

Abstract
We introduce iridium-based conditions for the conversion of primary alcohols to potassium carboxylates (or carboxylic acids) in the presence of potassium hydroxide and either [Ir(2-PyCH2(C4H5N2))(COD)]OTf (1) or [Ir(2-PyCH2PBu2t)(COD)]OTf (2). The method provides both aliphatic and benzylic carboxylates in high yield and with outstanding functional group tolerance. We illustrate the application of this method to a diverse variety of primary alcohols, including those involving heterocycles and even free amines. Complex 2 reacts with alcohols to form crystallographically-characterized catalytic intermediates [IrH(η1,η3-C8H12)(2-PyCH2PtBu2)] (2a) and [Ir2H3(CO)(2-PyCH2PtBu2){μ-(C5H3N)CH2PtBu2}] (2c). The unexpected similarities in reactivities of 1 and 2 in this reaction, along with synthetic studies on several of our iridium intermediates, enable us to form a general proposal of the mechanisms of catalyst activation that govern the disparate reactivities of 1 and 2, respectively in glycerol and formic acid dehydrogenation. Moreover, careful analysis of the organic intermediates in the oxidation sequence enable new insights into the role of Tishchenko and Cannizzaro reactions in the overall oxidation.
Keywords: Acceptorless dehydrogenation, Iridium, Metal hydride, Alcohol, Carboxylic acid
TOC image
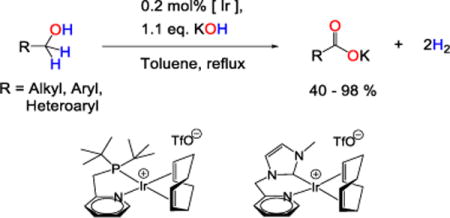
INTRODUCTION
Oxidation of primary alcohols to carboxylic acids is a quintessential transformation in organic chemistry, historically accomplished with a stoichiometric portion of a metal-based oxidant, such as potassium permanganate,1,2 chromium(VI) oxide,3 pyridinium dichromate,4 RuCl3/NaIO45, sodium hypochlorite,6 or the like. These evolved, ultimately to culminate in Lindgrin (and related) oxidation of aldehydes, the mild conditions of choice for complex molecule synthesis. Still, some forcing conditions to dehydrogenate primary alcohols directly to carboxylates have been known since the 19th century. For example, J. B. Dumas (1840) and later E. Reid et al.7 converted primary alcohols to carboxylate salts and H2 by heating with hydroxide at 350 °C. This approach has recently advanced to emerge as an elegant 21st-century replacement for the stoichiometric oxidant methods, which is important, because alcohol to carboxylate conversion remains a frequent operation in complex molecule synthesis.8–11
Carboxylate synthesis by acceptorless dehydrogenation presents a graceful approach with the recent development of new catalytic conditions, some enabling the reaction under very convenient conditions (25 – 120 °C, 1 atm). To date, homogeneous catalysts for this reaction include systems based on rhodium, ruthenium, and recently one iridium complex. The former include a diolefin amido tridentate ligand and catalyze hydrogen transfer from alcohols to acceptors like cyclohexanone,12 1-hexene,13 or O2/DMSO.14 Ruthenium complexes featuring tridentate PNN,15 PNP,16 and NHC-ligands,17 catalyze acceptorless dehydrogenation. The Cp*IrIII system enables reactions in neutral water.17a The former reactions take place in boiling NaOH solutions and give high yields of aliphatic carboxylates and benzoates. Such reactions also are possible in refluxing toluene18 and neat alcohol (150 °C).19
We recently reported very robust catalysts for acceptorless dehydrogenation of formic acid (based on 2, Figure 1)20 and glycerol (1, 4),21 with the latter giving an example of excellent selectivity for primary alcohol oxidation in the presence of other molecular complexity. In this study we report that these same iridium-based systems enable a method for acceptorless dehydrogenation of primary alcohols to carboxylic acids. This method gives good yields of many carboxylates and enables selectivity that is difficult to achieve with other catalytic systems. We show broad substrate scope, including reactions of alcohols with a secondary amino group or aryl halide, and report unanticipated features of the catalytic mechanism. Particularly, we show details of catalyst initiation and provide a unifying proposal of the mechanisms of catalyst activation that govern the disparate reactivities of 1 and 2, respectively in glycerol and formic acid dehydrogenation.
Figure 1.
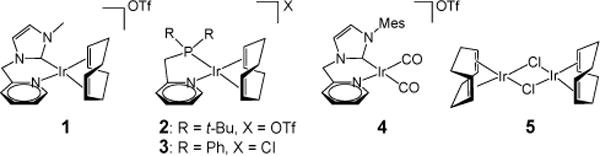
Iridium Complexes 1-5.
RESULTS AND DISCUSSION
Alcohol Oxidation
The discovery and optimization of our reaction is outlined in Table 1. The material balance of the reaction involves one equivalent of hydroxide. However, any base in the reaction will cause a parallel Guerbet self-condensation.7 For example,22 neat 1-octanol (6b) converts to potassium octanoate (7b, 72%) and 2-octyl-1-octanol (8b, 28%) in the presence of 1 eq. of potassium hydroxide and 1 (1 mol%) at 150 °C (yields calculated by NMR). In order to suppress this side-reaction, we conduct the dehydrogenation in a solvent of refluxing toluene: here, 1-octanol gives 99% of octanoate, 1% of Guerbet alcohol 8b after 40 h. This solvent choice contrasts many examples of primary alcohol dehydrogenation in the literature,15–17 in which reactions are typically run in water. Complex 1 is less effective in an aqueous medium, and the yield of octanoate is only 7% after 40 h. The origin of selectivity for carboxylate formation appears to be solubility: hydroxide’s limited solubility in toluene limits the total base concentration available for the Guerbet side reaction. In our hands, toluene is useful for most alcohols; diols and triols have limited solubility, and thus limited reactivity.
Table 1.
Selectivity Screening of Catalysts 1–5.a
|
| ||||
|---|---|---|---|---|
| Entry | [Ir] | R | Carboxylate, % | Guerbet Alcohol, % |
| 1 | 1 | Ethyl | > 99 | 0 |
| 2 | 2 | Ethyl | >99 | 0 |
| 3 | 1 | n-Hexyl | 99 | 1 |
| 4 | 2 | n-Hexyl | 95 | 5 |
| 5b | 1 | n-Tetradecyl | 90 | 10 |
| 6 | 2 | n-Tetradecyl | 92 | 8 |
| 7 | 3 | n-Tetradecyl | 77 | 23 |
| 8 | 4 | n-Tetradecyl | 77 | 23 |
| 9 | 5 | n-Tetradecyl | 76 | 24 |
Reaction conditions: a mixture of alcohol (2.1 mmol), KOH (2.4 mmol), catalyst (2.1 × 10−5 mol, 1 mol%), and toluene (4 mL) was stirred at reflux for 37 h (oil bath, 120 °C). The yields were derived from 1H NMR spectra.
0.1 mol% of 1 was used.
Table 1 shows the performance of our iridium pre-catalysts in alcohol dehydrogenation in toluene. The most effective catalyst, typically 1 or 2 in different cases, was determined among our available iridium(I) complexes (Figure 1) on the basis of product yield and selectivity. 1-Butanol (6a), 1-octanol (6b), and 1-hexadecanol (6c) were used as substrates. The selectivity for carboxylate synthesis decreases with increased molecular weight of the alcohol, which we believe to be an effect of base solubility.
Relative rates of catalysis for complexes 1, 2, and 5 were evaluated by recording hydrogen evolution in the conversion of benzyl alcohol (6f) to benzoate. Benzyl alcohol was chosen to avoid a Guerbet reaction. Kinetic profiles of hydrogen evolution demonstrate that complex 2 has the highest catalytic activity (Figure 2). Though 2 proved to be the fastest catalyst among the iridium compounds, complex 1 was also examined in substrate scope studies.
Figure 2.
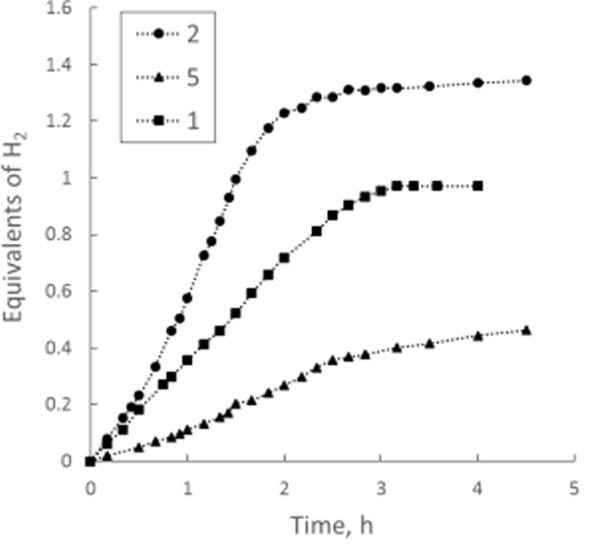
Hydrogen evolution profiles of 6f dehydrogenation with 1, 2, and 5. Conditions: a mixture of a catalyst (4 × 10−6 mol, 0.2 mol%), KOH (2.2 mmol), 6f (2.0 mmol), and toluene (10 mL) was actively stirred at reflux (oil bath, 120 °C).
Application of the optimized reaction conditions enabled an effective conversion of a variety of primary alcohols to potassium carboxylates (Table 2). Substrate scope includes aliphatic alcohols (entries 1 – 5), benzylic alcohols (entries 6 – 12), and even heteroatom functionalized systems like thioethers (entry 8), amino alcohols (entries 13 and 14) and heterocycles (entries 16 and 17). This latter class of substrates is unknown for other catalytic systems for this reaction, including the one based on iridium.
Table 2.
Substrate Scope for Dehydrogenation of Primary Alcohols Using Pre-Catalysts 1 and 2.a
| |||
|---|---|---|---|
| Entry | Alcohol | Product | Catalyst Time (h) Isolated yield (%) |
| 1 |
|
|
1 (0.2%) 40 96 |
| 2 |
|
|
1 (0.2%) 40 90 |
| 3 |
|
|
1 (0.2%) 40 85 |
| 4 |
|
|
2 (0.2%) 40 81 |
| 5 |
|
|
2 (0.4%) 75 77 |
| 6 |
|
|
2 (0.2%) 40 98 |
| 7 |
|
|
1 (0.4%) 36 79 |
| 8 |
|
|
1(0.1%) 40 74 |
| 9 |
|
|
2 (0.1%) 20 80 |
| 10 |
|
|
1 (0.1%) 15 40 |
| 11 |
|
|
2 (0.3%) 18 42 |
| 12 |
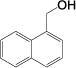
|
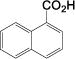
|
2(0.2%) 40 84 |
| 13 |
|
|
2 (0.2%) 40 82 |
| 14 |
|
|
2 (0.2%) 13 80 |
| 15 |
|
|
2 (0.2%) 40 80 |
| 16 |
|
|
1 (0.2%) 20 63 |
| 17 |
|
|
2 (0.2%) 40 65 |
| 18 |
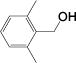
|
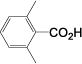
|
1 (0.1%) 15 0 |
Reaction conditions: a mixture of an alcohol (2.0 mmol), KOH (2.2 mmol), catalyst, and toluene (10 mL) was stirred at reflux.
Several generalizations can be drawn from these data. Unfunctionalized alkyl systems proceed smoothly (entries 1-3), and an adjacent strained ring is not derivatized in the reaction (entry 4). Sterically bulky systems can be problematic. For example, entries 5 and 18 demonstrate that a 1-adamantyl substituent (6e) slows the reaction, ca. half the rate, although it reaches completion. A doubly-blocked 2,6-dimethylbenzyl alcohol (6r) is unreactive.
Although aryl bromides and iodides are only moderately tolerated, entries 10 and 11 give examples of Ar-Br and Ar-I bonds surviving alcohol dehydrogenation conditions. We observe reduction (to Ar-H groups) as the major side reaction in these cases.18 For example, 4-bromobenzyl alcohol (6j) undergoes dehydrogenation to form both the corresponding 4-bromobenzoate (58%) and the reduced benzoate (9%) product. 4-Iodobenzyl alcohol (6k) afforded a larger amount of dehalogenated benzoate (32%) in addition to the halogenated product (42%). In contrast to aryl bromides and iodides, aryl chlorides are well tolerated (entry 9), and afford access to a growing number of metal-catalyzed coupling reactions.23
Secondary amines and azoles are tolerated. Whereas several iridium complexes catalyze alkylation of primary amines with alcohols,24,25 we were curious how an amino alcohol would fare in our conditions. We found that we can convert amino alcohol 6m efficiently to the corresponding amino acid without polymerization or other side reactions. This presents an unprecedented approach to amino acid synthesis. We further find that intramolecular oxidative cyclization of amino alcohols is possible: dehydrogenation of 6n results in cyclization, yielding indole 7n exclusively.24a
We observe good selectivity for carboxylate synthesis in cases where arene hydrogenation or reductive decarboxylation can take place. For example, alcohol 6l gives a high yield (84%, entry 12) of the corresponding carboxylic acid, however, we also observe trace quantities of naphthalene, 1,2-dihydronaphthalene, tetralin, and potassium formate in the reaction mixture. Quinoline 6q is even more susceptible to over-reduction, yet we see only a trace of the corresponding tetrahydroquinoline in the crude product mixture of 7q. By contrast, an attempt to dehydrogenate 2-(hydroxymethyl)thiophene and 3-phenylpropargyl alcohol resulted in product decarboxylation, giving potassium formate. Unfortunately, alkenes and nitro compounds are incompatible with our conditions and undergo uncontrolled reduction.
We showed that our catalytic systems can be applied to large-scale synthesis of carboxylic acids. Complexes 1 and 2 convert benzyl alcohol to benzoic acid with turnover numbers 16400 and 40600 respectively. Pre-catalyst 2 loading can be as low as 50 ppm to give up to 15 g of benzoic acid. Moreover, precipitation of the carboxylate during the reaction enables easy separation of the product by simple filtration. The catalyst-containing toluene solution can then be reused.
The catalytic method presented here is the second example we know for iridium based primary alcohol dehydrogenation. The previously reported method by Fujita utilizes IrIII pre-catalyst [Cp*Ir(NC)(H2O)](OTf)2 (NC – pyridylcarbene bidentate ligand).17a An advantage of the method is the possibility to conduct alcohol oxidation under base-free conditions, whereas our method requires stoichiometric KOH. Fujita’s method uses high catalyst loading (2 – 5 mol%) and the reaction scope is confined to simple benzyl alcohols, with reactions of aliphatic alcohols giving yields of <25%. Thus, our method is a useful complement to the Fujita’s chemistry.
Mechanism
The near-interchangeability of 1 and 2 in the conversions of 6 to 7 was not anticipated. We observe very different reactivity of these species respectively in glycerol21 and formic acid20 dehydrogenation: 1 works only in the former and 2 only in the latter. Carboxylate synthesis thus provides us with a platform from which to run comparative stoichiometric reactions of 1 and 2 (Scheme 1) to gain insight into both the mechanisms of catalyst initiation and the differences in their reactivities.
Scheme 1.
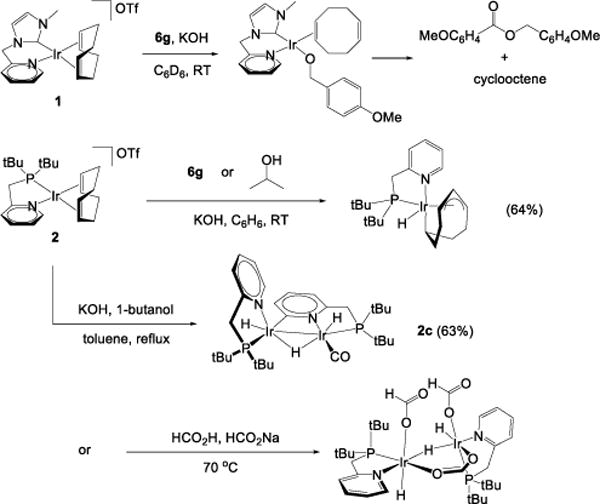
Reactions of Complexes 1 and 2.
Complex 1 reacts with neat alcohol 6g in the presence of potassium hydroxide to form iridium(I) alkoxide complex 1a, which can be extracted from potassium salts with C6D6. The structure of 1a was established by NMR, with the coordination geometry assigned by NOE analysis (see Supporting Information). The complex retains its bidentate N—C ligand without proton loss, and the aryl alkoxide ligand exchanges slowly (k ~ 1 s−1) with excess 6g in solution. Complex 1a is reactive in benzene solution in the presence of excess 6g, converting it to 4-methoxybenzyl-4-methoxybenzoate (1b). The iridium-containing species precipitates from solution leaving cyclooctene. These data are consistent with hydrogen transfer from 6g to coordinated 1,5-cyclooctadiene to give reduction of one its olefins. We observe the same of 1 in glycerol dehydrogenation.21 Importantly, we see no evidence of an iridium hydride or any other metallic intermediate in this sequence.
Complex 2 reacts with 6g or isopropanol to give iridium(III) complex 2b (Scheme 1), which can be isolated in 64% yield. The structure of 2b was established by single-crystal X-ray diffraction (Figure 3). It is analogous to the structure of [IrH(η1,η3-C8H12)(dppm)], reported by Werner and co-workers.26 We believe that an intermediate iridium alkoxide is involved in the formation of 2b, but unlike 1a, this converts to a stable iridium hydride species. We believe that this is the same initiation sequence observed for 2 in the dehydrogenation of formic acid, because 2b is easily converted to 2d, a form of the active catalyst of formic acid dehydrogenation initiating from 2.20
Figure 3.
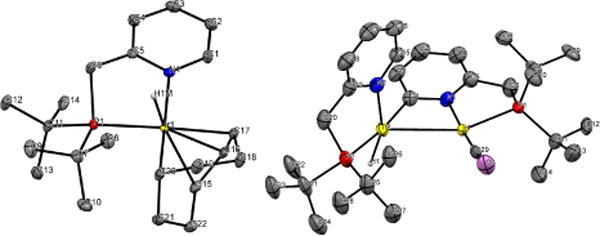
Molecular structures of 2b (left) and 2c (right) shown with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity, except for localized hydrides.
Complex 2 reacts with 1-butanol and potassium hydroxide in boiling toluene to form dinuclear iridium(II) complex 2c (Scheme 1), which is isolated in 63% yield. Its structure, shown in Figure 3, has two notable features: a single CO ligand and a bridging ortho-metalated pyridine fragment. The CO ligand derives from n-butanal,27–30 and several cases of pyridine ortho-metalation by iridium complexes have been described.31–33 1H NMR of 2c shows three hydride ligands; their arrangement in the coordination sphere was confirmed by COSY, NOESY/EXSY, and 1H – 31P HMBC experiments (see Supporting Information).
Complex 2c is stable in the solid state and in solution at room temperature in the absence of air, however it undergoes reversible isomerization to 2f in toluene at 110 °C (Scheme 2). According to 1H NMR data, heating the solution of pure 2c leads to disappearance of the two cis-hydride signals and reduction of types of tert-butyl groups from four to two. This indicates that 2c is involved in a fast dynamic equilibrium with a symmetrical species, which we propose to be the product of Ir–H–Ir bridge cleavage 2e. The next step is slow, and the system comes to equilibrium only after four hours (Keq = [2f]/[2c]= 0.626; ΔG°383 = 360 cal/mol; k1 = 1.4(1) × 10−4 s−1; and k−1 = 2.2(1) × 10−4 s−1). The equilibrated mixture at room temperature contains chemical shifts of pure 2c and 2f only.
Scheme 2.
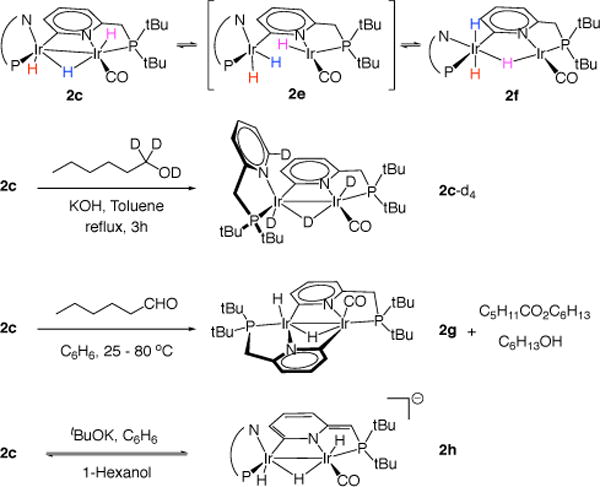
Reactions of Complex 2c.
The structure of 2f was established by NMR studies. 1H spectrum contains two hydride peaks in the ratio 1 : 2 at −6.64 (d, 2JPH = 150.4) and −8.98 (br. s) ppm respectively. Seven aromatic peaks indicate that 2f has two ligands and one of them is ortho-metalated. Two doublets of the four tert-butyl groups indicate the presence of a symmetry plane in the molecule. 1H – 31P HMBC experiment demonstrates coupling between different hydrides (−6.64, and −8.98 ppm) and different phosphorus nuclei (63.79, and 88.66 ppm respectively). Hence, the only reasonable molecular structure must have all three hydrides bound to one iridium center. We assign this structure as 2f in scheme 2.
Complex 2c reacts with 10 eq. of n-hexanal at room temperature. This initially produces a number of unidentified iridium hydride complexes. After two days at 80 °C the reaction reaches completion and all the transient iridium hydrides turn to a single complex, 2g (Scheme 2). Its structure was determined with NMR and MALDI-MS data. The 1H NMR spectrum of 2g contains two peaks of the hydride ligands at −8.44 (ddd) and −11.15 (ddd) ppm and six aromatic peaks, suggesting that both pyridine fragments are ortho-metalated. No chemical exchange (EXSY) was observed between the hydride ligands, which is consistent with their proposed trans configuration. Complex 2g has two hydrogen atoms fewer than 2c, meaning that it is a dehydrogenated form of 2c. The identified organic products in this reaction are 1-hexanol and hexyl hexanoate. 1-Hexanol is the product of hydrogen transfer from 2c to n-hexanal, and hexyl hexanoate is the product of Tishchenko dimerization. Hexanal remains present in the mixture after prolonged heating, showing that 2g does not catalyze its disproportionation; this is most likely accessed via one of the transient iridium hydrides mentioned above. Reduction of 2g back to 2c by 1-hexanol or H2 (1 atm) at 110 °C in toluene is not detected, even after 12 h.
When 2 reacts with 1-butanol and KOH in boiling toluene a mixture of iridium hydride complexes is formed, in which 2c, 2f, and 2g are the major components (Figure 4). Moreover, we showed that 2c is active in benzyl alcohol dehydrogenation with an identical kinetic profile to 2. These facts suggest that 2c, 2f, and 2g are the catalyst resting states which convert among each other during alcohol dehydrogenation with pre-catalyst 2.
Figure 4.
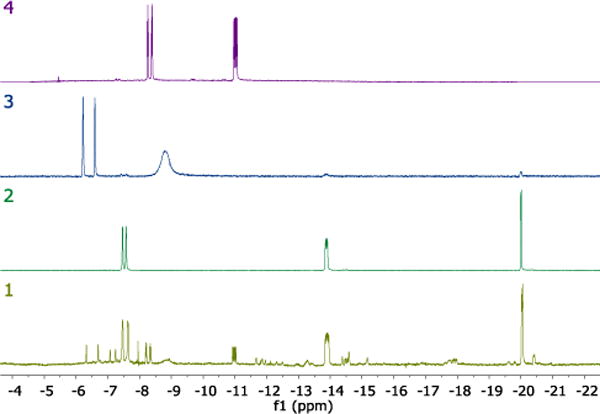
1H NMR spectra (600 MHz, C6D6) demonstrating IrH peaks: (1) mixture of complexes formed in the reaction between 2, 1-butanol, and KOH in boiling toluene; (2) 2c; (3) 2f; (4) 2g.
Whereas the interconversion of 2c and 2f is accessed at elevated temperature, it is unclear how 2c can convert reversibly to 2g in the catalytic process. Nevertheless, we showed this transformation to be involved in the catalytic mechanism by conducting deuteration experiment. Treatment of 2c with 1-hexanol-O,1,1-d3 (6s) under dehydrogenation conditions (Scheme 2) reveals partial deuteration of all three hydrides and the pyridine ortho-hydrogen in equal portions. Thus, all three hydrides are reversibly derivatized under the catalytic conditions. Since 2g contains two ortho-metalated pyridine fragments we propose it as the intermediate in the ortho-deuteration of the free pyridine fragment of 2c.
The linking methylene group of complex 2c has an acidity comparable to that of alcohols (Scheme 2), but we do not observe the deprotonated form as a major species in catalysis. For example, we observe that treating yellow solution of 2c with t-BuOK in benzene gives red-colored complex 2h (Scheme 2). A 1H NMR spectrum of 2h shows selective deprotonation of benzylic arm of the metalated ligand and dearomatization of the corresponding pyridine system. The hydride ligands remain intact. Similar reactivity of structurally related PNP pincer ligands with similar colorometric behavior was reported by Milstein.34 Complex 2h undergoes complete protonation by 1-hexanol to return initial 2c, indicating that 2c is more acidic than t-BuOH, but less acidic than 1-hexanol.
Scheme 3 illustrates the proposed mechanism of catalyst generation and catalytic alcohol dehydrogenation using complex 2 as a pre-catalyst. We believe that both 1 and 2 initiate through analogous sequences, but that the hydrides of 1 are high energy, and are thus not observed. We believe that the difference in hydride stabilities is a consequence of the subtle electronic differences between the respective carbene and phosphine groups of 1 and 2. The first step involves a nucleophilic attack of an alkoxide anion on 2, producing iridium(I) alkoxide 2a, which is the structural analogue of 1a. Species 2a undergoes β-hydride elimination to give an aldehyde and complex 2b, the mechanism of this transformation had been studied previously on a similar iridium complex.26
Scheme 3.
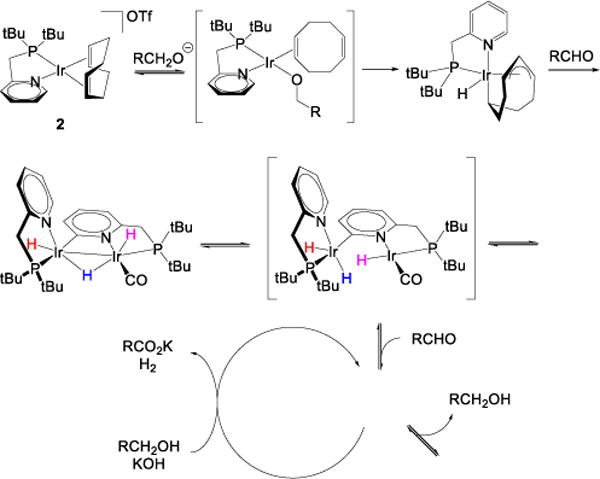
Mechanism of Pre-catalyst 2 Activation.a
a. Colored labels on iridium hydride groups are not intended to imply specificity in hydride transfer steps, but simply to guide the reader. In fact, these hydrides equilibrate rapidly.
Complex 2b is stable at room temperature, and subsequent intermediates were accessed at elevated temperature. We find that 2b forms complex mixtures of iridium hydride species when reacted with 1-butanol or n-hexanal at 80 °C, however, complex 2c was detected after reaction with n-hexanal. Hence, we conclude that 2b can be converted to 2c by an aldehyde produced in the conversion of 2a. This gives our system access to the 2c/2f equilibrium. Since we have shown that 2c reacts with n-hexanal rather than 1-hexanol, we believe that the next step of the mechanism involves reversible reaction between 2e and an aldehyde, ultimately leading to 2g. We suggest that one of the intermediates in this reaction is the jumping-off point for the catalytic process (“Active Catalyst”, scheme 3). We expect the catalytic cycle to proceed by a traditional β-hydride elimination from an intermediate iridium alkoxide. We expect this to be irreversible, because toluene solutions of 2c, 2f, and 2g do not react with H2 (1 atm) at either 25 or 110 °C. Although we could not fully characterize the chain of all iridium species involved, all of them that we have are dinuclear, starting from 2c.
Our studies on the organic intermediates involved in the alcohol dehydrogenation process turned up two unexpected observations (Scheme 4). First, formation of ester by-products rules in the possibility of Tishchenko like reaction of an intermediate aldehyde. Second, kinetic evidence necessitates Cannizzaro reaction in the mechanism of oxidation of benzyl alcohols.
Scheme 4.
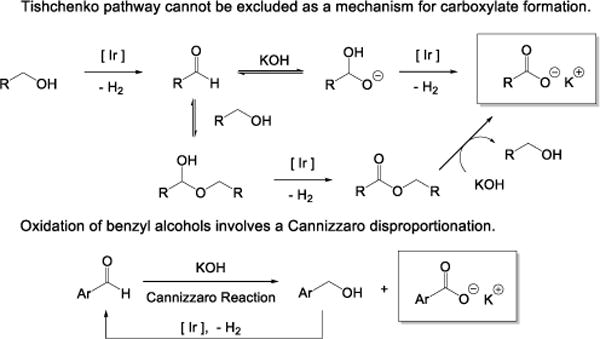
Mechanisms of Aldehyde and Carboxylate Formation.
Synthesis of 1a is illustrated in scheme 1. We find that upon exposure to our catalytic conditions at 25 °C, this material generates 4,4′-dimethoxybenzyl benzoate, which indicates that Tishchenko like reaction of 4-methoxybenzaldehyde is faster than its direct conversion to 7g. Moreover, formation of hexyl hexanoate in the reaction between 2c and n-hexanal shows that ester generation is also possible when pre-catalyst 2 is used. In order to verify our proposed Tishchenko pathway, we showed that a significant portion of an aldehyde exists in the hemiacetal form under catalytic conditions: NMR data show that n-hexanal reacts with an excess of 1-butanol in the presence of catalytic KOH in toluene to give the corresponding hemiacetal (1-butoxyhexan-1-ol). Thus, we must consider ester formation as a route in the mechanism of carboxylate synthesis (Scheme 4A).
Aromatic aldehydes undergo Cannizzaro reaction in the presence of KOH to give the corresponding benzyl alcohols and carboxylates (Scheme 4B). Such disproportionation is known under our conditions in the absence of iridium (1 h, KOH, toluene, 110 °C).18 In our conditions, the resulting alcohol converts to aldehyde via iridium catalyzed dehydrogenation. We are convinced that Cannizzaro reaction must be happening, because when benzaldehyde is used instead of an alcohol under typical dehydrogenation conditions, it takes ca. 30 min for hydrogen evolution to begin. If no Cannizzaro reaction was involved, aldehyde oxidation would initiate immediately. This delay is apparently benzaldehyde disproportionation that is required to generate a sufficient amount of benzyl alcohol to form the catalytic species. We expect that it is required to convert 1 to 1a (and 2 to 2a), and that its C—H groups are needed to convert on to 1b (or 2b): an aldehyde cannot fill this role. After hydrogen formation ceases, traces of benzyl alcohol can be detected in the reaction mixture by 1H NMR spectroscopy. These observations necessitate Cannizzaro reaction, but do not necessitate or exclude Tishchenko reaction in the sequence.
SUMMARY
In conclusion, complexes 1 and 2 are efficient pre-catalysts for the conversion of primary alcohols to potassium carboxylates. Under optimized the reaction conditions the method applicable to a wide range of substrates, including some (amino alcohols and some heterocycles) that are unknown for any catalytic conditions in this class. We show that 1 and 2 are both active in primary alcohol dehydrogenation, despite their previously investigated difference in catalytic activity towards glycerol and formic acid dehydrogenation. Upon catalysis initiation, complex 1 forms observable iridium(I) alkoxide 1a, which decomposes to relatively reactive iridium hydrides. On the contrary, complex 2 reacts with alcohols in the presence of KOH to form a number of stable iridium hydride complexes 2b, 2c, 2f, and 2g even though 2a is not observed. We propose that the jumping-off point for catalysis to be an intermediate in the equilibration of 2c and 2g.
Taken together with our prior work on glycerol and formic acid dehydrogenation, realized respectively with 1 and 2, these data help to frame a picture that explains the specify of 1 and 2 for those processes: both proceed through initiation sequences that are differentiated by the reactivities of intermediate iridium hydride intermediates that steer initiation to the respective active species. These stabilities are apparently governed by differences in metal-ligand bonding between NHC carbene and phosphine groups, thus illustrating dramatic consequences manifested by the subtle differences in these ligating moieties. More details on this generalization are forthcoming from our lab.
EXPERIMENTAL SECTION
Materials and Methods
Chloroform-d1, dimethyl sulfoxide-d6, methanol-d4, D2O, benzene-d6, and toluene-d8 were purchased from Cambridge Isotopes Laboratories. Benzene-d6, toluene-d8, and toluene were dried and distilled according to known procedures. Iridium complexes 1, 2, 3, and 4 were synthesized according to described procedures.20,21 Alcohols 6a – 6r, methanol, isopropanol, dichloromethane, ethyl acetate, methyl hexanoate, 18-crown-6, chloro(1,5-cyclooctadiene)iridium(I) dimer, and potassium hydroxide were purchased from commercial sources without further purification. Benzaldehyde was distilled under reduced pressure prior to use. All air and water sensitive procedures were carried out in a Vacuum Atmosphere glove box under nitrogen (2-10 ppm O2 for all manipulations). 1H, 13C, 31P NMR spectra were recorded on Varian Mercury 400 and VNMRS 600 spectrometers, and processed using MestReNova v11.0.2. All chemical shifts are reported in ppm and referenced to the residual 1H or 13C solvent peaks. Following abbreviations are used: (s) singlet, (bs s) broad singlet, (d) doublet, (t) triplet, (dd) double doublet, etc. NMR spectra of air-sensitive compounds were taken in 8″ J. Young tubes (Wilmad or Norell) with Teflon valve plugs. Infrared spectra were recorded on Bruker OPUS FTIR spectrometer. Samples of pure potassium carboxylates were treated with acetic acid in ethyl acetate followed by GC-MS analysis on Thermo Scientific Focus DSQ II instrument. MALDI-MS spectra were acquired on Bruker Autoflex Speed MALDI Mass Spectrometer. Elemental analyses were conducted on Flash 2000 CHNS Elemental Analyzer.
General Procedure for Alcohol Dehydrogenation
An alcohol (2.0 mmol), iridium complex 1 or 2 (see table 2), and potassium hydroxide (123 mg, 2.2 mmol) were mixed with dry toluene (10 mL). The suspension was stirred at reflux for required period of time (oil bath, 120 °C). After the reaction was over, the solvent was evaporated under reduced pressure affording crude potassium carboxylate.
Isolation method A
Potassium carboxylate was dissolved in deionized water (40 mL) and the resulting solution was washed with dichloromethane (2 × 10 mL). Then, the solution was acidified with 1 M HCl, and extracted with ethyl acetate (3 × 10 mL). The organic phase was separated, dried (Na2SO4) and evaporated in vacuum, giving pure carboxylic acid.
Isolation method B
Potassium carboxylate was dissolved in DI water (40 mL) and the resulting solution was washed with dichloromethane (2 × 10 mL). The aqueous solution was evaporated in vacuum to dryness and the residue was dissolved in methanol. The methanol solution was filtered, and the filtrate was evaporated to dryness, giving pure potassium carboxylate.
Potassium Butyrate (7a)
Potassium Butyrate (7a) was isolated by method B as a white powder (0.24 g, 96%). 1H NMR (600 MHz, D2O): δ 2.16 (t, J = 7.3 Hz, 2H, CH2), 1.57 (h, J = 7.3 Hz, 2H, CH2), 0.90 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (151 MHz, D2O): δ 184.05, 39.64, 19.35, 13.26. IR (PE film, cm−1): 2956, 2918, 1564, 1412, 1254, 888, 750. GC-MS: m/z calcd. for C4H8O2 [M]+ 88.05, found 88.1.
Potassium Octanoate (7b)
Potassium Octanoate (7b) was isolated by method B as a white powder (0.33 g, 90%). 1H NMR (400 MHz, D2O): δ 2.18 (t, J = 7.5 Hz, 2H, CH2), 1.63 – 1.47 (m, 2H, CH2), 1.38 – 1.20 (m, 8H, 4CH2), 0.88 (t, J = 6.7 Hz, 3H, CH3). 13C NMR (100 MHz, D2O): δ 184.13, 37.50, 30.88, 28.55, 28.09, 25.75, 21.83, 13.24. IR (KBr, cm−1): 2954, 2926, 2854, 1563, 1411, 914, 718, 694. GC-MS: m/z calcd. for C8H16O2 [M]+ 144.12, found 144.1.
Potassium Palmitate (7c)
After evaporation of toluene, crude 7c was washed with hexanes and dried under reduced pressure. White powder (0.50 g, 85%). 1H NMR (600 MHz, CD3OD): δ 2.15 (t, J = 8.6 Hz, 2H, CH2), 1.63 – 1.55 (m, 2H, CH2), 1.35 – 1.25 (m, 24H, 12CH2), 0.90 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, CD3OD): δ 183.12, 39.38, 33.07, 30.90, 30.79 (6CH2), 30.76, 30.68, 30.47, 27.85, 23.73, 14.45. IR (PE film, cm−1): 2925, 2850, 1561, 1472, 1331, 1104, 716. GC-MS: m/z calcd. for C16H32O2 [M]+ 256.24, found 256.3.
Potassium Cyclobutanecarboxylate (7d)
Potassium Cyclobutanecarboxylate (7d) was isolated by method B as a white powder (0.22 g, 81%). 1H NMR (600 MHz, D2O): δ 3.03 (p, J = 8.7 Hz, 1H, CH), 2.15 – 2.05 (m, 4H, 2CH2), 1.89 (h, J = 9.2 Hz, 1H, CH2), 1.78 – 1.70 (m, 1H, CH2). 13C NMR (151 MHz, D2O): δ 185.74, 40.92, 25.93, 17.29. IR (PE film, cm−1): 2975, 2941, 2859, 1656, 1553, 1409, 680. GC-MS: m/z calcd. for C5H8O2 [M]+ 100.05, found 100.1.
1-Adamantanecarboxylic Acid (7e)
Isolation by method A followed by recrystallization from hexane-ethanol mixture gave 7e as colorless crystals (0.34 g, 77%). 1H NMR (600 MHz, CDCl3): δ 2.00 – 2.05 (m, 3H, 3CH), 1.88 – 1.94 (m, 6H, 3CH2), 1.67 – 1.77 (m, 6H, 3CH2). 13C NMR (151 MHz, CDCl3): δ 184.45, 40.64, 38.71, 36.57, 27.97. IR (PE film, cm−1): 2928, 1693, 1450, 1410, 1284, 1085, 951, 744, 670, 531. GC-MS: m/z calcd. for C11H16O2 [M]+ 180.12, found 180.1.
Benzoic Acid (7f)
Isolation by method A followed by recrystallization from toluene gave 7f as colorless crystals (0.24 g, 98%). 1H NMR (600 MHz, DMSO-d6): δ 12.93 (br s, 1H, CO2H), 7.95 (d, J = 7.0 Hz, 2H, 2CH), 7.62 (t, J = 7.4 Hz, 1H, CH), 7.50 (t, J = 7.7 Hz, 2H, 2CH). 13C NMR (151 MHz, DMSO-d6): δ 167.33, 132.88, 130.76, 129.27, 128.58. IR (PE film, cm−1): 1689, 1455, 1426, 1327, 1294, 936, 709. GC-MS: m/z calcd. for C7H6O2 [M]+ 122.04, found 122.0.
4-Methoxybenzoic Acid (7g)
Isolation by method A followed by recrystallization from toluene-ethanol mixture gave 7g as colorless crystals (0.24 g, 79%). 1H NMR (600 MHz, DMSO-d6): δ 12.62 (br s, 1H, CO2H), 7.89 (d, J = 8.7 Hz, 2H, 2CH), 7.01 (d, J = 8.7 Hz, 2H, 2CH), 3.81 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6): δ 167.04, 162.86, 131.37, 123.00, 113.83, 55.45. IR (PE film, cm−1): 1683, 1603, 1427, 1301, 1263, 1168, 1026, 927, 844, 773, 617, 550. GC-MS: m/z calcd. for C8H8O3 [M]+ 152.05, found 152.0.
4-(Methylthio)benzoic Acid (7h)
Isolation by method A followed by recrystallization from toluene-ethanol mixture gave 7h as colorless crystals (0.25 g, 74%). 1H NMR (500 MHz, CD3OD): δ 7.92 (d, J = 7.9 Hz, 2H, 2CH), 7.30 (d, J = 8.1 Hz, 2H, 2CH), 2.52 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 169.62, 147.25, 131.07, 127.81, 125.94, 14.66. IR (KBr, cm−1): 1680, 1595, 1421, 1325, 1192, 757. GC-MS: m/z calcd. for C8H8O2S [M]+ 168.02, found 168.0.
4-Chlorobenzoic Acid (7i)
Isolation by method A followed by recrystallization from toluene-ethanol mixture gave 7i as colorless crystals (0.25 g, 80%). 1H NMR (400 MHz, DMSO-d6): δ 13.09 (br s, 1H, CO2H), 7.93 (d, J = 8.5 Hz, 2H, 2CH), 7.55 (d, J = 8.5 Hz, 2H, 2CH). 13C NMR (151 MHz, DMSO-d6): δ 166.49, 137.83, 131.16, 129.67, 128.75. IR (KBr, cm−1): 2924, 2955, 1680, 1322, 1284, 762. GC-MS: m/z calcd. for C7H5ClO2 [M]+ 156.00, 157.99; found 156.0, 158.0.
4-Bromobenzoic Acid (7j)
Isolation by method A followed by recrystallization from toluene-ethanol mixture gave 7j as colorless crystals (0.16 g, 40%). 1H NMR (600 MHz, DMSO-d6): δ 13.17 (s, 1H, CO2H), 7.86 (d, J = 7.1 Hz, 2H, 2CH), 7.70 (d, J = 7.1 Hz, 2H, 2CH). 13C NMR (151 MHz, DMSO-d6): δ 166.58, 131.68, 131.27, 130.00, 126.85. IR (PE film, cm−1): 1676, 1587, 1426, 1320, 1070, 1013, 758. GC-MS: m/z calcd. for C7H5BrO2 [M]+ 199.95, 201.95; found 199.9, 201.9.
4-Iodobenzoic Acid (7k)
Isolation by method A followed by recrystallization from toluene-ethanol mixture gave 7k as colorless crystals (0.21 g, 42%). 1H NMR (600 MHz, DMSO-d6): δ 13.12 (s, 1H, CO2H), 7.88 (d, J = 8.1 Hz, 2H, 2CH), 7.69 (d, J = 8.0 Hz, 2H, 2CH). 13C NMR (151 MHz, DMSO-d6): δ 166.88, 137.55, 131.04, 130.27, 101.14. IR (PE film, cm−1): 1675, 1427, 1009, 754. MALDI-MS: m/z calcd. for C7H5INaO2 [M + Na]+ 270.92, found 270.72.
1-Naphthoic Acid (7l)
Isolation by method A followed by recrystallization from toluene gave 7l as colorless crystals (0.29 g, 84%). 1H NMR (600 MHz, DMSO-d6): δ 13.14 (s, 1H, CO2H), 8.87 (d, J = 8.6 Hz, 1H, CH), 8.15 (d, J = 7.4 Hz, 2H, 2CH), 8.01 (d, J = 8.0 Hz, 1H, CH), 7.64 (t, J = 7.7 Hz, 1H, CH), 7.59 (t, J = 7.7 Hz, 2H, 2CH). 13C NMR (151 MHz, DMSO-d6): δ 168.65, 133.46, 132.92, 130.67, 129.84, 128.59, 127.71, 127.55, 126.17, 125.48, 124.87. IR (PE film, cm−1): 2916, 1674, 1593, 1306, 774. MALDI-MS: m/z calcd. for C11H8NaO2 [M + Na]+ 195.04, found 194.87.
Potassium Ethylaminoacetate (7m)
Potassium Ethylaminoacetate (7m) isolated by method B as a white powder (0.23 g, 82%). 1H NMR (600 MHz, D2O): δ 3.26 (s, 2H, CH2), 2.70 (q, J = 6.8 Hz, 2H, CH2), 1.13 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (151 MHz, D2O): δ 177.81, 51.11, 42.62, 12.90. IR (PE film, cm−1): 1597, 1407, 1383, 1283. MALDI-MS: m/z calcd. for C4H8K2NO2 [M + K]+ 179.98, found 180.00.
Indole (7n)
Complex 2 (2.7 mg, 4 μmol), 6n (274 mg, 2.0 mmol), and potassium hydroxide (123 mg, 2.2 mmol) were mixed with dry toluene (10 mL). The suspension was stirred at reflux for 13 hours (oil bath, 120 °C). After the reaction was over, the solvent was evaporated in vacuum, and the residue was stirred at reflux with hexanes (50 mL) and charcoal for 1 h. Then, the hexane solution was filtered and evaporated in vacuum giving 7n as a colorless liquid (0.19 g, 80%). 1H NMR (600 MHz, CDCl3): δ 8.11 (br s, 1H, NH), 7.68 (dd, J = 7.8, 0.7 Hz, 1H, CH), 7.41 (dd, J = 8.0, 0.7 Hz, 1H, CH), 7.25 – 7.19 (m, 2H, 2CH), 7.17 – 7.12 (m, 1H, CH), 6.60 – 6.56 (m, 1H, CH). 13C NMR (151 MHz, CDCl3): δ 135.88, 127.95, 124.25, 122.09, 120.84, 119.92, 111.14, 102.71. IR (PE film, cm−1): 3401, 1457, 1353, 1246, 1090, 932, 746, 612, 505, 430. GC-MS: m/z calcd. for C8H7N [M]+ 117.06, found 117.1.
3-Phenylpropanoic Acid (7o)
Isolation by method A gave 7o as a yellow liquid (0.24 g, 80%). 1H NMR (600 MHz, CDCl3): δ 11.57 (br s, 1H, CO2H), 7.34 – 7.29 (m, 2H, 2CH), 7.25 – 7.20 (m, 3H, 3CH), 2.99 (t, J = 7.8 Hz, 2H, CH2), 2.71 (t, J = 7.8 Hz, 2H, CH2). 13C NMR (151 MHz, CDCl3): δ 179.50, 140.27, 128.70, 128.39, 126.51, 35.77, 30.71. IR (PE film, cm−1): 3028, 1708, 1496, 1417, 1295, 1215, 936, 748, 699. GC-MS: m/z calcd. for C9H10O2 [M]+ 150.07, found 150.1.
Potassium Pyridine-2-carboxylate (7p)
Potassium Pyridine-2-carboxylate (7p) was isolated by method B as a white powder (0.20 g, 63%). 1H NMR (500 MHz, CD3OD): δ 8.58 (d, J = 4.4 Hz, 1H, CH), 8.02 (d, J = 7.7 Hz, 1H, CH), 7.85 (t, J = 8.1 Hz, 1H, CH), 7.46 – 7.32 (m, 1H, CH). 13C NMR (126 MHz, CD3OD): δ 172.94, 156.47, 149.55, 138.17, 125.93, 125.01. IR (KBr, cm−1): 2927, 1640, 1405, 702. GC-MS: m/z calcd. for C6H5NO2 [M]+ 123.03, found 123.0.
Potassium Quinoline-2-carboxylate (7q)
Potassium Quinoline-2-carboxylate (7q) was isolated by method B as a white powder (0.27 g, 65%). 1H NMR (600 MHz, D2O): δ 7.88 (d, J = 8.5 Hz, 1H, CH), 7.77 (d, J = 8.5 Hz, 1H, CH), 7.60 (d, J = 8.4 Hz, 1H, CH), 7.50 (t, J = 7.7 Hz, 1H, CH), 7.45 (d, J = 8.1 Hz, 1H, CH), 7.28 (t, J = 7.5 Hz, 1H, CH). 13C NMR (151 MHz, D2O): δ 172.84, 154.01, 145.89, 137.75, 130.12, 128.09, 127.96, 127.60, 127.38, 120.13. IR (KBr, cm−1): 3298, 1615, 1387, 802, 770. GC-MS: m/z calcd. for C9H7N [M – CO2]+ 129.16, found 129.1.
1-Hexanol-O,1,1-d3 (6s)
In a glovebox, a solution of methyl hexanoate (2.06 g, 15.8 mmol) in 10 mL of dry ether was added drop-wise to a stirred solution of LiAlD4 (0.67 g, 15.8 mmol) in 20 mL of ether. The mixture was stirred at room temperature for 24 h, and then carefully quenched with D2O at vigorous stirring till aluminum hydroxide separated from organic phase. The ethereal solution was separated, dried (Na2SO4), and evaporated in vacuum. The product was obtained as a colorless liquid (1.34 g, 81%). 1H NMR (600 MHz, CDCl3): δ 1.52 (t, J = 7.8 Hz, 2H, CH2), 1.34 – 1.24 (m, 6H, 3CH2), 0.87 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, CDCl3): δ 62.13 (p, 1JCD = 21.5 Hz), 32.58, 31.76, 25.49, 22.73, 14.10. IR (PE film, cm−1): 3331, 2957, 2859, 1467, 1160, 1127, 967. GC-MS: m/z calcd. for C6H10D2 [M – HDO]+ 86.11, found 86.1.
[Ir(2-PyCH2(C4H5N2))(η2-COD)(OCH2C6H4OMe)] (1a)
In a glovebox, complex 1 (40.0 mg, 6.4 × 10−5 mol), 6g (18.0 mg, 1.3 × 10−4 mol), potassium hydroxide (7.2 mg, 1.3 × 10−4 mol) and 2 – 3 drops of C6D6 were mixed together till the slurry turned yellow. Then, C6D6 (0.7 mL) was added resulting in a yellow solution of 1a and potassium triflate precipitate. The solution was immediately filtered and transferred to J. Young NMR tube. The structure of 1a was derived from NMR data. 1H NMR (600 MHz, C6D6): δ 8.33 (dq, J = 5.0, 0.9 Hz, 1H, Py), 7.39 (d, J = 7.8 Hz, 1H, Py), 7.35 (d, J = 8.6 Hz, 2H, C6H4), 7.08 (td, J = 7.7, 1.8 Hz, 1H, Py), 6.85 (d, J = 8.6 Hz, 2H, C6H4), 6.64 (d, J = 1.9 Hz, 1H, NHC), 6.58 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H, Py), 5.86 (d, J = 1.9 Hz, 1H, NHC), 5.73 (d, J = 14.7 Hz, 1H, NCH2), 5.42 (d, J = 14.7 Hz, 1H, NCH2), 5.24 (d, J = 13.8 Hz, 1H, OCH2), 5.10 (td, J = 7.7, 3.6 Hz, 1H, =CH), 5.00 – 5.06 (m, 1H, =CH), 4.85 (d, J = 13.7 Hz, 1H, OCH2), 3.40 (s, 3H, OCH3), 3.19 (s, 3H, NCH3), 2.49 (td, J = 7.0, 2.4 Hz, 1H, =CH), 2.41 (td, J = 7.4, 3.0 Hz, 1H, =CH), 2.28 – 2.37 (m, 3H, 2CH2), 2.11 – 2.17 (m, 1H, CH2), 1.62 – 1.83 (m, 4H, 2CH2).13C NMR (151 MHz, C6D6, derived from HSQC and HMBC): δ 183.25, 158.69, 156.75, 149.34, 141.45, 136.46, 127.26, 123.17, 122.46, 120.82, 120.56, 113.36, 84.78, 84.04, 75.30, 55.27, 54.66, 45.81, 45.44, 36.38, 34.54, 33.77, 29.90, 29.23. MALDI-MS: m/z calcd. for C26H32IrN3O2 [M]+ 611.21, found 611.14.
Decomposition of 1a to 1b and 1c
Complex 1a is unstable in a solution, and it completely decomposes in two days to 1b, 1c, and iridium-containing precipitate. 1b: 1H NMR (600 MHz, C6D6): δ 8.16 (d, J = 9.0 Hz, 2H, 2CH), 7.21 (d, J = 8.8 Hz, 2H, 2CH), 6.73 (d, J = 8.7 Hz, 2H, 2CH), 6.62 (d, J = 9.0 Hz, 2H, 2CH), 5.23 (s, 2H, CH2), 3.26 (s, 3H, CH3), 3.13 (s, 3H, CH3). GC-MS: m/z calcd. for C16H16O4 [M]+ 272.10, found 272.1. 1c: 1H NMR (600 MHz, C6D6): δ 5.62 – 6.67 (m, 2H, 2CH), 2.02 – 2.12 (m, 4H, 2CH2), 1.36 – 1.48 (m, 8H, 4CH2). GC-MS: m/z calcd. for C8H14 [M]+ 110.11, found 110.1.
[IrH(η1,η3-C8H12)(2-PyCH2PBu2t)] (2b)
In a glovebox, complex 2 (50.0 mg, 7.3 × 10−5 mol), isopropyl alcohol (20.0 mg, 3.3 × 10−4 mol), and potassium hydroxide (12.0 mg, 2.2 × 10−4 mol) were mixed with dry benzene (1.0 mL) and stirred at room temperature for two days. Then, the brown solution was filtered and the solvent was evaporated in vacuum to dryness. The residue was recrystallized twice from benzene affording the product as a pale-yellow crystalline powder (25.0 mg, 64%). Crystals suitable for X-ray analysis were obtained by slow evaporation of benzene solution. 1H NMR (600 MHz, C6D6): δ 7.77 (d, J = 5.6 Hz, 1H, Py), 6.65 (t, J = 7.3 Hz, 1H, Py), 6.44 (d, J = 7.5 Hz, 1H, Py), 6.05 (t, J = 6.4 Hz, 1H, Py), 5.20 – 5.13 (m, 1H, CH), 4.64 – 4.57 (m, 1H, CH), 3.91 (t, J = 7.8 Hz, 1H, CH), 2.96 – 2.90 (m, 1H, CH), 2.87 (dd, J = 16.6, 9.0 Hz, 1H, PCH2), 2.68 (dd, J = 16.6, 8.2 Hz, 1H, PCH2), 2.62 – 2.51 (m, 1H, CH2), 2.27 – 2.11 (m, 4H, 3CH2), 2.05 – 1.99 (m, 1H, CH2), 1.94 – 1.77 (m, 2H, 2CH2), 1.38 (d, J = 12.1 Hz, 9H, 3CH3), 1.10 (d, J = 12.3 Hz, 9H, 3CH3), −9.99 (d, J = 17.6 Hz, 1H, IrH). 13C NMR (151 MHz, C6D6): δ 163.78, 148.87, 133.73, 121.87, 121.10, 96.21, 76.00, 60.79, 56.43, 55.14, 38.75, 36.48, 35.29, 30.34, 29.06, 28.05, 25.33, 14.01. 31P{1H} NMR (243 MHz, C6D6): δ 56.20. IR (KBr, cm−1): 2915, 2871, 2800, 2043, 1472, 821, 762. MALDI-MS: m/z calcd. for C22H37IrNP [M]+ 539.23, found 539.31. Anal. calcd for C22H37IrNP: C 49.05, H 6.92, N 2.60. Found: C 48.11, H 6.96, N 2.76.
[Ir2H3(CO)(2-PyCH2PBu2t){μ-(C5H3N)CH2PtBu2}] (2c)
In a glovebox, complex 2 (50.0 mg, 7.3 × 10−5 mol), n-butanol (108.0 mg, 1.46 × 10−3 mol, 20 eq.), and potassium hydroxide (88.0 mg, 1.57 × 10−3 mol, 21.5 eq.) were mixed with dry toluene (5 mL) in a 20 mL Straus flask. The flask was charged with a stirring bar and sealed with a septum. Outside the glovebox, the flask was placed in an oil bath (120 °C), and the septum was immediately pierced with a syringe needle attached to eudiometer. In 15 min hydrogen evolution stopped, and the solution color turned from dark-violet to orange. The flask was brought back to the glovebox, and the precipitate (potassium butyrate) was filtered off and washed with toluene. The orange solution was evaporated to dryness under vacuum, and then hexane (3 mL) was added to the solid to form a yellow precipitate. The precipitate was filtered and washed with hexane. After crystallization from benzene-hexane mixture, 2c was obtained as a yellow crystalline powder (20 mg, 63%). Crystals suitable for X-ray analysis were obtained by slow addition of hexane to benzene solution. 1H NMR (600 MHz, C6D6): δ 8.58 (d, J = 5.6 Hz, 1H, ArH), 7.90 (dd, J = 7.7, 2.1 Hz, 1H, ArH), 6.86 (t, J = 7.6 Hz, 1H, ArH), 6.78 (td, J = 7.5, 1.3 Hz, 1H, ArH), 6.62 (d, J = 7.8 Hz, 1H, ArH), 6.51 (d, J = 7.4 Hz, 1H, ArH), 6.07 (t, J = 7.1 Hz, 1H, ArH), 3.19 (dd, J = 15.7, 7.7 Hz, 1H, CH2), 3.00 (dd, J = 15.8, 7.3 Hz, 1H, CH2), 2.58 – 2.43 (m, 2H, CH2), 1.50 (d, J = 12.2 Hz, 9H, 3CH3), 1.37 (d, J = 12.1 Hz, 9H, 3CH3), 1.18 (d, J = 13.6 Hz, 9H, 3CH3), 1.11 (d, J = 13.4 Hz, 9H, 3CH3), −7.51 (dd, J = 67.6, 7.5 Hz, 1H, IrH), −13.87 (ddd, J = 26.7, 13.7, 5.3 Hz, 1H, IrH), −19.99 (dt, J = 12.2, 4.1 Hz, 1H, IrH). 13C NMR (151 MHz, C6D6): δ 187.12 (d, J = 8.0 Hz), 178.55 (dd, J = 103.5, 4.4 Hz), 166.04 (d, J = 6.2 Hz), 164.98 (dd, J = 10.8, 6.1 Hz), 153.04 (d, J = 2.5 Hz), 144.85 (d, J = 7.7 Hz), 134.48, 133.03 (d, J = 4.6 Hz), 121.93 (d, J = 7.3 Hz), 120.35, 112.56 (d, J = 9.2 Hz), 39.69 (d, J = 16.1 Hz), 35.21 (d, J = 15.4 Hz), 35.06 (d, J = 13.5 Hz), 34.71 (d, J = 12.0 Hz), 33.14 (d, J = 19.0 Hz), 30.92 (d, J = 23.9 Hz), 30.59 – 30.08 (m), 29.87 (d, J = 4.6 Hz), 29.61 – 28.98 (m). 31P{1H} NMR (243 MHz, C6D6): δ 89.01, 62.75. IR (KBr, cm−1): 2942, 2896, 2106, 1914, 1582, 1473, 826. MALDI-MS: m/z calcd. for C29H50Ir2N2OP2 [M]+ 888.26, found 888.23. Anal. calcd for C29H50Ir2N2OP2: C 39.18, H 5.67, N 3.15. Found: C 39.91, H 5.71, N 3.19.
Conversion of 2b to 2d
A solution of 2b (10 mg) and sodium formate (10 mg) in formic acid (0.7 mL) was placed in a J. Young NMR tube. The tube was connected to eudiometer and heated in an oil bath at 70 °C for 30 min. About 15 mL of gas was produced during heating. The solvent was evaporated in vacuum and the resulting residue was left under vacuum for 1 h. The residue was then dissolved in formic acid-d2. 1H NMR spectrum of the residue contains peaks at −19.39, −25.04, and −27.11 ppm which were assigned to complex 2d.20
Isomerization of 2c to 2f
A solution of 2c (40 mg) in toluene-d8 (0.7 mL) was placed in a J. Young NMR tube and heated in an oil bath at 110 °C for 4 h. The resulting solution contained 2c and 2f in a 1.6 : 1 ratio (Keq = 0.626). The structure of 2f was derived from NMR data: 1H NMR (600 MHz, toluene-d8): δ 9.41 (d, J = 6.7 Hz, 1H, ArH), 7.26 (d, J = 8.0 Hz, 1H, ArH), 6.74 (td, J = 7.7, 1.6 Hz, 1H, ArH), 6.61 (d, J = 7.8 Hz, 1H, ArH), 6.55 (t, J = 7.3 Hz, 1H, ArH), 6.20 (d, J = 7.3 Hz, 1H, ArH), 5.98 (t, J = 7.2 Hz, 1H, ArH), 3.02 (d, J = 7.8 Hz, 2H, CH2), 2.52 (d, J = 8.7 Hz, 2H, CH2), 1.30 (d, J = 12.2 Hz, 18H, 6CH3), 1.23 (d, J = 13.4 Hz, 18H, 6CH3), −6.64 (d, J = 150.4 Hz, 1H, IrH), −8.98 (br s, 2H, 2IrH, resolves to a doublet at 100 °C with J = 46.2 Hz). 13C NMR (151 MHz, toluene-d8, derived from HSQC and HMBC): δ 164.19, 163.48, 162.13, 159.87, 140.85, 134.09, 131.10, 121.13, 120.70, 109.83, 39.65, 34.44, 34.03, 31.80. 31P{1H} NMR (243 MHz, toluene-d8): δ 88.66, 63.79.
Deuteration of 2c under Alcohol Dehydrogenation Conditions
In a glovebox, complex 2c (10.0 mg, 1.13 × 10−5 mol), 1-hexanol-O,1,1-d3 (59.0 mg, 5.62 × 10−4 mol, 50 eq.), and potassium hydroxide (31.0 mg, 5.62 × 10−4 mol, 50 eq.) were mixed with dry toluene (5 mL) in a 20 mL Straus flask. The following manipulations were the same as in the synthesis of 2c (reaction time was 3 h). Analysis of the recovered 2c-d4 by 1H NMR spectroscopy showed partial deuteration (up to 34%) of protons with chemical shifts at 6.62, −7.51, −13.87, and −19.99 ppm.
Conversion of 2c to 2g
A solution of 2c (20.0 mg, 2.25 × 10−5 mol) and freshly distilled n-hexanal (22.5 mg, 2.25 × 10−4 mol, 10 eq.) in C6D6 (0.5 mL) was placed in a J. Young NMR tube and heated in an oil bath at 80 °C for two days. The resulting solution contained 2g, n-hexanol, hexyl hexanoate, and unreacted n-hexanal. The solution was evaporated to dryness under vacuum giving crude 2g as a dark-yellow oil. The structure of 2g was derived from NMR data: 1H NMR (600 MHz, toluene-d8): δ 8.18 (dd, J = 4.7, 1.7 Hz, 1H, ArH), 7.43 (dd, J = 7.6, 2.6 Hz, 1H, ArH), 6.81 (t, J = 7.6 Hz, 1H, ArH), 6.76 (dd, J = 7.5, 1.8 Hz, 1H, ArH), 6.53 (d, J = 7.5 Hz, 1H, ArH), 6.36 (dd, J = 7.5, 4.7 Hz, 1H, ArH), 3.76 (dd, J = 16.6, 9.2 Hz, 1H, CH2), 3.60 (dd, J = 16.6, 9.5 Hz, 1H, CH2), 2.41 (d, J = 8.7 Hz, 2H, CH2), 1.37 (d, J = 13.1 Hz, 9H, 3CH3), 1.20 (d, J = 12.8 Hz, 9H, 3CH3), 1.06 (d, J = 13.9 Hz, 9H, 3CH3), 0.96 (d, J = 14.0 Hz, 9H, 3CH3), −8.44 (ddd, J = 52.5, 7.3, 5.2 Hz, 1H, IrH), −11.15 (ddd, J = 25.9, 12.6, 5.2 Hz, 1H, IrH). 31P{1H} NMR (243 MHz, toluene-d8): δ 88.18, 44.46. MALDI-MS: m/z calcd. for C29H48Ir2N2OP2 [M]+ 886.25, found 886.25.
Deprotonation of 2c
t-BuOK (12.6 mg, 1.13 × 10−4 mol, 5 eq.) was added to a solution of 2c (20.0 mg, 2.25 × 10−5 mol) in C6D6 (0.5 mL) which caused formation of 2h and instant color change from yellow to dark-red. Addition of n-hexanol (23.0 mg, 2.25 × 10−4 mol, 10 eq.) to the red solution converted 2h back to 2c. These transformations were monitored by 1H NMR (see Supporting Information).
Supplementary Material
Acknowledgments
This work is sponsored by the NSF (CHE-1566167), and the Hydrocarbon Research Foundation. We thank the NSF (DBI-0821671, CHE-0840366, CHE-1048807) and the NIH (S10 RR25432) for analytical instrumentation. We thank Prof. Ralf Haiges for help with X-ray crystallography. Fellowship assistance from USC Dornsife College is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/**.
Experimental procedures, graphical and tabular characterization information (PDF)
Notes
The authors declare no competing financial interests.
References
- 1.Fatiadi AJ. The Classical Permanganate Ion: Still a Novel Oxidant in Organic Chemistry. Synthesis. 1987;2:85–127. [Google Scholar]
- 2.Lee DG, Ribagorda M, Adrio J. “Potassium Permanganate” Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 3.Freeman F. “Chromic Acid” Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 4.Piancatelli G. “Pyridinium Dichromate” Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 5.Martin VS, Palazon JM, Rodriguez CM, Nevill CR, Hutchinson DK. “Ruthenium(VIII) Oxide” Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 6.Galvin JM, Jacobsen EN, Palucki M, Frederick MO. “Sodium Hypochlorite” Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 7.Reid EE, Worthington H, Larchar AW. The Action of Caustic Alkali and of Alkaline Salts on Alcohols. J Am Chem Soc. 1939;61:99–101. [Google Scholar]
- 8.Ciufolini MA, Swaminathan S. Synthesis of a Model Depsipeptide Segment of Luzopeptins (BBM 928), Potent Antitumor and Antiretroviral Antibiotics. Tetrahedron Lett. 1989;30:3027–3028. [Google Scholar]
- 9.Crimmins MT, DeBaillie AC. Enantioselective Total Synthesis of Bistramide A. J Am Chem Soc. 2006;128:4936–4937. doi: 10.1021/ja057686l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salunke GB, Shivakumar I, Gurjar MK. Total Synthesis of Verbalactone: an Efficient, Carbohydrate-Based Approach. Tetrahedron Lett. 2009;50:2048–2049. [Google Scholar]
- 11.Fuwa H. Chapter 5 - (−)-Lyngbyaloside B, a Marine Macrolide Glycoside: Total Synthesis and Stereochemical Revision. Strategies and Tactics in Organic Synthesis. 2016;12:143–168. [Google Scholar]
- 12.Zweifel T, Naubron J, Grützmacher H. Catalyzed Dehydrogenative Coupling of Primary Alcohols with Water, Methanol, or Amines. Angew Chem Int Ed. 2009;48:559–563. doi: 10.1002/anie.200804757. [DOI] [PubMed] [Google Scholar]
- 13.Trincado M, Grutzmacher H, Vizza F, Bianchini C. Domino Rhodium/Palladium-Catalyzed Dehydrogenation Reactions of Alcohols to Acids by Hydrogen Transfer to Inactivated Alkenes. Chem Eur J. 2010;16:2751–2757. doi: 10.1002/chem.200903069. [DOI] [PubMed] [Google Scholar]
- 14.Annen S, Zweifel T, Ricatto F, Grutzmacher H. Catalytic Aerobic Dehydrogenative Coupling of Primary Alcohols and Water to Acids Promoted by a Rhodium(I) Amido N-Heterocyclic Carbene Complex. ChemCatChem. 2010;2:1286–1295. [Google Scholar]
- 15.Balaraman E, Khaskin E, Leitus G, Milstein D. Catalytic Transformation of Alcohols to Carboxylic Acid Salts and H2 Using Water as the Oxygen Atom Source. Nature Chemistry. 2013;5:122–125. doi: 10.1038/nchem.1536. [DOI] [PubMed] [Google Scholar]
- 16.Choi J, Heim LE, Ahrens M, Prechtl MHG. Selective Conversion of Alcohols in Water to Carboxylic Acids by in situ Generated Ruthenium Trans Dihydrido Carbonyl PNP Complexes. Dalton Trans. 2014;43:17248–17254. doi: 10.1039/c4dt01634c. [DOI] [PubMed] [Google Scholar]
- 17.Malineni J, Keul H, Möller M. A Green and Sustainable Phosphine-Free NHC-Ruthenium Catalyst for Selective Oxidation of Alcohols to Carboxylic Acids in Water. Dalton Trans. 2015;44:17409–17414. doi: 10.1039/c5dt01358e. [DOI] [PubMed] [Google Scholar]; 17a Fujita K, Tamura R, Tanaka Y, Yoshida M, Onoda M, Yamaguchi R. Dehydrogenative Oxidation of Alcohols in Aqueous Media Catalyzed by a Water-Soluble Dicationic Iridium Complex Bearing a Functional N-Heterocyclic Carbene Ligand without Using Base. ACS Catal. 2017;7:7226–7230. [Google Scholar]
- 18.Santilli C, Makarov IS, Fristrup P, Madsen RJ. Dehydrogenative Synthesis of Carboxylic Acids from Primary Alcohols and Hydroxide Catalyzed by a Ruthenium N-Heterocyclic Carbene Complex. Org Chem. 2016;81:9931–9938. doi: 10.1021/acs.joc.6b02105. [DOI] [PubMed] [Google Scholar]
- 19.Dai Z, Luo Q, Meng X, Li R, Zhang J, Peng T. Ru(II) Complexes Bearing 2,6-Bis(benzimidazole-2-yl)pyridine Ligands: a New Class of Catalysts for Efficient Dehydrogenation of Primary Alcohols to Carboxylic Acids and H2 in the Alcohol/CsOH System. J Organomet Chem. 2017;830:11–18. [Google Scholar]
- 20.Celaje JJA, Lu Z, Kedzie EA, Terrile NJ, Lo JN, Williams TJ. A Prolific Catalyst for Dehydrogenation of Neat Formic Acid. Nature Com. 2016;7:11308. doi: 10.1038/ncomms11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Z, Demianets I, Hamze R, Terrile NJ, Williams TJ. A Prolific Catalyst for Selective Conversion of Neat Glycerol to Lactic Acid. ACS Catal. 2016;6:2014–2017. [Google Scholar]
- 22.This dimerization is known to be catalyzed by [IrCl(COD)]2 and [Cp*IrCl2]2 in the presence of a strong base. See; Matsu-ura T, Sakaguchi S, Obora Y, Ishii Y. Guerbet Reaction of Primary Alcohols Leading to β-Alkylated Dimer Alcohols Catalyzed by Iridium Complexes. J Org Chem. 2006;71:8306–8308. doi: 10.1021/jo061400t. [DOI] [PubMed] [Google Scholar]
- 23.Mako TL, Byers JA. Recent Advances in Iron-Catalysed Cross Coupling Reactions and Their Mechanistic Underpinning. Inorg Chem Front. 2016;3:766–790. [Google Scholar]
- 24.Kawahara R, Fujita K, Yamaguchi R. N-Alkylation of Amines with Alcohols Catalyzed by a Water-Soluble Cp*Iridium Complex: An Efficient Method for the Synthesis of Amines in Aqueous Media. Adv Synth Catal. 2011;353:1161–1168. [Google Scholar]; 24a Fujita K, Yamamoto K, Yamaguchi R. Oxidative Cyclization of Amino Alcohols Catalyzed by a Cp*Ir Complex. Synthesis of Indoles, 1,2,3,4-Tetrahydroquinolines, and 2,3,4,5-Tetrahydro-1-benzazepine. Org Lett. 2002;4:2691–2694. doi: 10.1021/ol026200s. [DOI] [PubMed] [Google Scholar]
- 25.Berliner MA, Dubant SPA, Makowski T, Ng K, Sitter B, Wager C, Zhang Y. Use of an Iridium-Catalyzed Redox-Neutral Alcohol-Amine Coupling on Kilogram Scale for the Synthesis of a GlyT1 Inhibitor. Org Process Res Dev. 2011;15:1052–1062. [Google Scholar]
- 26.Esteruelas MA, Olivan M, Oro LA, Schulz M, Sola E, Werner H. Synthesis, Molecular Structure and Reactivity of the Octahedral Iridium(III) Compound [IrH(η1,η3-C8H12)(dppm)] [dppm = bis(diphenylphosphino)methane] Organometallics. 1992;11:3659–3664. [Google Scholar]
- 27.Olsen EPK, Madsen R. Iridium-Catalyzed Dehydrogenative Decarbonylation of Primary Alcohols with the Liberation of Syngas. Chem Eur J. 2012;18:16023–16029. doi: 10.1002/chem.201202631. [DOI] [PubMed] [Google Scholar]
- 28.Olsen EPK, Singh T, Harris P, Andersson PG, Madsen R. Experimental and Theoretical Mechanistic Investigation of the Iridium-Catalyzed Dehydrogenative Decarbonylation of Primary Alcohols. J Am Chem Soc. 2015;137:834–842. doi: 10.1021/ja5106943. [DOI] [PubMed] [Google Scholar]
- 29.Melnick JG, Radosevich AT, Villagran D, Nocera DG. Decarbonylation of Ethanol to Methane, Carbon Monoxide and Hydrogen by a [PNP]Ir Complex. Chem Commun. 2010;46:79–81. doi: 10.1039/b914083b. [DOI] [PubMed] [Google Scholar]
- 30.Kloek SM, Heinekey DM, Goldberg KI. Stereoselective Decarbonylation of Methanol to Form a Stable Iridium(III) trans-Dihydride Complex. Organometallics. 2006;25:3007–3011. [Google Scholar]
- 31.Cotton FA, Poli R. Ortho Metalation of Pyridine at a Diiridium Center. Synthesis and Spectroscopic and Crystallographic Characterization of NC5H4- and N,N′-Di-p-tolylformamidinato-Bridged Complexes of Diiridium(II) Organometallics. 1987;6:1743–1751. [Google Scholar]
- 32.Takahashi Y, Nonogawa M, Fujita K, Yamaguchi R. C–H Activation on a Diphosphine and Hydrido-Bridged Diiridium Complex: Generation and Detection of an Active IrII–IrII Species [(Cp*Ir)2(μ-dmpm)(μ-H)]+ Dalton Trans. 2008:3546–3552. doi: 10.1039/b803626h. [DOI] [PubMed] [Google Scholar]
- 33.Iali W, Green GGR, Hart SJ, Whitwood AC, Duckett SB. Iridium Cyclooctene Complex That Forms a Hyperpolarization Transfer Catalyst before Converting to a Binuclear C–H Bond Activation Product Responsible for Hydrogen Isotope Exchange. Inorg Chem. 2016;55:11639–11643. doi: 10.1021/acs.inorgchem.6b02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zell T, Milstein D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal–Ligand Cooperation by Aromatization/Dearomatization. Acc Chem Res. 2015;48:1979–1994. doi: 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.