to the unperturbed ellipsoid case for larger gap sizes.
Flow cytometry
The BD Accuri C6 Flow Cytometer and FlowJo were used to characterize cell uptake of cyanine-3 (488 nm excitation and 585/40 filter) and FITC-dextran or GFP RNA or plasmid (488 nm excitation and 533/30 filter). The viability of the cells was tested by staining with 2 μM EthD-1 (Molecular Probes Inc.) solution [34,35] (640 nm excitation and 670 LP filter). Fluorescence intensity was normalized with respect to the highest intensity group. The gap size study was normalized to ~75 to reflect lower delivery due to fewer ridges (only 7 ridges), while all other studies were normalized to ~100. Each different molecule size and molecule concentration condition had a different No device control, but the same negative control. Therefore, the negative control appears on the overlayed flow cytometry histograms to provide for a clear and unchanging metric for comparison. A threshold fluorescence intensity set to include the brightest 10% of the No device control was used to gate for positive delivery, unless otherwise stated.
Confocal microscopy
Confocal microscopy of live cells with tetramethylrhodamine (TRITC)-dextran (555 nm excitation and 560–800 nm detection) was performed using the Zeiss LSM 700. A 63X Apochromat oil lens was used to image fixed K562 cells stained with Hoechst (405 nm excitation and 300–629 nm detection) per manufacturer protocol and delivered with Cy5-mRNA (639 nm excitation and 629–800 nm detection) by cell VECT. The Zeiss 710 NLO with a 40X water lens was used to image live K562 cells with 100 nm nanoparticles (514 nm excitation and 527–601 nm detection) (ThermoFisher).
Atomic force microscopy
Measurements of the viscous relaxation of individual cells during repeated compressions were performed using an MFP-3D AFM (Asylum Research) in concert with an inverted optical microscope (Nikon Ti) to optically align the AFM probe with the center of each cell. The probes used in this study were MLCT-O10-D probes with a nominal spring constant of 0.03 N/m. The AFM cantilever interacted with the cells via a 15 μm diameter PMMA microsphere. Cantilever calibration was performed using the thermal vibration method against a glass surface. K562 cells in culture media were adhered to the surface of a glass Fluorodish using Cell-Tak (Corning). The indentation depth was chosen to be 10 μm to simulate the strain imposed by a 5 μm gap in a microfluidic channel. The cell relaxation constant was extracted from the decay of viscous forces acting on the probe while maintaining constant indentation for 2 seconds after compression.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Mark R. Prausnitz for helpful discussions, Seung Joon Paik for device fabrication and analysis, and Wilbur Lam lab for providing discarded human blood samples. This work was supported by the NSF Stem Cell Biomanufacturing IGERT, the Wallace H. Coulter Translational Partnership Research Award, the Achievement Rewards for College Scientists (ARCS) Scholars Award, NIH award 1R21CA191243-01A1, and the NSF Graduate Research Fellowship under Grant No. DGE-1650044. We also acknowledge the core facilities at the Parker H. Petit Institute for Bioengineering and Bioscience and Institute for Electronics and Nanotechnology at the Georgia Institute of Technology for the use of their shared facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
A.L., M.I., N.S., V.V., E.W., A.A., and T.S. designed and conducted experiments and recorded footage for volume change analysis. A.L., M.I., N.S., J.J., S.B., and P.Q performed computational analyses. A.L. designed and conducted the mRNA and confocal microscopy experiments. M.I. performed the primary leukocyte experiments. N.S. designed and performed AFM experiments and developed the mathematical model. J.J., S.B., and P.Q. performed volume change video analyses. A.L., M.I., N.S., P.Q., A.A., and T.S prepared the figures and manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Bongiorno T, Chojnowski Jena L, Lauderdale James D, et al. Cellular Stiffness as a Novel Stemness Marker in the Corneal Limbus. Biophysical Journal. 2016;8(111):1761. doi: 10.1016/j.bpj.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng YX, Davis SP, Yang F, et al. Inertial Microfluidic Cell Stretcher (iMCS): Fully Automated, High-Throughput, and Near Real-Time Cell Mechanotyping. Small. 2017;28(13) doi: 10.1002/smll.201700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans EA. Structural model for passive granulocyte behaviour based on mechanical deformation and recovery after deformation tests. Kroc Found Ser. 1984;16:53. [PubMed] [Google Scholar]
- 4.Gabriele S, Benoliel AM, Bongrand P, et al. Microfluidic Investigation Reveals Distinct Roles for Actin Cytoskeleton and Myosin II Activity in Capillary Leukocyte Trafficking. Biophysical Journal. 2009;10(96):4308. doi: 10.1016/j.bpj.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lokhandwalla M, Sturtevant B. Mechanical haemolysis in shock wave lithotripsy (SWL): I. Analysis of cell deformation due to SWL flow-fields. Phys Med Biol. 2001;2(46):413. doi: 10.1088/0031-9155/46/2/310. [DOI] [PubMed] [Google Scholar]
- 6.Sharei A, Cho N, Mao S, et al. Cell squeezing as a robust, microfluidic intracellular delivery platform. J Vis Exp. 2013;81:e50980. doi: 10.3791/50980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran-Son-Tay R, Needham D, Yeung A, et al. Time-dependent recovery of passive neutrophils after large deformation. Biophys J. 1991;4(60):856. doi: 10.1016/S0006-3495(91)82119-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mokbel M, Mokbel D, Mietke A, et al. Numerical Simulation of Real-Time Deformability Cytometry To Extract Cell Mechanical Properties. ACS Biomaterials Science & Engineering. 2017 doi: 10.1021/acsbiomaterials.6b00558. [DOI] [PubMed] [Google Scholar]
- 9.Guo Q, Park S, Ma H. Microfluidic micropipette aspiration for measuring the deformability of single cells. Lab Chip. 2012;15(12):2687. doi: 10.1039/c2lc40205j. [DOI] [PubMed] [Google Scholar]
- 10.Needham D, Hochmuth RM. Rapid flow of passive neutrophils into a 4 microns pipet and measurement of cytoplasmic viscosity. J Biomech Eng. 1990;3(112):269. doi: 10.1115/1.2891184. [DOI] [PubMed] [Google Scholar]
- 11.Zheng Y, Nguyen J, Wei Y, et al. Recent advances in microfluidic techniques for single-cell biophysical characterization. Lab Chip. 2013;13(13):2464. doi: 10.1039/c3lc50355k. [DOI] [PubMed] [Google Scholar]
- 12.Sharei A, Poceviciute R, Jackson EL, et al. Plasma membrane recovery kinetics of a microfluidic intracellular delivery platform. Integr Biol (Camb) 2014;4(6):470. doi: 10.1039/c3ib40215k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallow DM, Seeger RA, Kamaev PP, et al. Shear-induced intracellular loading of cells with molecules by controlled microfluidics. Biotechnol Bioeng. 2008;4(99):846. doi: 10.1002/bit.21651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharei A, Trifonova R, Jhunjhunwala S, et al. Ex vivo cytosolic delivery of functional macromolecules to immune cells. PLoS One. 2015;4(10):e0118803. doi: 10.1371/journal.pone.0118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szeto GL, Van Egeren D, Worku H, et al. Microfluidic squeezing for intracellular antigen loading in polyclonal B-cells as cellular vaccines. Sci Rep. 2015;5:10276. doi: 10.1038/srep10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding X, Stewart MP, Sharei A, et al. High-throughput nuclear delivery and rapid expression of DNA via mechanical and electrical cell-membrane disruption. Nature Biomedical Engineering. 2017;1:0039. doi: 10.1038/s41551-017-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saung MT, Sharei A, Adalsteinsson VA, et al. A Size-Selective Intracellular Delivery Platform. Small. 2016 doi: 10.1002/smll.201601155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G, Crawford K, Turbyfield C, et al. Microfluidic cellular enrichment and separation through differences in viscoelastic deformation. Lab Chip. 2015;2(15):532. doi: 10.1039/c4lc01150c. [DOI] [PubMed] [Google Scholar]
- 19.Wang G, Mao W, Byler R, et al. Stiffness dependent separation of cells in a microfluidic device. PLoS One. 2013;10(8):e75901. doi: 10.1371/journal.pone.0075901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang G, Turbyfield C, Crawford K, et al. Cellular enrichment through microfluidic fractionation based on cell biomechanical properties. Microfluidics and Nanofluidics. 2015;4(19):987. doi: 10.1007/s10404-015-1608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Islam M, Brink H, Blanche S, et al. Microfluidic Sorting of Cells by Viability Based on Differences in Cell Stiffness. Scientific Reports. 2017;1(7):1997. doi: 10.1038/s41598-017-01807-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirschman JL, Bhosle S, Vanover D, et al. Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA-protein correlations at the level of single cells. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharei A, Zoldan J, Adamo A, et al. A vector-free microfluidic platform for intracellular delivery. Proc Natl Acad Sci U S A. 2013;6(110):2082. doi: 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoelzle DJ, Varghese BA, Chan CK, et al. A Microfluidic Technique to Probe Cell Deformability. Journal of Visualized Experiments : JoVE. 2014;91:51474. doi: 10.3791/51474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moeendarbary E, Valon L, Fritzsche M, et al. The cytoplasm of living cells behaves as a poroelastic material. Nat Mater. 2013;3(12):253. doi: 10.1038/nmat3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachs F, Sivaselvan MV. Cell volume control in three dimensions: Water movement without solute movement. The Journal of General Physiology. 2015;5(145):373. doi: 10.1085/jgp.201411297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zorko M, Langel Ü. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Advanced Drug Delivery Reviews. 2005;4(57):529. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Osumi N, Inoue T. Gene Transfer into Cultured Mammalian Embryos by Electroporation. Methods. 2001;1(24):35. doi: 10.1006/meth.2001.1154. [DOI] [PubMed] [Google Scholar]
- 29.Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Analytical and Bioanalytical Chemistry. 2010;8(397):3173. doi: 10.1007/s00216-010-3821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meacham JM, Durvasula K, Degertekin FL, et al. Physical methods for intracellular delivery: practical aspects from laboratory use to industrial-scale processing. J Lab Autom. 2014;1(19):1. doi: 10.1177/2211068213494388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. 2005;4(6):299. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 32.Joliot A, Prochiantz A. Transduction peptides: from technology to physiology. Nat Cell Biol. 2004;3(6):189. doi: 10.1038/ncb0304-189. [DOI] [PubMed] [Google Scholar]
- 33.Boissel L, Betancur M, Wels WS, et al. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;9(33):1255. doi: 10.1016/j.leukres.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barron JA, Ringeisen BR, Kim HS, et al. Application of laser printing to mammalian cells. Thin Solid Films. 2004;453:383. [Google Scholar]
- 35.Zhou HM, Du TF, Shen Y, et al. In vitro cytotoxicity of calcium silicate-containing endodontic sealers. J Endod. 2015;1(41):56. doi: 10.1016/j.joen.2014.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.