

Abstract
Background:
High-dose factor VIIa (FVIIa) is routinely used as an effective bypassing agent to treat hemophilia patients with inhibitory antibodies that compromise factor replacement. However, the mechanism by which FVIIa binds activated platelets to promote hemostasis is not fully understood. FVIIa-DVQ is an analog of FVIIa with enhanced tissue factor (TF)-independent activity and hemostatic efficacy relative to FVIIa. Our previous studies have shown that FVIIa-DVQ exhibits greater platelet binding, thereby suggesting that features in addition to lipid composition contribute to platelet-FVIIa interactions.
Objectives:
Endothelial cell protein C receptor (EPCR) also functions as a receptor for FVIIa on endothelial cells. We therefore hypothesized that an interaction with EPCR might play a role in platelet-FVIIa binding.
Methods/Results:
In the present study, we used flow cytometric analyses to show that platelet binding of both FVIIa and FVIIa-DVQ is partially inhibited in the presence of excess Protein C or an anti-EPCR antibody. This decreased binding results in a corresponding decrease in the activity of both molecules in factor Xa and thrombin generation assays. Enhanced binding to EPCR was sufficient to account for the increased platelet binding of FVIIa-DVQ compared to wild type FVIIa. As EPCR protein expression has not previously been shown in platelets, we confirmed the presence of EPCR in platelets using immunofluorescence, flow cytometry, immunoprecipitation, and mass spectrometry.
Conclusions:
This work represents the first demonstration that human platelets express EPCR, and suggests that modulation of EPCR binding could be utilized to enhance the hemostatic efficacy of rationally designed FVIIa analogs.
Keywords: coagulation, endothelial protein c receptor, factor VIIa, hemophilia, hemostasis
Introduction
Recombinant Factor VIIa (FVIIa) is routinely used to promote hemostasis in hemophilia patients who have developed inhibitory antibodies that render traditional factor replacement ineffective [1, 2]. Following injury, FVIIa/tissue factor (TF) complexes initiate hemostasis by activating factor X (FX). At pharmacologic concentrations, FVIIa also binds to activated platelets, which do not express TF [3], to enhance FXa and thrombin generation in a dose-dependent manner [4]. This TF-independent activation of FX on the platelet surface is thought to be the dominant mechanism by which high-dose FVIIa facilitates hemostasis [5–9].
While effective, FVIIa therapy does not completely normalize hemostatic function [10], and efforts to design more effective FVIIa variants have been hampered by an incomplete understanding of the mechanism(s) by which FVIIa enhances hemostasis [4–9].
The FVIIa analog V158D/E296V/M298Q-FVIIa (FVIIa-DVQ) was designed to improve the therapeutic potential of FVIIa [11]. While FVIIa-DVQ and wild-type FVIIa have similar activity in the presence of TF, FVIIa-DVQ expresses significantly greater TF-independent activity [12, 13], due to the amino acid changes that facilitate a conformation similar to that of TF-bound FVIIa [14]. As such, FVIIa-DVQ showed improved hemostatic efficacy in mouse models of hemophilia [15] and better sustained bleeding control in hemophilia patients with inhibitors [16]. Surprisingly, even though FVIIa-DVQ and FVIIa bind similarly to phospholipid vesicles, FVIIa-DVQ binds to a greater number of sites per platelet than FVIIa [17], suggesting that platelet membrane features, in addition to lipid composition, contribute to this binding.
Only a fraction of activated platelets can bind high levels of coagulation factors to provide the primary sites of thrombin generation [18–22]. These platelets, often called COATed platelets [23], appear to play a critical role in both hemostasis and thrombosis [19]. COATed platelet levels are elevated in patients with transient ischemic attack [24] and non-lacunar stroke [25]. Conversely, low levels are associated with an increased risk of spontaneous intracranial hemorrhage and hemorrhagic transformation of ischemic stroke [26–28]. COATed platelets also bind the highest levels of FVIIa [29] and FVIIa-DVQ [17] in a Gla-domain-dependent manner.
Endothelial cell protein C receptor (EPCR) is a transmembrane glycoprotein belonging to the CD1/major histocompatibility complex superfamily [30]. Gla-domain-dependent binding of Protein C (PC) to EPCR localizes it to the endothelial surface and enhances its conversion to activated protein C (APC) by the thrombin-thrombomodulin complex [31, 32]. APC inactivates factors Va and VIIIa to down-regulate thrombin production, while APC binding to EPCR mediates anti-inflammatory and cytoprotective effects in a variety of diseases including stroke, coronary reperfusion injury, and diabetes [33, 34]. FVIIa also binds to EPCR via its Gla-domain with an affinity similar to that of PC/APC [35, 36]. This binding supports TF-independent endothelial signaling by FVIIa to promote vascular barrier integrity [37].
Our current data indicate that human platelets are capable of expressing EPCR, and that binding to EPCR is responsible for the increased platelet binding of FVIIa-DVQ compared to wild-type FVIIa. While the development of FVIIa-DVQ was discontinued due to clinical immunogenicity [38], its characterization provides important insights into the mechanism of platelet-FVIIa interactions and suggests future approaches for improving FVIIa efficacy.
Materials and Methods
Reagents
FVIIa, FVIIa-DVQ, and thrombin: generous gifts from Dr. Egon Persson (Novo Nordisk, Denmark). Non-blocking anti-EPCR mAb (JRK-1500) [39]: generous gift from Dr. Charles Esmon (OMRF, Oklahoma City, OK). Rabbit-anti-β1-tubulin: generous gift from Dr. Joseph Italiano (Harvard, Boston, MA). Factor X and Protein C: Haematologic Technologies (Essex Junction, VT). Convulxin: Enzyme Research Laboratories (South Bend, IN). Collagen related peptide: prepared as described [20]. Pefachrome FXa and Pefachrome TH: Pentapharm (Basel, Switzerland). RIPA Lysis Buffer, Rabbit-anti-hEPCR (sc-28978) and phycoerythrin (PE)-conjugated rat-anti-hEPCR (RCR-49): Santa Cruz Biotechnology (Dallas, TX). Human dermal microvascular endothelial cells (HDMVEC): Lonza (Basel, Switzerland). Poly-L-lysine coverslips and Triton-X-100: Sigma Aldrich (St. Louis, MO). Protein A/G beads: Thermo Scientific (Rockford, IL). Affi-Gel 10: Bio-Rad (Hercules, CA). Goat-anti-hEPCR (AF2245), Mouse-anti-hEPCR (MAB22451), and isotype controls: R&D Systems (Minneapolis, MN).
Donkey-anti-sheep/goat IgG conjugated to horseradish peroxidase (HRP) or fluorescein isothiocyanate (FITC): AbD Serotec (Raleigh, NC). Goat-anti-mouse IgG coupled to Alexa Fluor 488 and goat-anti-rabbit IgG coupled to Alexa Fluor 568: Cell Signaling Technology (Danvers, MA). Sheep-anti-FVII: Affinity Biologicals (Ancaster, Canada).
Platelet Isolation
Whole blood was collected from healthy adult volunteers under a protocol approved by the DVAMC Institutional Review Board. Platelets were isolated by density gradient centrifugation and gel filtration as detailed previously [40], and analyzed by flow cytometry to ensure minimal leukocyte contamination (< 1:10,000 events).
Measurement of FVIIa and FVIIa-DVQ binding to platelets
Platelet binding was detected by flow cytometry as previously described [17]. Platelets (1×108/mL) were activated with thrombin (1nM) ± convulxin (100ng/mL) and incubated with FVIIa or FVIIa-DVQ (10nM-2μM). Following fixation (1% paraformaldehyde), platelets were incubated with sheep anti-hFVII. For some experiments, 250nM PC (the highest achievable concentration using a commercial preparation), or 1μg/ml rat anti-EPCR/isotype control were added prior to FVIIa/FVIIa-DVQ. Binding was detected with FITC-conjugated donkey-anti-sheep/goat and analyzed using a FACSCAN flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Platelet surface activity of FVIIa and FVIIa-DVQ
FXa and thrombin generation on platelets were measured as described [17]. Dual-activated platelets (1.5×108/ml) were incubated with 100nM FVIIa-DVQ ± 250nM PC or anti-EPCR/isotype control. Plasma levels of FX (135nM) were added and FXa generation assessed by measuring cleavage of Pefachrome® FXa (1mM) at 405nm. Alternatively, 135nM FX, 5nM Factor Va (FVa), and 1.2μM prothrombin were added to assess thrombin generation using Pefachrome® TH (1mM).
FVIIa-dependent FXa generation was measured using a prothrombinase detection system [17]. Dual-activated platelets were incubated with 100nM FVIIa ± anti-EPCR antibody (250nM blocking or 500nM non-blocking). FX (135nM) was added and subsamples transferred at timed intervals to a solution containing phospholipid vesicles (50μM), FVa (5nM), and Prothrombin (500nM). The FXa produced on the platelet surface assembles with FVa on the phospholipid vesicles to produce thrombin which was assayed by monitoring Pefachrome® TH (1mM) cleavage at 405nm.
Measurement of EPCR expression on platelets
Platelet expression of EPCR was analyzed by flow cytometry. Activated and unactivated platelets (1×108/mL) were incubated with a PE-conjugated rat anti-EPCR mAb. Following fixation (1% paraformaldehyde), some samples were prepared for two-color analyses using a sheep anti-FVII antibody. The anti-EPCR antibody did not recognize EPCR on platelets that were fixed prior to staining.
RT-PCR
Total RNA was purified from platelets as previously described [41]. RNA was isolated from PMBC controls using the Ribopure kit (Life Technologies, Carlsbad, CA). Reverse transcription utilized the High Capacity RNA-to-cDNA kit (Life Technologies, Carlsbad, CA) per manufacturer instructions. PCR was performed using Platinum Taq (Clontech, Mountain View, CA) and the following forward (CTCCTACTTCCGCGACCCCTATCA) and reverse (CGGCTTGTTTGGCTCCCTTTCGTG) primers.
Purification of Platelet-derived EPCR
EPCR was purified from platelet and HDMVEC lysates by immunoprecipitation. Lysates were pre-cleared using 2μg/ml non-immune goat IgG and protein A/G beads. The pre-cleared lysate was incubated with a goat anti-EPCR antibody coupled to Affi-Gel 10 matrix. Proteins were eluted by heating to 95°C in SDS sample buffer.
SDS-PAGE and Western Blotting
Eluates were subjected to SDS-PAGE and visualized with Coomassie Brilliant Blue R (Thermo Scientific, Rockford, IL) or subjected to immunoblotting using a goat anti-EPCR antibody detected with a HRP-conjugated donkey anti-sheep/goat IgG.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
LC-MS/MS analyses were performed on tryptic peptides from SDS-PAGE gels using a Synapt G2 QToF mass spectrometer (Waters, Milford, MA). Acquired spectra were submitted to independent Mascot searches (Matrix Science, Boston, MA) against a SwissProt database (Human taxonomy). Scoring thresholds were set to a peptide false discovery rate of 0% using the PeptideProphet algorithm [42, 43].
Immunofluorescence Microscopy
Platelet immunofluorescence was performed as described [44]. Unactivated platelets were fixed (4% formaldehyde) and centrifuged onto 1μg/mL poly-L-lysine-coated coverslips. Samples were permeabilized (0.5% Triton-X-100) and incubated overnight in blocking buffer (1% BSA, 0.05% NaN3, 10% FCS in 1xPBS) prior to labeling with a murine anti-EPCR mAb and/or a rabbit anti-β1-tubulin antibody [45]. Samples were visualized with secondary antibodies conjugated to Alexa Fluor 488/568. Non-specific and background fluorescence were assessed by incubation with secondary antibody alone. Samples were examined with a Nikon TE-2000-E Microscope (Nikon, Tokyo, Japan) equipped with a 100x (N.A. = 0.3) Plan-Fluor objective. Images were obtained using a charged coupled device (CCD) camera (Hamamatsu Photonics, Bridgewater, NJ).
Statistical Analysis
Data are expressed as mean±SD. Dependent t-tests were used to calculate 2-tail probability values. Differences were considered significant at P < 0.05.
Results
Protein C competes with FVIIa and FVIIa-DVQ for binding to platelets
Platelets were activated with thrombin plus the glycoprotein (GP) VI agonist convulxin to generate COATed platelets as previously described [18]. The platelet binding at various concentrations (≤ 2μM) of FVIIa or FVIIa-DVQ was subsequently determined by flow cytometry (Figure 1A). Consistent with our previous results [17], the binding of FVIIa-DVQ was significantly higher than that of FVIIa, suggesting that FVIIa-DVQ binds to a greater number of sites per platelet.
Figure 1. Protein C competes with FVIIa and FVIIa-DVQ for platelet binding in an EPCR-dependent manner.
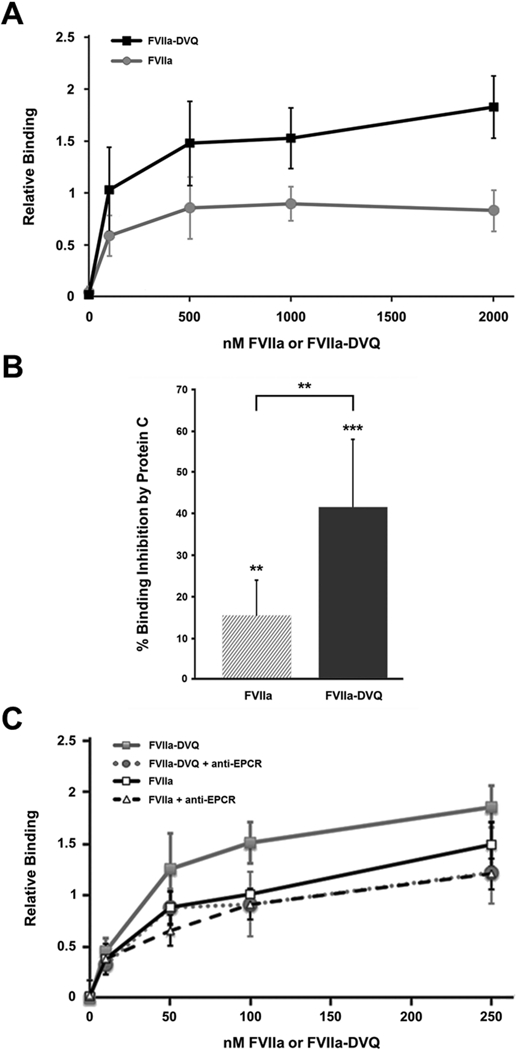
(A) Platelets activated with thrombin plus convulxin were incubated with the indicated concentrations of FVIIa or FVIIa-DVQ. The platelet binding of each molecule was analyzed by flow cytometry as described. Binding was normalized to the highest level of FVIIa binding in each experiment (n=3). (B) Similar experiments were performed to determine the platelet binding of 100nM FVIIa or FVIIa-DVQ in the presence and absence of excess (250nM) Protein C. Data shown represents the mean ± SD (n=7). (C) The platelet binding of various concentrations of FVIIa or FVIIa-DVQ was similarly analyzed in the presence and absence of an anti-EPCR antibody (blocking). Binding was normalized to the binding of FVIIa at 100 nM (=100%). No difference was seen in the binding of either molecule in the presence of an isotype control antibody. Data shown represents the mean ± SD (n=3). ** P < 0.01, *** P < 0.001 by paired t-test.
Since the Gla-domain is critical for the platelet binding of FVIIa [29] and FVIIa-DVQ [17], it is possible that other Gla-domain containing proteins, such as prothrombin or PC, may be able to compete for platelet binding. However, previous work has shown that FVIIa binding to COATed platelets was not completely inhibited by a 1000-fold excess of prothrombin [29]. Therefore, we determined the binding of FVIIa and FVIIa-DVQ (100 nM) in the presence and absence of excess (250 nM) PC. As shown in Figure 1B, the addition of PC inhibited the binding of both FVIIa (15.3 ± 8.7%, P < 0.01) and FVIIa-DVQ (41.5 ± 16.6%, P < 0.001). However, the binding of FVIIa-DVQ was inhibited to a greater extent than that of FVIIa (P < 0.01). Similar analyses with multiple platelet donors (n=7) showed no correlation with donor age or gender. These data suggest that: (1) FVIIa-DVQ binding comprises at least two distinct platelet binding sites, one of which is shared with PC, and (2) FVIIa-DVQ exhibits enhanced binding to this shared site.
Antibodies to EPCR inhibit FVIIa and FVIIa-DVQ binding to platelets
The inhibition of FVIIa/FVIIa-DVQ binding by PC led us to hypothesize a role for EPCR in platelet-FVIIa interactions. As shown in Figure 1C, the presence of a blocking antibody against EPCR inhibited both FVIIa and FVIIa-DVQ binding to COATed platelets (P < 0.01 at 100 nM). Binding of FVIIa-DVQ was again inhibited to a greater extent (36.5 ± 6.5%, P < 0.01) than FVIIa (18.5 ± 5.5%, P < 0.01). The presence of the anti-EPCR antibody abolished the difference in binding between FVIIa and FVIIa-DVQ, while an isotype control antibody had no such effect. These data confirm that EPCR is responsible for a portion of FVIIa binding to COATed platelets, and that enhanced binding to EPCR is responsible for the increased platelet binding of FVIIa-DVQ.
Protein C competition reduces platelet surface activity of FVIIa-DVQ
To determine whether competition by PC could interfere with FVIIa-DVQ activity, FXa generation was assayed on COATed platelets. In these experiments, FXa generation was inhibited by 23.3 ± 9.0% in the presence of PC. This decrease is comparable to the decrease seen in FVIIa-DVQ binding. Assays with different platelet donors (n=6) indicated that FXa generation was inhibited from 9.5% to 40.2% depending on the donor. This is consistent with donor-dependent variation in the percentage of COATed platelets, which ranges from 8% to 65% [19].
Thrombin generation assays were also performed under similar conditions after adding prothrombin (1.2 μM) and FVa (5 nM). The presence of PC decreased thrombin generation by 20.5 ± 1.5%, thereby confirming that EPCR-mediated binding of FVIIa-DVQ to COATed platelets contributes to procoagulant activity.
Antibodies to EPCR reduce platelet surface activity of FVIIa and FVIIa-DVQ
To confirm that a specific interaction with platelet-associated EPCR contributes to the hemostatic efficacy of FVIIa/FVIIa-DVQ, similar FXa generation assays were performed in the presence and absence of an anti-EPCR antibody.
In these studies, the rate of FXa generation by FVIIa-DVQ was inhibited by 35.0 ± 9.0% (P < 0.05, n=6) in the presence of the anti-EPCR antibody (Figure 2A-B). This effect was proportional to the decrease in platelet binding, and confirmed that these findings are specifically dependent on interactions with EPCR. No differences in activity were seen in the presence of an isotype control or a non-blocking anti-EPCR antibody. As with PC, analyses of multiple platelet donors showed similar donor-dependent variation (12.4% to 76.5% inhibition, n=6).
Figure 2. The platelet surface activity of FVIIa and FVIIa-DVQ is dependent on binding to EPCR.
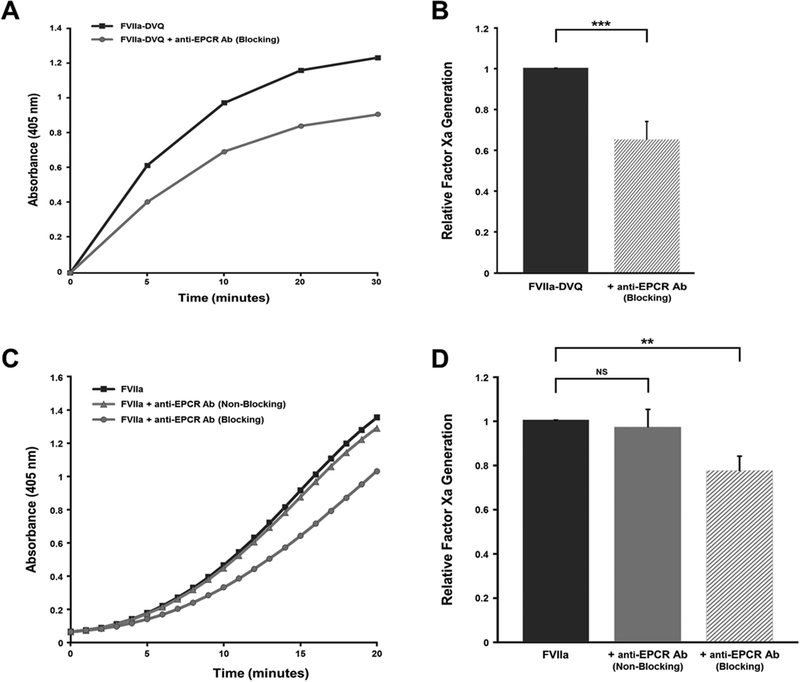
(A) Freshly isolated platelets were activated with thrombin plus convulxin and incubated with 100 nM FVIIa-DVQ in the presence and absence of excess (250 nM) anti-EPCR antibody (blocking). Plasma levels of FX (135 nM) were added and FXa generation assessed by continuously measuring cleavage of a chromogenic substrate. Data shown is a representative plot of actual FXa generation rate as a function of optical density at 405 nm over time. (B) FXa generation rates from multiple donors and experiments were normalized to the rate of FXa generation in the absence of antibody (=100%). Data shown represents the mean ± SD (n=6). No difference was seen in the rate of FXa generation in the presence of either an isotype control or a non-blocking anti-EPCR antibody. (C) FVIIa-dependent FXa generation was assessed using a prothrombinase detection system as described in ‘Methods’. Activated platelets were incubated with 100 nM FVIIa in the presence and absence of excess (250nM) anti-EPCR (blocking or non-blocking). Thrombin generation was assessed by continuously measuring cleavage of a chromogenic substrate. Data shown is a representative plot of actual thrombin generation rate as a function of optical density at 405 nm over time. (D) FXa generation rates were determined for each experiment and normalized to the rate of FXa generation in the absence of antibody (=100%). Data shown represents the mean ± SD (n=7). ** P < 0.01, *** P < 0.001 by paired t-test, NS = not significant.
In additional experiments, collagen related peptide replaced convulxin as the GPVI agonist for platelet activation. Under these conditions, the rate of FXa generation was similarly inhibited (32.5 ± 11.5%, P < 0.05, n=6) and the decrease in activity remained proportional to the decrease in FVIIa-DVQ binding.
Since FVIIa-dependent FXa generation on platelets is too low for accurate detection at low concentrations of FVIIa, these experiments were performed using a previously described prothrombinase detection system [17]. In these studies, the rate of FXa generation was inhibited by 22.8 ± 8.3% (P < 0.01, n=7) in the presence of a blocking anti-EPCR antibody (Figure 2C-D). This rate of inhibition is consistent with the observed decrease in FVIIa binding, and is significantly less than the inhibition seen with FVIIa-DVQ (P < 0.01). There was no difference in FVIIa activity in the presence of a non-blocking anti-EPCR antibody. Taken together, these data confirm that the hemostatic activity of FVIIa and FVIIa-DVQ is partially dependent on a specific interaction with EPCR, and that this interaction is enhanced by the alterations in FVIIa-DVQ.
EPCR is expressed on the surface of COATed platelets
While mRNA encoding EPCR has been reported in genome-wide sequencing of the human platelet transcriptome [46], EPCR protein expression has not previously been shown in platelets. We therefore conducted flow cytometric analyses to determine the ability of platelets to express EPCR. As shown Figure 3, EPCR is not expressed on unactivated platelets (A), nor is it expressed on non-COATed platelets, despite their activation as evidenced by P-selectin (CD62) expression (B). Conversely, COATed platelets express significant amounts of EPCR on their surface (C).
Figure 3. Procoagulant (COATed) platelets express EPCR.
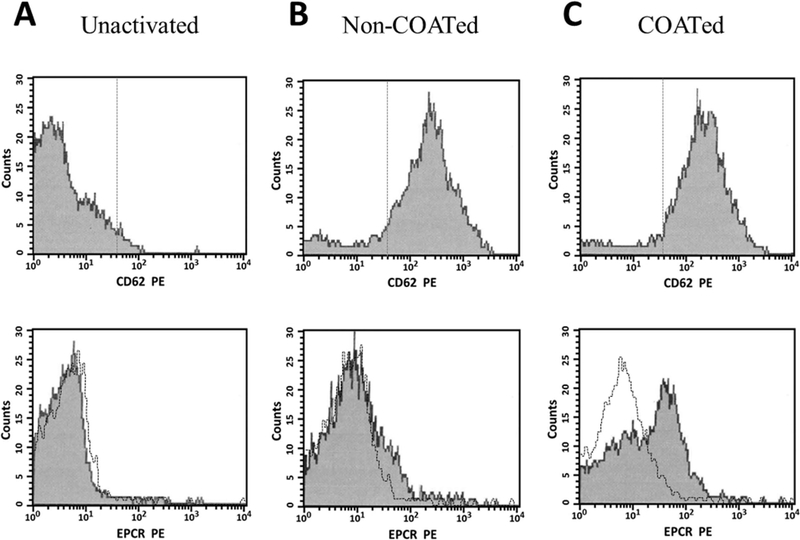
Human platelets were isolated as described, and evaluated prior to (unactivated) or following activation with thrombin (non-COATed) or thrombin plus convulxin (COATed) for their ability to express EPCR by flow cytometry. Following fixation, platelets were stained with either PE-conjugated anti-CD62P to identify activated platelets (top panel, x-axes) or specific PE-conjugated antibodies to detect EPCR (bottom panel, x-axes). For the overlay (bottom panel), transparent histograms indicate the binding of an isotype matched IgG control (black line). The fluorescence for 5,000 platelets was analyzed for each sample.
EPCR expression defines the platelet subpopulation capable of binding FVIIa
Dual-labeling studies were performed to assess platelet EPCR expression and FVIIa binding simultaneously. As seen in Figure 4, neither FVIIa binding nor EPCR expression were seen on the surface of unactivated platelets (A). In addition, only 3.7 ± 0.4% (n=3) of the non-COATed platelets showed high levels of both FVIIa binding and EPCR expression (B). However, 85.2 ± 6.6% of platelets that express high levels of EPCR were capable of binding high levels of FVIIa (C). These platelets represent the COATed platelet subpopulation. Conversely, only 44.5 ± 6.5% of platelets capable of binding FVIIa showed high levels of EPCR expression, consistent with our data suggesting that EPCR may provide one of two potential binding sites for FVIIa on the platelet membrane.
Figure 4. Platelets that express EPCR also bind the highest levels of FVIIa.
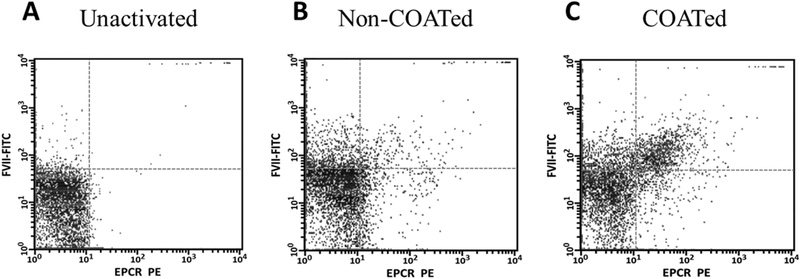
Human platelets were isolated and activated as described in Figure 3. Following fixation, platelets were simultaneously labeled with PE-conjugated anti-EPCR (x-axes) and FITC-conjugated anti-FVIIa (y-axes) antibodies to quantify FVIIa binding to platelets capable of expressing EPCR. The right upper quadrants in each panel indicate EPCR expression with concomitant FVIIa binding. The fluorescence for 5,000 platelets was analyzed for each sample.
EPCR is stored in unactivated platelets
The rapid expression of EPCR following platelet activation suggests that pre-formed protein is stored in unactivated platelets. However, platelets also contain mRNA encoding EPCR [46] and are capable of pre-mRNA splicing and protein synthesis following activation [47]. To evaluate these potential mechanisms for EPCR expression, we first confirmed the presence of EPCR pre-mRNA in unactivated platelets by RT-PCR (Figure 5A). To determine whether unactivated platelets contain pre-formed EPCR protein, we subjected platelet and endothelial cell lysates to immunoprecipitation using an anti-EPCR antibody. The presence of the immuno-purified protein was then determined by western blot analysis (Figure 5B).
Figure 5. EPCR is stored in unactivated platelets.
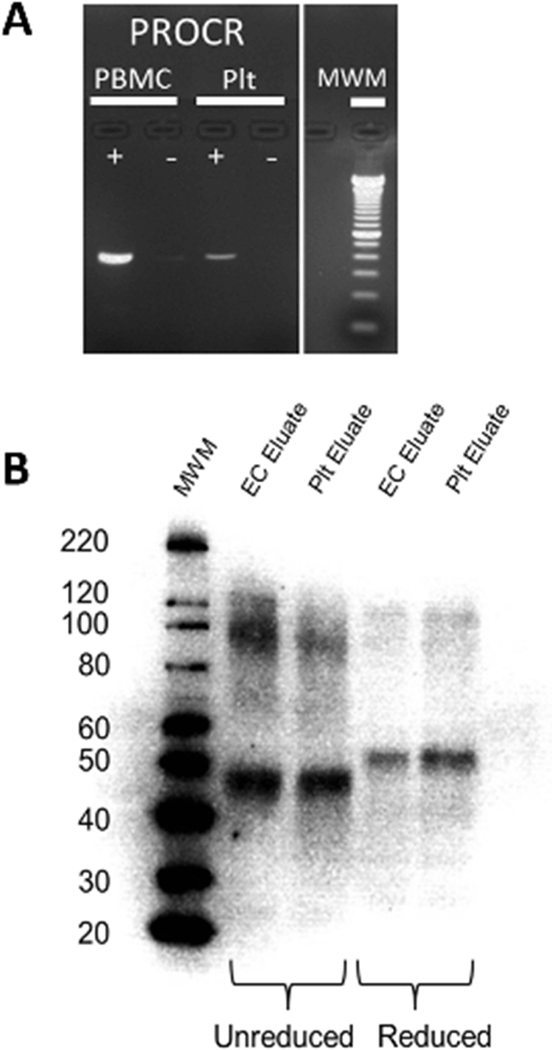
(A) Total RNA was purified from unactivated human platelets (Plt) and peripheral blood mononuclear cell (PMBC) controls prior to cDNA conversion as described. The presence of EPCR pre-mRNA (PROCR) was then confirmed in each of these samples by RT-PCR performed in the presence (+) and absence (−) of a Taq polymerase. Molecular weight markers are indicated on the right. (B) Affinity purification of EPCR was performed on platelet (Plt) and endothelial cell (EC) lysates using a goat anti-EPCR antibody as described. Samples were analyzed by western blotting under reducing (lane 4–5) and non-reducing (lane 2–3) conditions as shown. Molecular weight markers are indicated on the left.
The identity of the EPCR protein purified from platelet and endothelial sources was confirmed by mass spectrometric analyses. Figure 6 shows a representative MS/MS spectrum corresponding to the [M+3H]3+ parent ion of the EPCR peptide 82TQSGLQSYLLQFHGLVR98 (m/z = 649.68) isolated from platelet lysate. Three additional peptides unique to EPCR (28LHMLQISYFR37, 105TLAFPLTIR113, 180EFLEDTCVQYVQK192) were also identified in platelet lysate samples, representing ~20% coverage of the molecule.
Figure 6. MS/MS analysis of EPCR in human platelets.
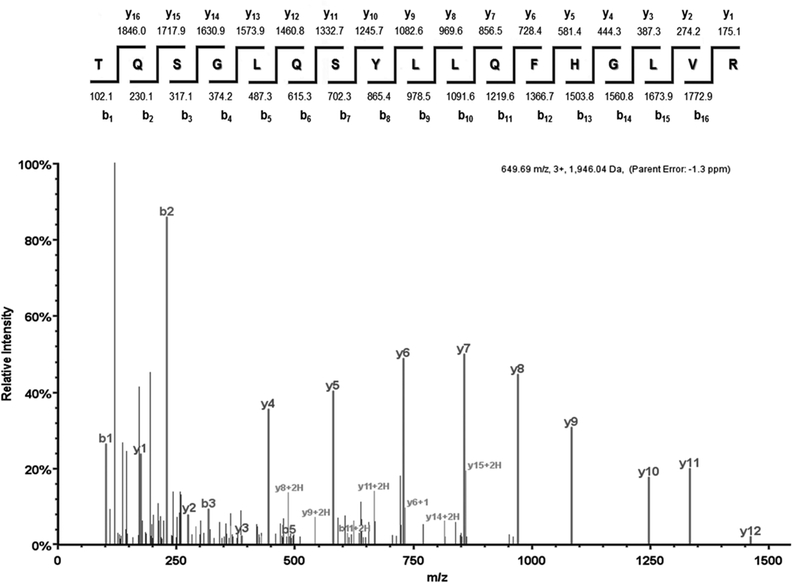
EPCR was purified from human platelet lysates as in Figure 5. Eluates were subjected to SDS-PAGE and bands of interest were excised prior to mass spectrometric analysis as described. In this MS/MS spectrum the [M+3H]3+ parent ion at m/z = 649.68 was fragmented, producing the displayed spectrum. The b or y ion designation of each peptide fragment is indicated above the corresponding peak. The peptide sequence and molar mass of each expected peptide fragment are indicated above. This spectrum represents one of the four exclusive peptides unique to EPCR that were identified in these studies.
To confirm the intracellular localization of EPCR in platelets and ensure that our previous results were not simply due to leukocyte contamination, resting human platelets were examined by immunofluorescence. To confirm the platelets remained unactivated, the peripheral β1-tubulin coil was labeled, and found to be intact (Figure 7A-B, red). Samples were also probed for EPCR (Figure 7A, green). Non-specific staining was assessed by labeling in the absence of the primary antibody (Figure 7B, lower right panel). Immunofluorescence microscopy confirmed that human platelets contain EPCR (Figure 7A, merge) in a punctate pattern, suggesting a granular localization. Taken together, these data unambiguously confirm the presence of pre-formed EPCR in unactivated platelets. Therefore, de novo synthesis is unlikely to be the primary mechanism of EPCR expression by COATed platelets.
Figure 7. Human platelets contain pre-formed EPCR.
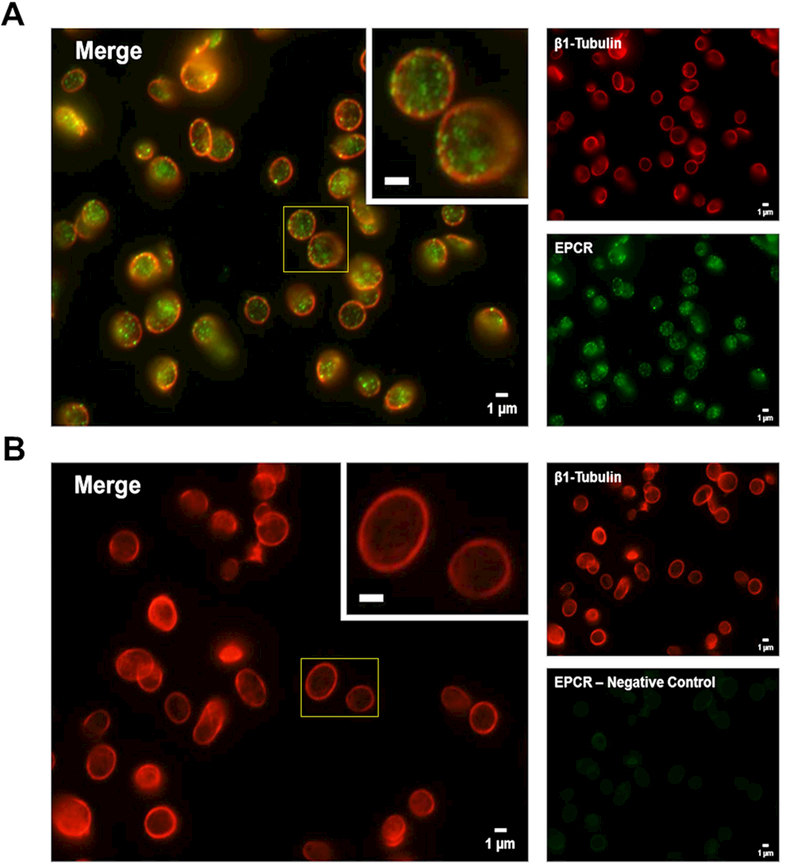
(A and B) Freshly isolated human platelets (unactivated) were fixed in 4% formaldehyde and spun down onto poly-L-lysine-coated glass coverslips. Samples were permeabilized with 0.5% Triton-X-100 and incubated overnight in blocking buffer prior to probing for EPCR and β1-tubulin using appropriate primary and secondary antibody pairs conjugated to Alexa Fluor 488 or 568. Samples were examined with a Nikon TE-2000-E Microscope (Nikon, Tokyo, Japan) equipped with a 100x (N.A. = 0.3) Plan-Fluor objective. Images were obtained using a charged coupled device (CCD) camera (Hamamatsu Photonics, Bridgewater, NJ). Background fluorescence was subtracted and image brightness/contrast was adjusted linearly for each micrograph. Larger panels (left) represent a merged image of the two smaller panels (right) for each image. Insets represent a magnified region outlined by the yellow box for each image (inset bars = 1μm). (A) β1-tubulin (red) was used to label the platelet cytoskeleton while EPCR (green) is predominantly localized in distinct, punctate granules within the platelets. (B) Non-specific staining was assessed by labeling in the absence of the primary anti-EPCR antibody (green secondary antibody alone).
Discussion
This work represents the first demonstration that human platelets can express EPCR which plays a modest role in platelet-FVIIa binding. In addition, a specific interaction with platelet-associated EPCR mediates the increased binding and activity of FVIIa-DVQ, suggesting that modulation of EPCR binding can be utilized to increase the hemostatic efficacy of rationally designed FVIIa variants.
We found that EPCR is localized to discrete granules within unactivated platelets and specifically expressed on the surface of COATed platelets. However, EPCR cannot be a component of all alpha-granules, since it is not expressed on all platelets that express the alpha-granule protein P-selectin [48]. It is therefore possible that EPCR may be stored in a novel subpopulation of granules that is differentially released during transition to the COATed state. This mechanism would certainly be supported by the well-described heterogeneity in platelet granule content and release [44, 49]. Additional studies are needed to confirm whether this differential expression is due to a lack of EPCR in non-COATed platelets, or to a unique ability of COATed platelets to translocate EPCR to their surface.
We have previously shown that the Gla-domain of FVIIa and FVIIa-DVQ is essential for platelet binding, and that the binding of these molecules is identical on phospholipid vesicles [17]. Based on these studies, we initially felt that platelet-FVIIa binding was primarily mediated by phospholipid [50, 51]. However, FVIIa-DVQ binds to a greater number of sites per platelet than FVIIa, suggesting the involvement of a non-lipid component in the platelet membrane. Our current studies clearly identify EPCR as the membrane component responsible for this phenomenon. It is therefore likely that the amino acid substitutions in FVIIa-DVQ lead to conformational changes that not only mimic the effects of binding to TF [52], but may also enhance binding to EPCR in the platelet membrane microenvironment.
While FVIIa-EPCR binding on platelets is novel, a physiologically relevant interaction between FVIIa and EPCR on vascular endothelial cells is well established [35]. FVIIa binding to endothelial EPCR triggers TF-independent signaling [53] that promotes vascular barrier integrity [37]. Endothelial EPCR also facilitates the transport of FVIIa into the extravascular space [54], thereby providing a potential mechanism for the prolonged efficacy of prophylactic FVIIa therapy [55, 56].
Pavani et al suggested a direct role for EPCR in FVIIa-mediated hemostasis [57, 58]. They engineered a murine FVIIa (mFVIIa) analog with three mutations (L4F/L8M/W9R) that allow it, unlike wild-type mFVIIa, to bind murine EPCR. This analog exhibited a 3-fold increase in hemostatic activity compared to wild-type mFVIIa [57]. More recently, the L4F substitution was found to be solely responsible for the EPCR-dependent increase in hemostatic activity [58]. They concluded that binding to EPCR on the endothelium may augment the procoagulant effect of FVIIa. It is unknown whether murine platelets can express EPCR, or to what extent platelet-associated EPCR may have contributed to these results.
An alternative explanation was suggested by Sundaram et al who showed that pretreatment of hemophilia A mice with a blocking antibody against EPCR led to effective hemostasis at lower doses of FVIIa than were otherwise required [59]. Similar studies in hemophilia A mice showed that co-administration of active-site-inhibited FVIIa also led to effective hemostasis at lower doses of wild-type FVIIa [60], suggesting that down-regulation of the PC/APC pathway by FVIIa competition for EPCR on endothelial cells may contribute to its hemostatic activity.
In contrast, the present study focuses on interactions with the human platelet surface and is the first to address the role of EPCR in platelet-dependent hemostasis. This work identifies platelet-expressed EPCR as an additional, and possibly synergistic, mechanism contributing to the pharmacologic effect of high-dose FVIIa. Importantly, this expression may also provide a strategy to more efficiently target future FVIIa variants to the platelet surface.
The mechanism of platelet-FVIIa binding has been the subject of multiple studies. Although quite controversial, very low amounts of TF have been reported in platelets [61, 62]. However, previous studies from our laboratory and others have shown that blocking antibodies against TF do not significantly reduce the binding [29] or activity [9] of FVIIa on platelet surfaces. It is therefore unlikely that platelet-associated TF contributes significantly to the pharmacologic effect of FVIIa.
Weeterings et al suggested that interactions with the GPIb/IX/V complex may make a small contribution to platelet surface FVIIa activity [63]. In these studies, CHO-cells transfected with GPIb/IX/V readily adhered to immobilized FVIIa. A potential interaction between FVIIa and GPIbα is further supported by studies showing that platelet uptake and internalization of FVIIa is decreased by ~77% in platelets isolated from patients with Bernard-Soulier Syndrome as compared to healthy controls. However, the control platelets showed only a 15–20% decrease in FVIIa uptake in the presence of an antibody against GPIbα [64]. The potential contribution of such an interaction therefore remains unclear, particularly since thrombin, which also binds GPIbα [65, 66], is unable to compete with FVIIa or FVIIa-DVQ for platelet binding [17].
While our data clearly indicate that binding to EPCR is responsible for a portion of FVIIa binding to COATed platelets, this cannot explain the entirety of platelet-FVIIa interactions. Instead, our data suggests that there is at least one additional binding site for FVIIa on platelets and could therefore be consistent with several different binding models. This paradigm involving simultaneous interaction with multiple platelet receptors has been suggested as a mechanism for the binding of other proteins including PC [67], β2-glycoprotein-I [68], and Factor XI [69].
It is therefore likely that platelet-FVIIa binding is dependent on simultaneous or coordinate interactions with multiple binding partners which may include phospholipids, GPIbα, ApoER2, or other membrane proteins. This is supported by studies showing only partial inhibition of FVIIa internalization by platelets in the presence of APC, annexin V, or antibodies to GPIbα or GPVI [64]. While the affinity of FVIIa for each of these partners may be relatively weak, simultaneous binding to multiple partners could synergistically increase FVIIa affinity for platelet surfaces. However, the fairly weak nature of each individual interaction may make it difficult to determine the extent of contribution to overall binding. In addition, expression levels of these potential partners may vary between individuals just as the percentage of COATed platelets varies between donors [19]. These variations almost certainly contribute to the clinically observed differences in patient responsiveness to FVIIa as a therapeutic agent [70].
While platelet-associated EPCR plays a role in FVIIa-mediated hemostasis, its potential role in PC/APC-mediated anticoagulation remains unclear. Previous studies have shown that platelets can bind to immobilized PC/APC, suggesting the presence of a specific platelet receptor for PC/APC [67]. However, this interaction was not dependent on the Gla-domain of PC, which is required for binding to EPCR, and therefore cannot be explained by the current studies. In addition, while human platelets contain a small amount of active thrombomodulin [71], the high local concentration of platelet-factor-4 at sites of activation may prevent APC generation [72]. Indeed, studies using knockout mice have shown that PC activation is primarily facilitated by endothelial EPCR with little contribution from EPCR on hematopoietic cells [73].
Even if APC is generated on platelet surfaces, platelet-bound FVa is resistant to inactivation by APC [74]. It is therefore unlikely that platelet-associated EPCR plays a major role in APC-mediated anticoagulation on the platelet surface. In contrast, platelet microparticles can support FVa inactivation by APC [75], and platelet-derived EPCR may contribute to counterbalancing the procoagulant effects of microparticles.
The current study shows that the significantly increased platelet binding and hemostatic activity of FVIIa-DVQ is directly facilitated by a specific interaction with platelet-associated EPCR. Therefore, manipulation of the interaction between FVIIa and platelet-associated EPCR represents a potentially exciting new pathway for the development of better therapeutic agents that will improve the treatment and quality of life for patients requiring emergency hemostasis.
Essentials.
Factor VIIa binds activated platelets to promote hemostasis in hemophilia patients with inhibitors
The interactions and sites responsible for platelet-FVIIa binding are not fully understood
Endothelial Cell Protein C Receptor (EPCR) is expressed on activated human platelets
EPCR binding enhances the efficacy of a FVIIa variant and could impact design of new therapeutics
Acknowledgements
We are grateful to Dr. Charles Esmon and Dr. Joseph Italiano for providing key reagents; Angela Crabtree and Dr. Joelle Romac for their technical support; and Dr. Gowthami Arepally and Dr. “Mac” Monroe for providing valuable insights into experimental design and critical reading of the manuscript.
This work was supported by National Institutes of Health through the National Heart, Lung, and Blood Institute (T32HL007057, K12HL087097, U54HL112307 [A.M. Fager], 5F32HL118865 [K.R. Machlus]) and the National Institute of Diabetes and Digestive and Kidney Diseases (K01DK111515 [K.R. Machlus]); The American Heart Association (16SDG29090007 [K.R. Machlus]); and the Bayer Hemophilia Awards Program (ECI Award [A.M. Fager]). M. Hoffman was supported by the US Department of Veterans Affairs.
Footnotes
Addendum
A.M. Fager designed and performed research, analyzed data, and wrote the manuscript. K.R. Machlus performed research. M. Ezban helped design initial studies and provided the FVIIa-DVQ. M. Hoffman oversaw the direction of experiments and analyzed data. All authors edited the final manuscript.
Disclosure of Conflict of Interests
A.M. Fager received funding from NIH NHLBI, as well as a competitive grant from the Bayer Hemophilia Awards Program. M. Hoffman received investigator-initiated funding from Novo Nordisk, serves on the advisory board of Roche and Dova, and consuls for Alnylan. M. Ezban is an employee of Novo Nordisk. K.R. Machlus declares no conflict of interest.
REFERENCES
- 1.Berntorp E, Shapiro AD. Modern haemophilia care. Lancet. 2012; 379: 1447–56. [DOI] [PubMed] [Google Scholar]
- 2.Kempton CL, White GC 2nd, How we treat a hemophilia A patient with a factor VIII inhibitor. Blood. 2009; 113: 11–7. [DOI] [PubMed] [Google Scholar]
- 3.Osterud B, Olsen JO. Human platelets do not express tissue factor. Thrombosis research. 2013; 132: 112–5. [DOI] [PubMed] [Google Scholar]
- 4.Monroe DM, Hoffman M, Oliver JA, Roberts HR. A possible mechanism of action of activated factor VII independent of tissue factor. Blood Coagul Fibrinolysis. 1998; 9 Suppl 1: S15–20. [PubMed] [Google Scholar]
- 5.Keshava S, Sundaram J, Rajulapati A, Pendurthi UR, Rao LV. Pharmacological concentrations of recombinant factor VIIa restore hemostasis independent of tissue factor in antibody-induced hemophilia mice. J Thromb Haemost. 2016; 14: 546–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng D, Whinna H, Monroe D, Stafford DW. FVIIa as used pharmacologically is not TF dependent in hemophilia B mice. Blood. 2014; 123: 1764–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman M, Monroe DM 3rd. The action of high-dose factor VIIa (FVIIa) in a cell-based model of hemostasis. Seminars in hematology. 2001; 38: 6–9. [DOI] [PubMed] [Google Scholar]
- 8.Weeterings C, Lisman T, de Groot PG. Tissue factor-independent effects of recombinant factor VIIa on hemostasis. Seminars in hematology. 2008; 45: S12–5. [DOI] [PubMed] [Google Scholar]
- 9.Monroe DM, Hoffman M, Oliver JA, Roberts HR. Platelet activity of high-dose factor VIIa is independent of tissue factor. British journal of haematology. 1997; 99: 542–7. [DOI] [PubMed] [Google Scholar]
- 10.Gomperts ED, Astermark J, Gringeri A, Teitel J. From theory to practice: applying current clinical knowledge and treatment strategies to the care of hemophilia a patients with inhibitors. Blood reviews. 2008; 22 Suppl 1: S1–11. [DOI] [PubMed] [Google Scholar]
- 11.Persson E, Kjalke M, Olsen OH. Rational design of coagulation factor VIIa variants with substantially increased intrinsic activity. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98: 13583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen OH, Clausen N, Persson E, Ezban M, Ingerslev J, Sorensen B. Whole blood coagulation in children with thrombocytopenia and the response to platelet replacement, recombinant factor VIIa, and a potent factor VIIa analogue. British journal of haematology. 2009; 144: 99–106. [DOI] [PubMed] [Google Scholar]
- 13.Allen GA, Persson E, Campbell RA, Ezban M, Hedner U, Wolberg AS. A variant of recombinant factor VIIa with enhanced procoagulant and antifibrinolytic activities in an in vitro model of hemophilia. Arteriosclerosis, thrombosis, and vascular biology. 2007; 27: 683–9. [DOI] [PubMed] [Google Scholar]
- 14.Rand KD, Andersen MD, Olsen OH, Jorgensen TJ, Ostergaard H, Jensen ON, Stennicke HR, Persson E. The origins of enhanced activity in factor VIIa analogs and the interplay between key allosteric sites revealed by hydrogen exchange mass spectrometry. The Journal of biological chemistry. 2008; 283: 13378–87. [DOI] [PubMed] [Google Scholar]
- 15.Tranholm M, Kristensen K, Kristensen AT, Pyke C, Rojkjaer R, Persson E. Improved hemostasis with superactive analogs of factor VIIa in a mouse model of hemophilia A. Blood. 2003; 102: 3615–20. [DOI] [PubMed] [Google Scholar]
- 16.Lentz SR, Ehrenforth S, Abdul Karim F, Matsushita T, Weldingh KN, Windyga J, Mahlangu JN, adept i. Recombinant factor VIIa analog in the management of hemophilia with inhibitors: results from a multicenter, randomized, controlled trial of vatreptacog alfa. J Thromb Haemost. 2014; 12: 1244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman M, Volovyk Z, Persson E, Gabriel DA, Ezban M, Monroe DM. Platelet binding and activity of a factor VIIa variant with enhanced tissue factor independent activity. J Thromb Haemost. 2011; 9: 759–66. [DOI] [PubMed] [Google Scholar]
- 18.Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000; 95: 1694–702. [PubMed] [Google Scholar]
- 19.Fager AM, Wood JP, Bouchard BA, Feng P, Tracy PB. Properties of procoagulant platelets: defining and characterizing the subpopulation binding a functional prothrombinase. Arteriosclerosis, thrombosis, and vascular biology. 2010; 30: 2400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kempton CL, Hoffman M, Roberts HR, Monroe DM. Platelet heterogeneity: variation in coagulation complexes on platelet subpopulations. Arteriosclerosis, thrombosis, and vascular biology. 2005; 25: 861–6. [DOI] [PubMed] [Google Scholar]
- 21.Panteleev MA, Ananyeva NM, Greco NJ, Ataullakhanov FI, Saenko EL. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J Thromb Haemost. 2005; 3: 2545–53. [DOI] [PubMed] [Google Scholar]
- 22.Mazepa M, Hoffman M, Monroe D. Superactivated platelets: thrombus regulators, thrombin generators, and potential clinical targets. Arteriosclerosis, thrombosis, and vascular biology. 2013; 33: 1747–52. [DOI] [PubMed] [Google Scholar]
- 23.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost. 2005; 3: 2185–92. [DOI] [PubMed] [Google Scholar]
- 24.Prodan CI, Vincent AS, Dale GL. Coated-platelet levels are elevated in patients with transient ischemic attack. Translational research : the journal of laboratory and clinical medicine. 2011; 158: 71–5. [DOI] [PubMed] [Google Scholar]
- 25.Prodan CI, Joseph PM, Vincent AS, Dale GL. Coated-platelets in ischemic stroke: differences between lacunar and cortical stroke. J Thromb Haemost. 2008; 6: 609–14. [DOI] [PubMed] [Google Scholar]
- 26.Prodan CI, Stoner JA, Cowan LD, Dale GL. Lower coated-platelet levels are associated with early hemorrhagic transformation in patients with non-lacunar brain infarction. J Thromb Haemost. 2010; 8: 1185–90. [DOI] [PubMed] [Google Scholar]
- 27.Prodan CI, Vincent AS, Dale GL. Coated platelet levels correlate with bleed volume in patients with spontaneous intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2010; 41: 1301–3. [DOI] [PubMed] [Google Scholar]
- 28.Prodan CI, Vincent AS, Padmanabhan R, Dale GL. Coated-platelet levels are low in patients with spontaneous intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2009; 40: 2578–80. [DOI] [PubMed] [Google Scholar]
- 29.Kjalke M, Kjellev S, Rojkjaer R. Preferential localization of recombinant factor VIIa to platelets activated with a combination of thrombin and a glycoprotein VI receptor agonist. J Thromb Haemost. 2007; 5: 774–80. [DOI] [PubMed] [Google Scholar]
- 30.Fukudome K, Esmon CT. Molecular cloning and expression of murine and bovine endothelial cell protein C/activated protein C receptor (EPCR). The structural and functional conservation in human, bovine, and murine EPCR. The Journal of biological chemistry. 1995; 270: 5571–7. [DOI] [PubMed] [Google Scholar]
- 31.Regan LM, Mollica JS, Rezaie AR, Esmon CT. The interaction between the endothelial cell protein C receptor and protein C is dictated by the gamma-carboxyglutamic acid domain of protein C. The Journal of biological chemistry. 1997; 272: 26279–84. [DOI] [PubMed] [Google Scholar]
- 32.Taylor FB Jr. Peer GT, Lockhart MS, Ferrell G, Esmon CT. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood. 2001; 97: 1685–8. [DOI] [PubMed] [Google Scholar]
- 33.Esmon CT. Protein C anticoagulant system--anti-inflammatory effects. Seminars in immunopathology. 2012; 34: 127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiler H Regulation of inflammation by the protein C system. Critical care medicine. 2010; 38: S18–25. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. The Journal of biological chemistry. 2007; 282: 11849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preston RJ, Ajzner E, Razzari C, Karageorgi S, Dua S, Dahlback B, Lane DA. Multifunctional specificity of the protein C/activated protein C Gla domain. The Journal of biological chemistry. 2006; 281: 28850–7. [DOI] [PubMed] [Google Scholar]
- 37.Sundaram J, Keshava S, Gopalakrishnan R, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa binding to endothelial cell protein C receptor protects vascular barrier integrity in vivo. J Thromb Haemost. 2014; 12: 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahlangu JN, Weldingh KN, Lentz SR, Kaicker S, Karim FA, Matsushita T, Recht M, Tomczak W, Windyga J, Ehrenforth S, Knobe K, adept I, adept I. Changes in the amino acid sequence of the recombinant human factor VIIa analog, vatreptacog alfa, are associated with clinical immunogenicity. J Thromb Haemost. 2015; 13: 1989–98. [DOI] [PubMed] [Google Scholar]
- 39.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93: 10212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffman M, Monroe DM, Roberts HR. A rapid method to isolate platelets from human blood by density gradient centrifugation. American journal of clinical pathology. 1992; 98: 531–3. [DOI] [PubMed] [Google Scholar]
- 41.Amisten S A rapid and efficient platelet purification protocol for platelet gene expression studies. Methods in molecular biology. 2012; 788: 155–72. [DOI] [PubMed] [Google Scholar]
- 42.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry. 2002; 74: 5383–92. [DOI] [PubMed] [Google Scholar]
- 43.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry. 2003; 75: 4646–58. [DOI] [PubMed] [Google Scholar]
- 44.Italiano JE Jr., Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Shor S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008; 111: 1227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, Ehrlicher A, Larson MK, Hoffmeister K, Hartwig JH, Italiano JE Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. The Journal of cell biology. 2010; 191: 861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011; 118: e101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005; 122: 379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmerman GA, McIntyre TM, Prescott SM. Thrombin stimulates neutrophil adherence by an endothelial cell-dependent mechanism: characterization of the response and relationship to platelet-activating factor synthesis. Annals of the New York Academy of Sciences. 1986; 485: 349–68. [DOI] [PubMed] [Google Scholar]
- 49.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE Jr. T granules in human platelets function in TLR9 organization and signaling. The Journal of cell biology. 2012; 198: 561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bom VJ, Bertina RM. The contributions of Ca2+, phospholipids and tissue-factor apoprotein to the activation of human blood-coagulation factor X by activated factor VII. The Biochemical journal. 1990; 265: 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao LV, Rapaport SI. Factor VIIa-catalyzed activation of factor X independent of tissue factor: its possible significance for control of hemophilic bleeding by infused factor VIIa. Blood. 1990; 75: 1069–73. [PubMed] [Google Scholar]
- 52.Persson E Engineering FVIIa Variants with Enhanced Activity. Blood. 2013; 122: SCI–7. [Google Scholar]
- 53.Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011; 117: 3199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clark CA, Vatsyayan R, Hedner U, Esmon CT, Pendurthi UR, Rao LV. Endothelial cell protein C receptor-mediated redistribution and tissue-level accumulation of factor VIIa. J Thromb Haemost. 2012; 10: 2383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konkle BA, Ebbesen LS, Erhardtsen E, Bianco RP, Lissitchkov T, Rusen L, Serban MA. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007; 5: 1904–13. [DOI] [PubMed] [Google Scholar]
- 56.Mariani G, Konkle BA, Ingerslev J. Congenital factor VII deficiency: therapy with recombinant activated factor VII -- a critical appraisal. Haemophilia : the official journal of the World Federation of Hemophilia. 2006; 12: 19–27. [DOI] [PubMed] [Google Scholar]
- 57.Pavani G, Ivanciu L, Faella A, Marcos-Contreras OA, Margaritis P. The endothelial protein C receptor enhances hemostasis of FVIIa administration in hemophilic mice in vivo. Blood. 2014; 124: 1157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pavani G, Zintner SM, Ivanciu L, Small JC, Stafford KA, Szeto JH, Margaritis P. One amino acid in mouse activated factor VII defines its endothelial protein C receptor (EPCR) binding and modulates its EPCR-dependent hemostatic activity in vivo. J Thromb Haemost. 2017; 15: 507–12. [DOI] [PubMed] [Google Scholar]
- 59.Sundaram J, Pendurthi UR, Esmon CT, Rao LV. Blockade of endothelial cell protein C receptor augments factor VIIa hemostatic effect in hemophilia treatment. Blood. 2014; 124: 3031–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keshava S, Sundaram J, Rajulapati A, Esmon C, Pendurthi U, Rao LVM. Factor VIIa interaction with EPCR modulates the hemostatic effect of rFVIIa in hemophilia therapy: Mode of its action. Blood Adv. 2017; 1: 1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bach RR. Tissue factor encryption. Arteriosclerosis, thrombosis, and vascular biology. 2006; 26: 456–61. [DOI] [PubMed] [Google Scholar]
- 62.Brambilla M, Facchinetti L, Canzano P, Rossetti L, Ferri N, Balduini A, Abbonante V, Boselli D, De Marco L, Di Minno MN, Toschi V, Corsini A, Tremoli E, Camera M. Human megakaryocytes confer tissue factor to a subset of shed platelets to stimulate thrombin generation. Thrombosis and haemostasis. 2015; 114: 579–92. [DOI] [PubMed] [Google Scholar]
- 63.Weeterings C, de Groot PG, Adelmeijer J, Lisman T. The glycoprotein Ib-IX-V complex contributes to tissue factor-independent thrombin generation by recombinant factor VIIa on the activated platelet surface. Blood. 2008; 112: 3227–33. [DOI] [PubMed] [Google Scholar]
- 64.Lopez-Vilchez I, Hedner U, Altisent C, Diaz-Ricart M, Escolar G, Galan AM. Redistribution and hemostatic action of recombinant activated factor VII associated with platelets. The American journal of pathology. 2011; 178: 2938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Candia E, De Cristofaro R, De Marco L, Mazzucato M, Picozzi M, Landolfi R. Thrombin interaction with platelet GpIB: role of the heparin binding domain. Thrombosis and haemostasis. 1997; 77: 735–40. [PubMed] [Google Scholar]
- 66.Okumura T, Hasitz M, Jamieson GA. Platelet glycocalicin. Interaction with thrombin and role as thrombin receptor of the platelet surface. The Journal of biological chemistry. 1978; 253: 3435–43. [PubMed] [Google Scholar]
- 67.White TC, Berny MA, Tucker EI, Urbanus RT, de Groot PG, Fernandez JA, Griffin JH, Gruber A, McCarty OJ. Protein C supports platelet binding and activation under flow: role of glycoprotein Ib and apolipoprotein E receptor 2. J Thromb Haemost. 2008; 6: 995–1002. [DOI] [PubMed] [Google Scholar]
- 68.van Lummel M, Pennings MT, Derksen RH, Urbanus RT, Lutters BC, Kaldenhoven N, de Groot PG. The binding site in {beta}2-glycoprotein I for ApoER2’ on platelets is located in domain V. The Journal of biological chemistry. 2005; 280: 36729–36. [DOI] [PubMed] [Google Scholar]
- 69.White-Adams TC, Berny MA, Tucker EI, Gertz JM, Gailani D, Urbanus RT, de Groot PG, Gruber A, McCarty OJ. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2). Arteriosclerosis, thrombosis, and vascular biology. 2009; 29: 1602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Puetz J Optimal use of recombinant factor VIIa in the control of bleeding episodes in hemophilic patients. Drug design , development and therapy. 2010; 4: 127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki K, Nishioka J, Hayashi T, Kosaka Y. Functionally active thrombomodulin is present in human platelets. Journal of biochemistry. 1988; 104: 628–32. [DOI] [PubMed] [Google Scholar]
- 72.Kowalska MA, Zhao G, Zhai L, David G 3rd, Marcus S, Krishnaswamy S, Poncz M. Modulation of protein C activation by histones, platelet factor 4, and heparinoids: new insights into activated protein C formation. Arteriosclerosis, thrombosis, and vascular biology. 2014; 34: 120–6. [DOI] [PubMed] [Google Scholar]
- 73.Zheng X, Li W, Song Y, Hu Y, Ferrell GL, Esmon NL, Esmon CT. Non-hematopoietic EPCR regulates the coagulation and inflammatory responses during endotoxemia. J Thromb Haemost. 2007; 5: 1394–400. [DOI] [PubMed] [Google Scholar]
- 74.Camire RM, Kalafatis M, Simioni P, Girolami A, Tracy PB. Platelet-derived factor Va/Va Leiden cofactor activities are sustained on the surface of activated platelets despite the presence of activated protein C. Blood. 1998; 91: 2818–29. [PubMed] [Google Scholar]
- 75.Somajo S, Koshiar RL, Norstrom E, Dahlback B. Protein S and factor V in regulation of coagulation on platelet microparticles by activated protein C. Thrombosis research. 2014; 134: 144–52. [DOI] [PubMed] [Google Scholar]