

Abstract
Glioblastoma multiforme (GBM) is a highly aggressive and heterogeneous form of primary brain tumors, driven by a complex repertoire of oncogenic alterations, including the constitutively active epidermal growth factor receptor (EGFRvIII). EGFRvIII impacts both cell-intrinsic and non-cell autonomous aspects of GBM progression, including cell invasion, angiogenesis and modulation of the tumor microenvironment. This is, at least in part, attributable to the release and intercellular trafficking of extracellular vesicles (EVs), heterogeneous membrane structures containing multiple bioactive macromolecules. Here we analyzed the impact of EGFRvIII on the profile of glioma EVs using isogenic tumor cell lines, in which this oncogene exhibits a strong transforming activity. We observed that EGFRvIII expression alters the expression of EV-regulating genes (vesiculome) and EV properties, including their protein composition. Using mass spectrometry, quantitative proteomic analysis and Gene Ontology terms filters, we observed that EVs released by EGFRvIII-transformed cells were enriched for extracellular exosome and focal adhesion related proteins. Among them, we validated the association of pro-invasive proteins (CD44, BSG, CD151) with EVs of EGFRvIII expressing glioma cells, and downregulation of exosomal markers (CD81 and CD82) relative to EVs of EGFRvIII-negative cells. Nano-flow cytometry revealed that the EV output from individual glioma cell lines was highly heterogeneous, such that only a fraction of vesicles contained specific proteins (including EGFRvIII). Notably, cells expressing EGFRvIII released EVs double positive for CD44/BSG, and these proteins also colocalized in cellular filopodia. We also detected the expression of homophilic adhesion molecules and increased homologous EV uptake by EGFRvIII-positive glioma cells. These results suggest that oncogenic EGFRvIII reprograms the proteome and uptake of GBM-related EVs, a notion with considerable implications for their biological activity and properties relevant for the development of EV-based cancer biomarkers.
Keywords: Oncogenes, Exosomes, Cancer Biology, Glioblastoma, Secretome, EGFRvIII, extracellular vesicles, heterogeneity, invasion, nano-flow cytometry
Glioblastoma multiforme (GBM)1 is the most common, highly invasive astrocytic brain tumor type associated with poor prognosis, rapid cell proliferation, extensive angiogenesis, and therapeutic resistance (1, 2). Deregulation of the epidermal growth factor receptor (EGFR) is commonly associated with specific GBM subtypes and contributes to aggressive features of tumor cells as well as alterations in the tumor microenvironment (3, 4). EGFR is a transmembrane receptor kinase which upon ligand-dependent activation and autophosphorylation forms signaling multimers that engage several intracellular regulatory pathways leading to changes in gene expression and altered cellular phenotype (5). Genomic and expression changes affecting EGFR are observed in 40–70% of GBMs (6–8), including EGFR gene amplification often coupled with the expression of an oncogenic mutant, known as EGFR variant III (EGFRvIII). EGFRvIII harbors a truncation in the ligand binding domain comprising exons 2–7 and exhibits constitutive ligand-independent transforming activity (9, 10), demonstrable through gene transfer and targeting experiments (3).
EGFRvIII expression imparts aggressive and proliferative properties upon GBM cells themselves, but also acts in a non-cell-autonomous manner by deregulating pathways of intercellular communication. This includes changes in the expression of proteins involved in angiogenic, coagulant, inflammatory, immune and paracrine processes crucial for GBM progression (3, 11). Moreover, the EGFR/EGFRvIII pathway has also emerged as an important regulator of the biogenesis, intercellular trafficking and biological effects of extracellular vesicles (EVs) (12).
EVs are heterogeneous structures ranging from 30 nm to over 1 μm in diameter and composed of a lipid bilayer that encases and protects a diverse molecular cargo (lipids, proteins and nucleic acids). Larger EVs form through cellular surface budding (ectosomes, microvesicles), whereas smaller vesicles (30–150 nm) usually originate within the endosomal compartments of various cells, from which they are released as exosomes, into extracellular space through processes involving tetraspanins, Rab proteins, elements of the endosomal sorting complexes required for export (ESCRT), syntenin and other regulators (13). Due to their unique ability to transfer bioactive cargo between cellular populations EVs have been implicated in homeostatic processes, as well as in pathology including cancer, where they may impact cell growth, directional migration (14), angiogenesis (15), and metastasis (16, 17). Indeed, GBM-derived EVs are known to stimulate cellular proliferation (18) and angiogenesis (19), upregulate invasion-related proteins including proteases (e.g. Cathepsin D) and alter the expression of proteins involved in cell-extracellular matrix (ECM) interactions (e.g. integrin beta-1) (20). Although complex changes in the EV proteome may harbor possible clues as to the state of donor cells and the nature of the EV activity (19, 21–23), the causative events upstream remain largely unknown.
In this regard, EGFRvIII is of interest as its expression triggers changes in the EV biogenesis, as well as their profile and biological activity (12). EGFRvIII oncoprotein and mRNA are found within the cargo of GBM-related EVs, an observation of considerable interest for biomarker development and given the general role of EVs as reservoirs of mutant oncoproteins in biofluids (12, 18). In this context a better understanding of molecular properties associated with GBM EVs could serve as means to improve diagnostic profiling of EVs extracted from biofluids of cancer patients and to understand (and oppose) the pathogenetic potential of these vesicles (12). For example, whether the reported changes in the phenotype of cells that have taken up EVs from EGFRvIII expressing glioma cells are attributable to a direct influence of this oncoprotein or are related to the concomitant transfer of additional proteins remains largely unknown.
In this study, we employed quantitative proteomics to analyze EVs derived from indolent parental U373 glioma cells and their EGFRvIII-expressing isogenic aggressive counterparts (U373vIII). EVs were purified using iodixanol density gradient ultracentrifugation and analyzed with UHPLC-Orbitrap Fusion Tribrid mass spectrometer. Compilation of three experimental replicates revealed remarkable EGFRvIII-related changes in the expression profiles of EV-associated proteins. Using a label-free quantitation approach, we identified a total of 1059 proteins in EV preparations from both cell lines, including 254 (24.0%) proteins significantly affected by EGFRvIII activation. This included 177 upregulated proteins in U373vIII EVs. These altered EV proteins were significantly associated with ontology terms such as focal adhesion, cell junction, cytosol, cell adhesion, and plasma membrane. Moreover, we found that U373vIII cells secrete EVs containing high levels of invasion-promoting proteins including proteases, ECM, and cell adhesion proteins. EGFRvIII transformation also caused a switch in EV tetraspanin markers (e.g. loss of CD82) (24), and possibly altered some aspects of their biogenesis. Importantly, these changes were not ubiquitous but specific to individual EVs, their subpopulations and related subcellular domains. In conclusion, our results suggest that oncogenic EGFRvIII impacts the proteome of EVs released by GBM cells, and this may influence their biological activities beyond the content of EGFRvIII oncoprotein itself.
EXPERIMENTAL PROCEDURES
In Vivo Mouse Tumor Model
All in vivo experiments were performed as described earlier (3) according to the Animal Use Protocol (AUP) approved by the Institutional Animal Facility Care Committee and following Guidelines of the Canadian Council of Animal Care (CCAC). Female mice, 22–24-week-old either wild type or harboring yellow fluorescent protein transgene on the background of severe combined immunodeficiency (SCID or YFP/SCID; Charles River, Saint-Constant, QC, Canada or own colony, respectively) were inoculated subcutaneously with 5 × 106 of U373 or U373vIII cells in 0.2 ml of Matrigel HC (BD Biosciences, San Jose, CA). Tumor volume (mm3) was calculated as (width)2 × (length) × 0.5.
Cell Culture
U373 (human astrocytoma) and their EGFRvIII expressing variant (U373vIII) were described earlier (3). U373vIII cells harbor Tet-off regulated EGFRvIII gene introduced by transfection, and their characteristics, generation, and maintenance were described previously (12). For standard culture the cells were grown in Dulbecco's modified essential medium (DMEM; Wisent, Canada) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Wisent) and 1% penicillin-streptomycin (Gibco-Life Technologies, Grand Island, NY) at 37 °C in 5% CO2.
Isolation of EVs
Isolation of EVs was performed as previously described (25). Briefly, the conditioned medium (CM) was collected from cells grown for 72 h in culture media containing 10% of EV-depleted FBS (generated by centrifugation at 150,000 × g for 18 h at 4 °C). Cell viabilities were checked by trypan blue staining with three biological replicates being analyzed. Cell proliferation in 10% EV-depleted FBS media was analyzed by MTS reaction, using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) according to the manufacturer's instructions. CM was centrifuged one time at 400 × g and then passed through 0.8 μm pore-size filter. The resulting filtrate was concentrated using Amicon Ultra-15 Centrifugal Filter Unit (EMD Millipore, Billerica, MA) with 100,000 NMWL molecular cut off. The concentrate was mixed with 50% of iodixanol solution (Sigma, St. Louis, MO) and processed for density gradient ultracentrifugation at 200,000 × g for 2 h (25). The EV-enriched fraction of iodixanol was collected (at the density of ∼1.10 g/ml) and particles were confirmed to carry CD81, and other established exosome markers (24). The concentration of EV proteins was quantified using the BCA assay (Pierce Biotechnology, Rockford, IL). For concentration and size distribution of EVs nanoparticle tracking analysis (NTA) was carried out with each collected iodixanol fraction using NanoSight NS500 instrument (NanoSight Ltd., UK). Three recordings of 30 s at 37 °C were obtained and processed using NTA software (version 3.0). All experiments were carried out in three biological replicates.
Experimental Design and Statistical Rationale for EV Proteomics
For proteomics, EVs were purified independently at three different times with parallel isolation on different days, each from 200 ml of conditioned media of U373 and their EGFRvIII-expressing isogenic counterpart, U373vIII. For each of the three biological replicates of LC-MS/MS, the same protein amount of EV preparation (9 μg) was desalted with SDS-PAGE, loaded onto the stacking gel followed by staining and destaining. The in-gel trypsin digestion was carried out under reducing conditions afforded by DTT, and alkylation was achieved using iodoacetic acid as previously described (26). The lyophilized peptides were resolubilized in 0.1% aqueous formic acid/2% acetonitrile, the peptides were loaded onto a Thermo Acclaim Pepmap (75 μm inner diameter × 2 cm, C18, 3 μm particle size, 100 Å pore size) (Thermo Fisher Scientific, San Jose, CA) pre-column and the onto an Acclaim Pepmap Easyspray (75 μm inner diameter × 15 cm, C18, 2 μm particle size, 100 Å pore size) (Thermo Fisher Scientific) analytical column. Separation was achieved using a Dionex Ultimate 3000 uHPLC at 220 nL/min with a gradient of 2–35% organic solvents (0.1% formic acid in acetonitrile) over 3 h. Peptides were analyzed using a Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) operating at 120,000 resolution (FWHM in MS1, 15,000 for MS/MS) with higher-energy collisional dissociation sequencing of all peptides with a charge of 2+ or greater. The raw data were converted into *.mgf format (Mascot generic format) by MSConvert (ProteinWizard, version 3.0.6150), and searched using Mascot 2.5.1 against SwissProt (http://www.uniprot.org) human protein database (release 2016_03, 20200 entries). The tolerance was 5 ppm monoisotopic for precursor ions and 0.1 Da for fragment ions. The permission of two potential missed cleavages was selected for trypsin digestion. The following modifications were used: fixed modification for the carbamidomethylation of cysteine (58 Da) and variable modification for the oxidation of methionine (16 Da) and the deamidation of asparagine and glutamine (1 Da). The database search results were further analyzed by Scaffold Q+ software (version 4.8.4, www.proteomesoftware.com/products/scaffold/) (Proteome Sciences, Portland) (Protein threshold > 0.95%, Peptide threshold > 0.95%, and 2 of minimum number of unique peptides) (Peptide FDR: 0.8%, Protein FDR: 5.0%, Protein and peptide FDR were determined by Scaffold Q+ using the probabilistic method used by the Trans-proteomic pipeline (see http://proteome-software.wikispaces.com/FAQ-Statistics)). From three biologically replicated MS data sets of both proteomes, from U373 and U373vIII EVs, we quantified the relative protein abundance by total ion chromatogram (TIC) and calculated the p value by student's t test using Scaffold Q+ software. Proteins with less than 0.05 of p value were considered as significantly changed. The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE (27) partner repository with the data set identifier PXD008311 and 10.6019/PXD008311.
Western Blotting
Cell lysates and EV proteins obtained by density gradient ultracentrifugation were resolved by SDS-PAGE and then transferred to a polyvinylidene difluoride membrane. The membrane was blocked, incubated with primary antibody followed by the secondary antibody conjugated with horseradish peroxidase, and subjected to the enhanced chemiluminescence. All images were acquired by a ChemiDoc MP imager (Bio-Rad, Hercules, CA). Rabbit anti-CD82, rabbit anti-EGFR, rabbit anti-ITGA6, rabbit anti-ITGB4, and goat anti-rabbit antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit anti-CANX, rabbit anti-CD44, mouse anti-CD81, rabbit anti-CD9, rabbit anti-GAPDH, rabbit anti-SDCB1 antibodies were purchased from Abcam (Cambridge, MA). Mouse anti-actin and goat anti-mouse IgG were purchased from Sigma.
Gene Ontology (GO) Analyses
The lists of identified proteins were imported into the DAVID Bioinformatics database (http://david.abcc.ncifcrf.gov) and assigned to their GO annotations (cellular component and biological process), KEGG pathway, UniProt tissue expression, and UniProt keyword with statistical analyses (28).
EV Labeling With Antibodies and Analyses by Nano-flow Cytometry
The conditioned medium from cells grown for 72 h in culture media containing 10% of EV-depleted FBS was centrifuged one time at 400g and then filtered through a 0.8 μm pore-size mesh. The supernatant was concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (EMD Millipore) with 100,000 NMWL molecular cut-off. The particles in the concentrated supernatant were quantified by NTA and diluted with PBS up to 1 × 1011 particles/ml. EVs were incubated with the indicated fluorophore-conjugated antibodies for 2 h at room temperature in the dark. To remove unbound antibodies, EVs were isolated using qEV size exclusion chromatography (SEC) column (Izon Science, UK) according to the manufacturer's instructions. After collecting the fractions of 0.5 ml each, the EV-enriched fraction was identified by NTA. These experiments were conducted together with isotype controls matched with the corresponding antibodies. All fluorophore-conjugated antibodies were purchased from BioLegend (UK), including mouse anti-BSG (Alexa 488), mouse anti-CD9 (FITC), mouse anti-CD44 (APC), mouse anti-CD81 (APC), mouse anti-CD151 (PE), and mouse anti-EGFR (APC). Nano flow cytometry was performed using CytoFLEX system (Beckman Coulter, Pasadena, CA) equipped with 3 lasers (405, 488, and 640 nm wavelength). The 405 nm violet laser for SSC (V-SSC) was selected with 1800 of manual threshold settings in V-SSC height channel. Samples were loaded and run for 2 min until the event rate became stable, and then 20 s acquisitions were saved. Data were acquired and analyzed using Cytexpert 2.0 software (Beckman Coulter).
Quantitative Real-time PCR Array
Total RNA was isolated from the cells using RNeasy Plus Mini Kit (Qiagen, Valencia, CA) following manufacturer's instructions. Cell lysates were passed through a gDNA eliminator column to remove genomic DNA, and high-quality RNA was eluted with 30 μl of water. Aliquots of 500 ng of total RNA were used to synthesize cDNA using RT2 First Strand Kit (Qiagen) according to the manufacturer's recommendations. Quantitative real-time PCR analysis was performed using the LightCycler 96 system (Roche Diagnostics, Germany) with SYBR Green PCR Master Mix (Qiagen). The thermal profile consisted of an initial step of 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Simultaneously the expression of 96 genes (including housekeeping genes) was analyzed using a custom made RT2 Profiler PCR arrays (Qiagen), (supplemental Table S1). PCR reactions were run in triplicate and transcript levels were normalized to the indicated housekeeping genes. Data analysis was performed using the comparative Ct (ΔΔCt) method, essentially as recommended by the manufacturer, SA biosciences; http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php.
EV Labeling With PKH67 and Uptake Measurement by Flow Cytometry
The conditioned media from cells grown for 72 h in culture media supplemented with 10% of EV-depleted FBS were centrifuged once at 400g, passed through 0.8 μm pore-size filter and concentrated as described above. The concentrated supernatants were diluted 4-fold with PBS and ultracentrifuged at 110,000 × g for 1 h. The resulting pellets were resuspended in 500 μl of diluent C (Sigma) each, mixed with 500 μl of 2 μm PKH67 (Sigma) and incubated for 5 min at room temperature as per manufacturer's instructions. To stop the labeling, 1 ml of EV-depleted FBS was added, incubated for 1 min and then diluted with 8-fold excess of PBS. Labeled EVs were purified by ultracentrifugation at 110,000 × g for 1 h. EVs were resuspended in PBS, quantified by NTA, and added to recipient cells at 2 × 108 particles/ml for 4 h. The cells were analyzed by flow cytometry using FacsCalibur instrument (BD) for the PKH67 fluorescence transfer.
Confocal Microscopy
Cells were plated in the μ-Slide 8-Well ibiTreat chambered coverslips (ibidi, Germany) for 24 h. For immunofluorescence, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% tween-20 in PBS. The cells were blocked and incubated with fluorophore-conjugated antibodies for 2 h, as indicated, whereas the nuclei were labeled with Nucblue DNA binding dye (Thermo Fisher Scientific). Images were collected using LSM780 confocal microscope (Carl Zeiss, Thornwood, NY) with the 63×/1.40 objective.
Transmission Electron Microscopy (TEM)
U373 EVs and U373vIII EVs were fixed with a solution containing 2.5% glutaraldehyde in 0.1 m sodium cacodylate (pH 7.4) for 2 h at 4 °C. Fixed EVs were loaded on carbon-coated copper grids for 3 min. The grids were rinsed with droplets of deionized water for 15 s, four times, and stained with 1% uranyl acetate for 45 s. Electron micrographs were recorded using Tecnai 12 BioTwin (Philips, Netherlands) 120 kV TEM instrument. Assistance was provided by Jennie Mui at the Facility for Electron Microscope Research (FEMR) unit of McGill University.
Data Analysis
Unless otherwise indicated all experiments were carried out in at least three independent biological replicates and processed for statistical significance as indicated. Whenever applicable, numerical values were plotted as mean ± S.D.
RESULTS
Changes in Characteristics of Glioma Cells and Their EVs Following EGFRvIII-Dependent Oncogenic Transformation
As reported in our previous study (12), the expression of the oncogenic EGFRvIII mutant in indolent U373 glioma cells resulted in formation of an aggressive variant (U373vIII) endowed with a highly tumorigenic phenotype in xenograft model systems (supplemental Fig. S1A) (3). This transition also impacted cellular morphology including cell shape and the appearance of the plasma membrane (supplemental Fig. S1B). Thus, bright-field images of U373 cells revealed well defined ruffle structures formed by distinct plasma membrane domains. Instead, U373vIII cells formed multiple filopodia around the entire cell perimeter, as well as ruffle structures, suggestive of a more migratory and invasive phenotype (supplemental Fig. S1B) (29), and often coupled with increased membrane budding and EV formation activity (30) (vesiculation). Indeed, we observed earlier that both cell lines release ample exosome-like EVs into their conditioned media (supplemental Fig. S1C). Although the cumulative protein content in cultures of EGFRvIII expressing cells tends to be higher than that of their parental counterparts (12, 31) the NTA normalized numbers of particles and their associated proteins are comparable between both cell lines (supplemental Fig. S2). However, the influence of EGFRvIII on the nature and composition of this material were not studied in detail.
To better understand the link between EGFRvIII status and cellular vesiculation, we used the U373/U373vIII model to perform EV isolation and detailed proteomic characterization (Fig. 1). To this end we designed a work flow (Fig. 1A) where conditioned media from parental U373 cells and from their EGFRvIII-positive (U373vIII) isogenic counterparts were collected upon 3-day culture in the presence of EV-depleted FBS (10%). Cell proliferation of both cell populations was not affected by EV- depletion of the FBS (supplemental Fig. S2A) in that cell viability as measured by trypan blue staining was 97.0 ± 0.6% and 96.7 ± 0.3% for U373 and U373vIII cells, respectively (supplemental Fig. S2B). The residual cell debris was removed from conditioned media by standard 400g centrifugation followed by filtration (0.8 μm) and concentration steps (100 kDa molecular cut-off), to enable subsequent purification and resolution of EV fractions using the iodixanol density gradient ultracentrifugation (200,000 × g for 2 h) (25). The successive fractions (1 ml each) were collected, starting from the top, to locate the iodixanol density with the highest EV content. This was accomplished through testing fractions for particle counts and sizes by NTA, and by detection of canonical EV marker proteins, such as CD81 (24), using Western blotting (Fig. 1B–1D) and through the analysis of EV morphology using TEM (supplemental Fig. S1C). According to this protocol, fraction F3 was found to contain the bulk of particles along with the strongest signal for CD81 (Fig. 1D), and for other canonical exosomal maker proteins including ALIX, TSG101, and SDCBP1 (supplemental Fig. S3). Therefore, this fraction was chosen for further analysis. Indeed, F3 preparations from U373 and U373vIII cells contained an appreciable number of EVs (median of 2.0–2.5 × 107 particles/ml of fraction) with sizes ranging between 215.9 ± 1.1 nm and 237.6 ± 2.0 nm (Fig. 1C, supplemental Fig. S1C), respectively. Based on the design of this protocol, iodixanol floatation density, EV morphology (supplemental Fig. S1C) and molecular marker distribution (supplemental Fig. S4–S5) the majority of EVs released by both glioma cell lines resemble exosomes (32). However, these EVs were also somewhat larger than typical exosomes emitted by other cellular populations (24), and therefore they will be further referred to as EVs.
Fig. 1.

Workflow design for isolation and proteome analysis of glioma cell-derived EVs. A, Diagram shows the schematic workflow of the purification and quantitative proteomics of EVs with the number of identified proteins in each EV subset. B. The particle concentration of each fraction was determined by NTA. C, The size distribution of EVs as measured by NTA indicating an average diameter of 215.9 ± 1.1 nm and 237.6 ± 2.0 nm for U373 EVs and U373vIII EVs, respectively. D, Fractions of Optiprep density gradients in U373 and U373vIII EVs were analyzed by Western blotting. CD81, marker protein of EVs, was detected mainly in Fraction 3; purified EVs contained in fraction 3 were further analyzed for proteomics.
Oncogenic EGFRvIII Impacts the Expression of Genes and Proteins Associated With EV Biogenesis and Biological Activity
Although some of the proteins involved in vesiculation are often thought of as generic and constitutive markers of EVs, or their subsets (e.g. exosomes) (24), their regulation may be affected by oncogenic transformation (17). To understand whether such influence is exerted by EGFRvIII, we profiled U373 and U373vIII cells for the expression of 91 vesiculation-related transcripts using a customized RT2 array (Fig. 2A; supplemental Table S1). This comparison revealed multiple EGFRvIII-dependent differences, including upregulation of certain tetraspanins (TSPAN8, CD151) linked to exosome-mediated intercellular communication (33). Conversely, EGFRvIII expression also resulted in downregulation of several other EV-related genes, notably including CD81 and CD82, markers and effectors of exosome biogenesis (24, 34, 35). Remarkable differences in the expression of some of these genes were also observed at the protein level, as revealed by multicolor immunofluorescence and confocal microscopy (Fig. 2B and 2C). Although U373 cells stained brightly for CD82, especially in the plasma membrane and as perinuclear puncta, their U373vIII counterparts were devoid of such signal and exhibited strong membrane and perinuclear staining for CD151. They were also weakly positive for CD81. These assays confirmed that U373vIII cells and EVs were virtually negative for CD82 and expressed lower levels of CD81, both known as EV markers.
Fig. 2.

The expression of genes and proteins associated with EV biogenesis and biological activity (vesiculome) are affected by oncogenic EGFRvIII. A, Fold change in mRNA of U373vIII cells over U373 cells. Among a total of 91 vesiculation-related transcripts included in the customized RT2 array (supplemental Table S1), top 10 significantly changed proteins (p value < 0.05) are indicated for both U373 and U373vIII cells. B and C, Confocal images showing remarkable differences in expression of selected EV-associated proteins. U373vIII cells are largely devoid of CD82 expression with low levels of CD81 relative to U373 cells. High levels of CD151 expressed by U373vIII cells versus their U373 counterparts in agreement with the corresponding levels of mRNA (see text).
Western blot analysis of both cellular and EV lysates revealed similar EGFRvIII-related changes in the expression of vesiculation-related proteins (supplemental Fig. S4–S5). Interestingly, EV preparations were enriched for EGFRvIII oncoprotein, CD81, CD9, and SDCB1 (syntenin-1), but not CANX (calnexin) compared with cellular content of these proteins. The absence of CANX in EV preparations also documents the purity of EV isolates as this protein is a marker of cellular cytoplasm and is normally excluded from the EV cargo (24). Cells expressing EGFRvIII produced EVs with lower levels of CD81 and SDCB1 and no CD82, but with somewhat higher CD9 expression compared with EVs derived from their EGFRvIII-negative counterparts. Especially, the downregulation of CD81 and CD82 in EVs was correlated in their lower cellular expression in Fig. 2. These observations suggest that our protocol is effective at isolating EVs free from cellular impurities and indicate that oncogenic EGFRvIII expression imposes marked changes in the protein composition of GBM-derived EVs.
The Global Proteome of Glioma Cell-derived EVs
To further understand the impact of EGFRvIII on the EV proteome we applied mass spectrometry (MS). A total of 9 μg of EV proteins was analyzed by LC-MS/MS with three biological replicates (Fig. 1A). Raw MS files were analyzed with Mascot and the Scaffold programs. Finally, 962 and 1014 proteins (protein threshold > 95.0%, peptide threshold > 95.0%, and 2 of minimum number of unique peptides) were identified in U373 EVs and U373vIII EVs, respectively (supplemental Table S2). Among 1059 proteins reliably identified in tumor EVs, 45 (4.2%) and 97 (9.2%) proteins were unique to either U373 EVs or U373vIII EVs, respectively, whereas 917 (86.6%) proteins were overlapping (Fig. 3A). We compared our EV protein lists with citations in Vesiclepedia (http://www.microvesicles.org) (36) and EVpedia (http://evpedia.info) (37), which are the largest web-based databases of EV proteins, mRNAs, miRNAs, and lipids (supplemental Table S2). As indicated in Fig. 3B, the majority of proteins identified in proteomes we analyzed were previously known to be EV-associated (Vesiclepedia). Notably, at the time of this writing, 25 proteins (2.4% of total) in our list were previously unreported as EV cargo. Most of these proteins were detected at low spectral numbers and low total TIC, implying low abundance. On the other hand, some of these unique EV-associated proteins such as TENM2 (Teneurin-2) (38), NTM (Neurotrimin) (39), GPM6B (Neuronal membrane glycoprotein M6-b) (40), were readily identified despite moderate spectral number and total TIC (supplemental Table S2). Interestingly, these novel proteins are relatively tissue specific and are associated with neuronal cells and the brain rather than having a broad expression. Thus, tissue or cell-type specific proteins can be sorted into brain tumor-derived EVs in agreement with their cellular identity. Moreover, the majority of the top 100 EV proteins, defined as frequently included in the EV cargo by EVpedia (41), were also detected in brain tumor cell-derived EV proteomes we examined (Fig. 3C), with an exception of PSMB3, Proteasome subunit beta type-3. These results are in good agreement with the expected protein profiles identified in EVs and brain cell populations and support the validity and reproducibility of our analysis.
Fig. 3.

The global proteome of U373 EVs and U373vIII EVs. A, Venn diagram shows the common and unique proteins identified in EVs of U373 and U373vIII cells. B, Comparison of identified proteins with Vesiclepedia database (http://www.microvesicles.org) (36); the search indicates that the majority of proteins identified in glioma EV proteomes were previously known to be EV-associated. C, Top 100 identified glioma EV proteins match EVpedia database (http://evpedia.info) (37); PSMB3 represents the only protein not listed in this dataset and found in the EV proteome generated by our study (see text).
To glean possible clues as to the functional role of glioma EV-associated proteins, we analyzed our datasets against publicly available databases for the expected tissue expression, GO cellular components, GO biological processes, and KEGG pathways analyses (Fig. 4). As expected (and mentioned previously), EV proteomes we studied were enriched for signals of neural system related cells and tissues including such categories as Cajal-Retzius cells, fetal brain cortex, and brain (Fig. 4A). In GO cellular components (Fig. 4B), cell membrane-related proteins such as those linked to focal adhesion and adherence junction were highly represented according to the enrichment analyses of the DAVID database (42), along with cytosol and ECM proteins. With respect to GO biological process analysis (Fig. 4C), we identified categories such as: cell adhesion, translation, signaling, and protein folding, as processes that were enriched in the EV cargo. Also, actin cytoskeleton, adhesion, endocytosis, and invasion according to KEGG pathways were enriched in glioma EVs (Fig. 4D). These features of tumor EV proteomes suggest a preferential inclusion of proteins related to EV biogenesis, targeting, and biological functions involved in recognition of cells and structures in the brain microenvironment. It should be noted that the global proteome composition was relatively similar between EVs originating from U373 and U373vIII cells as indicated by appreciable contribution of common proteins (86.6%). As these cells are isogenic the differences observed are related to the expression of the EGFRvIII oncogene.
Fig. 4.
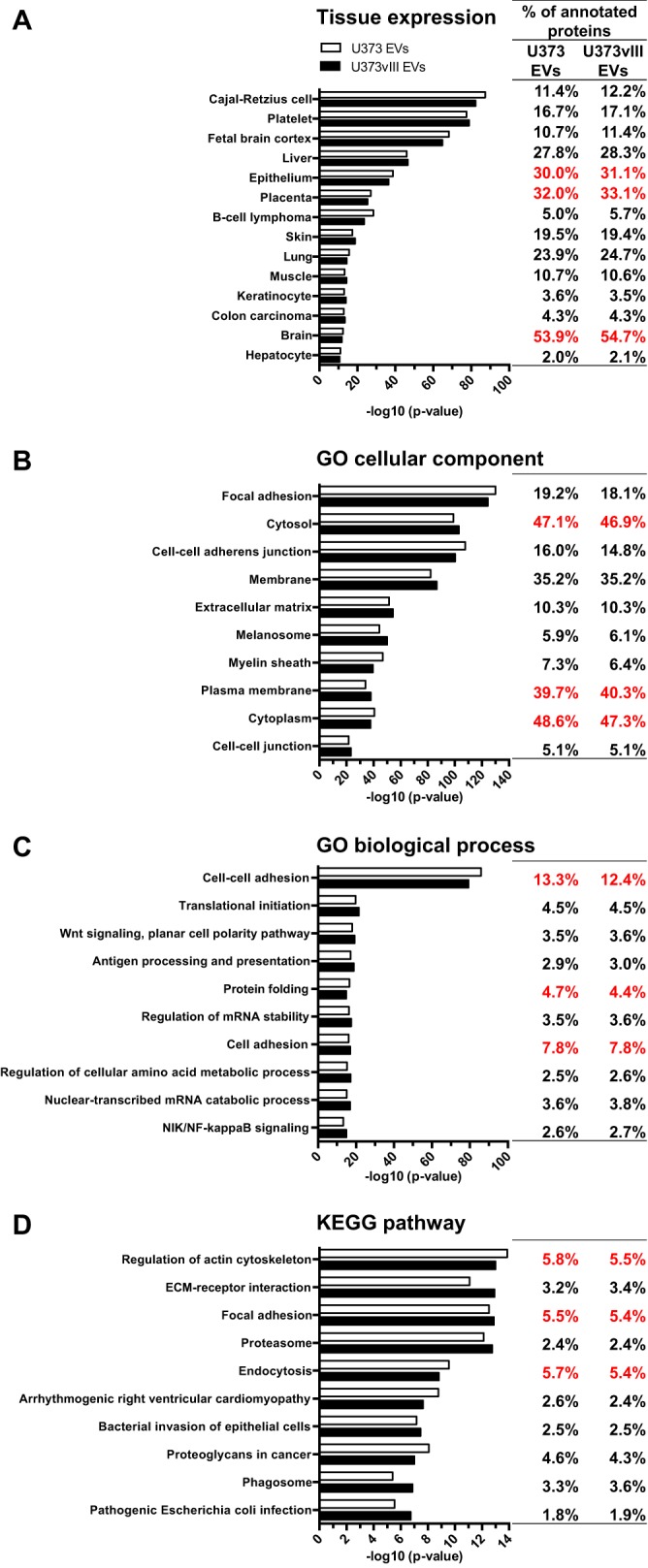
Functional characteristics of glioma EV proteins. GO analyses for UniProt (A) tissue expression (B), cellular component (C), biological process (D), and KEGG pathway. The analysis was conducted on U373 EVs and U373vIII EVs using DAVID database (42). Top 3 terms with highest numbers are indicated in red.
The Impact of EGFRvIII Expression on the Protein Composition of Glioma EVs
We next chose to compare in more detail the protein composition of EVs released by the EGFRvIII-negative (U373) and -positive (U373vIII) glioma cell lines. The abundance of each protein was calculated by the Scaffold program as total TIC (Fig. 5A, 5B). Levels of selected proteins were also verified by Western blotting (supplemental Fig. S4–S5). Importantly, quantitative proteomic analysis of glioma EVs revealed large numbers of significantly changed proteins (p value < 0.05) between U373 and U373vIII cellular donors (Fig. 5A, 5B and supplemental Table S2). A total of 254 differentially expressed EV proteins were identified of which 77 (7.3%) and 177 (16.7%) were significantly upregulated in either U373 or U373vIII EVs, respectively, relative to their counterparts (Fig. 5B). Top 10 of such significantly upregulated proteins found in EVs of either U373 or U373vIII cell lines were indicated by names in Fig. 5A.
Fig. 5.
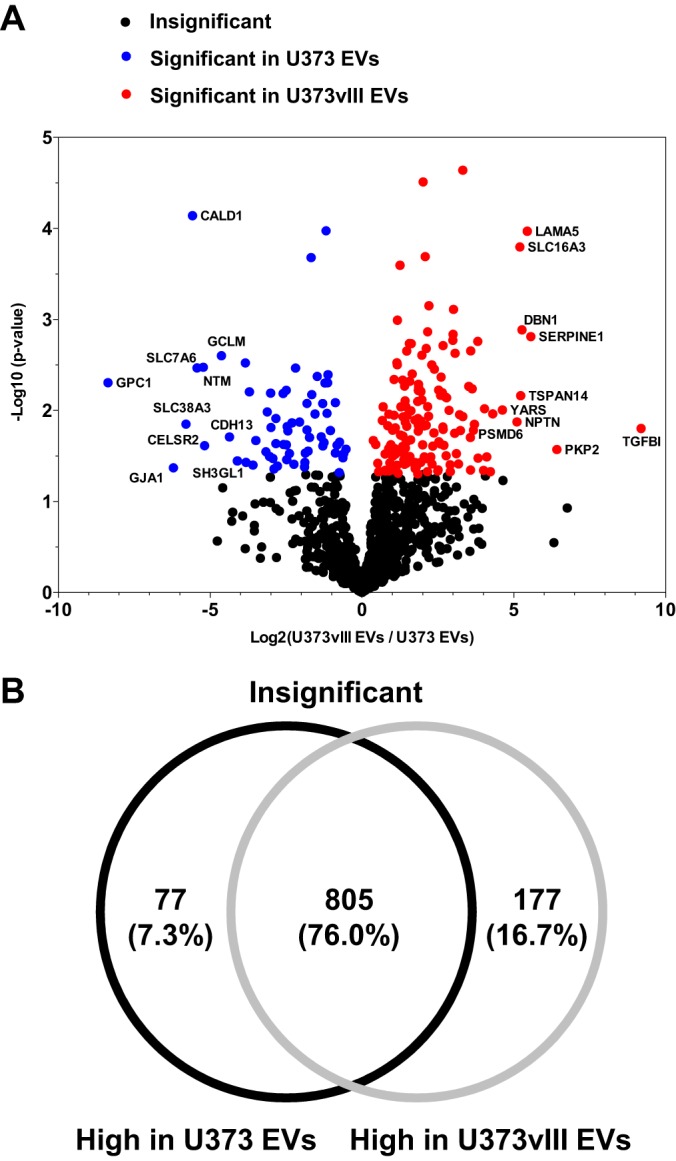
Quantitative protein profiles of glioma EVs. A, Relative abundances of EV proteins were calculated based on total TIC by the Scaffold program. Volcano plot shows the significantly changed proteins in EV proteomes being affected by the EGFRvIII-dependent transformation. Top 10 proteins with highest p value are indicated with gene symbol on the plot. B, Venn diagram shows the differentially expressed proteins in U373 EVs and U373vIII EVs (p value < 0.05).
Several of these changes were in good agreement with the results of Western blotting (supplemental Fig. S4–S5) with the aforementioned enrichment in EGFRvIII, CD9 and CD151 in EVs from U373vIII cells and concomitant depletion of exosomal markers (CD81-low, CD82-absent). Interestingly, proteins regarded as having a housekeeping role, such as actin and GAPDH were also expressed at different levels between EVs emanating from these respective cell lines, which may suggest a differential cargo packing mechanism and questions their utility as loading controls for EVs. Notably, several surface integral membrane proteins such CD44, ITGA6, and ITGB4 with functional roles in invasion and metastasis (16), were upregulated in U373vIII EVs in keeping with the aggressive phenotype of this cell line (supplemental Fig. S5 and supplemental Fig. S1A).
To further explore these plausible functional connections, we used GO algorithm to distinguish cellular components and biological processes linked to significantly changed (rather than globally expressed) EV proteins (Fig. 6). This analysis revealed EV proteins related to focal adhesion, cell-cell adherens junction, cytosol, and cell-cell adhesion as highly enriched in both proteomes. Interestingly, plasma membrane-related proteins were more uniquely enriched in U373 EVs (Fig. 6B), whereas ECM-related proteins were more robustly represented in U373vIII EVs (Fig. 6C). These differences may suggest, once again, a link to differential aggressiveness of these respective cell lines in vivo (3) and of GBMs harboring the EGFRvIII mutation (1, 43).
Fig. 6.
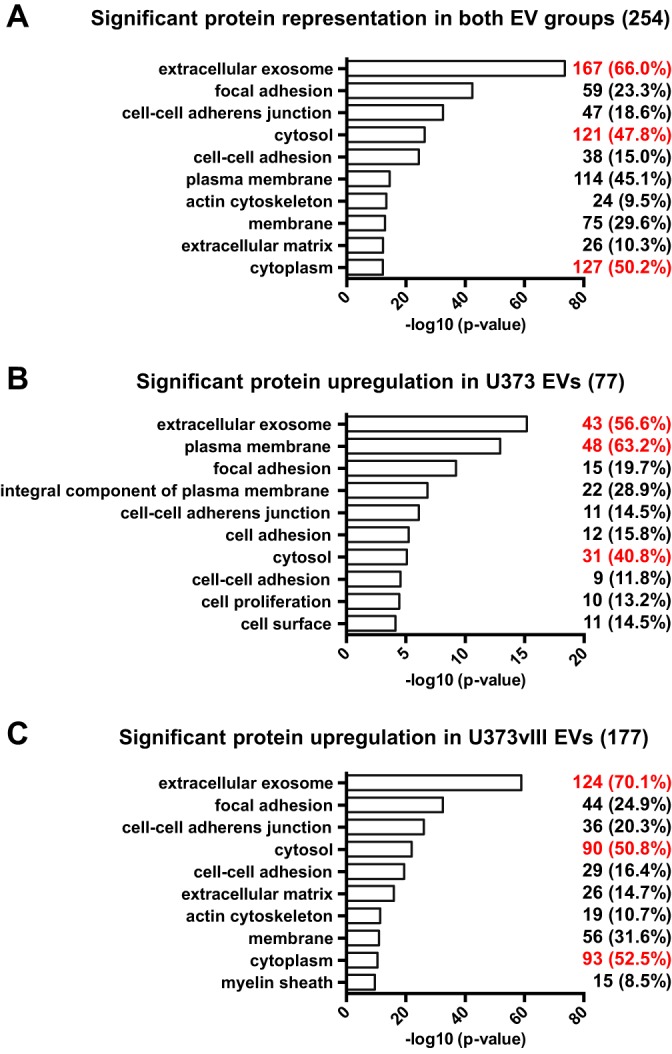
Functional characteristics of proteins differentially incorporated into glioma EVs as a function of EGFRvIII transformation. Significantly changed protein categories (p value < 0.05) in both EV groups (A), protein categories preferentially upregulated in U373 EVs (B), protein categories preferentially upregulated in U373vIII EVs (C). The analysis was conducted using GO cellular component and biological process terms. Top 3 terms with highest numerical change are indicated in red.
Oncogenic EGFRvIII Alters the Expression of Invasion Related Proteins in the EV Cargo
Molecular regulators of tumor invasion are often detectable in the cellular secretome (44), EVs (20), and cellular plasma membrane in GBM and other cancers (45). EVs also act as regulators of cellular motility and dissemination (16, 46). Moreover, the GO analyses of enriched proteins in U373vIII EVs (Fig. 6C) suggest a close relation to cellular adhesion, actin cytoskeleton regulation, and ECM, which is consistent with the cellular invasive phenotype (47). This profile is also in line with the emerging functional role of tumor EVs in cellular invasiveness (48, 49). Because the impact of EGFRvIII on this aspect of the EV bioactivity in GBM remains relatively unknown, we selected the significantly enriched U373vIII EV proteins (p value < 0.05) and surveyed them in silico for their predicted pro-invasive properties. Thus, EV proteins were categorized into proteases, ECM, and cell adhesion molecules according to keyword terms of UniProt database and the published literature pertaining to tumor invasion (Table I). Interestingly, we observed that several ECM proteins were overexpressed in U373vIII EVs including laminins (LAMA5, LAMB1, LAMC1), collagens (COL5A1, COL18A1), basement membrane proteins (NID1), enzymes (LOXL2), tenascins (TNC) and binding proteins (LGALS3BP, TGFBI). Of note, TGFBI (Transforming growth factor-beta-induced protein ig-h3) is a marker of epithelial-to-mesenchymal transition (50) and is known to be related to cancer invasion, extravasation, and dissemination (51). Also, TNC (Tenascin C) stimulates angiogenesis and invasion in GBM (52). Among EV-associated proteases, PLAT (Tissue-type plasminogen activator, known as tPA) and PLAU (Urokinase-type plasminogen activator, known as uPA) share plasminogen as the major substrate, and are significantly increased in U373vIII EVs. It is known that EGFR activation stimulates the overexpression of these proteins involved in GBM invasion (53), and degradation of intravascular fibrin. Importantly, some ECM-binding adhesion proteins CD44, MCAM, THBS1, ITGA6, and ITGB4 are highly overexpressed in U373vIII EVs. Especially ITGA6, ITGB4, and MCAM are of interest as receptors for laminins and they may contribute to the role of EVs in GBM cell dissemination (14).
Table I. Invasion-related proteins enriched in U373vIII EVs (p value < 0.05).
| Name | Gene symbol | Fold | p value | Total TIC (average) |
|
|---|---|---|---|---|---|
| U373 | U373vIII | ||||
| Protease | |||||
| Calpain-2 catalytic subunit | CAPN2 | 5.22 | 0.0035 | 7.99E+05 | 4.18E+06 |
| Tissue-type plasminogen activator | PLAT | U373vIII EV only | 0.0043 | 0.00E+00 | 3.94E+06 |
| Urokinase-type plasminogen activator | PLAU | 8.29 | 0.018 | 5.64E+05 | 4.68E+06 |
| Aminopeptidase B | RNPEP | 11.96 | 0.0022 | 2.31E+05 | 2.76E+06 |
| Extracellular matrix (ECM) | |||||
| Collagen alpha-1(XVIII) chain | COL18A1 | 5.64 | 0.046 | 3.90E+06 | 2.20E+07 |
| Collagen alpha-1(V) chain | COL5A1 | 5.69 | 0.045 | 5.55E+05 | 3.16E+06 |
| Laminin subunit alpha-5 | LAMA5 | 43.52 | 0.00011 | 7.08E+05 | 3.08E+07 |
| Laminin subunit beta-1 | LAMB1 | 11.92 | 0.02 | 9.05E+05 | 1.08E+07 |
| Laminin subunit gamma-1 | LAMC1 | 10.02 | <0.00010 | 1.43E+06 | 1.43E+07 |
| Galectin-3-binding protein | LGALS3BP | 4.34 | 0.015 | 1.35E+07 | 5.84E+07 |
| Lysyl oxidase homolog 2 | LOXL2 | U373vIII EV only | 0.042 | 0.00E+00 | 1.45E+06 |
| Nidogen-1 | NID1 | 3.36 | 0.043 | 1.46E+06 | 4.92E+06 |
| Transforming growth factor-beta-induced protein ig-h3 | TGFBI | 585.12 | 0.016 | 1.86E+05 | 1.09E+08 |
| Tenascin | TNC | 5.17 | 0.018 | 2.04E+08 | 1.05E+09 |
| Cell adhesion | |||||
| Sodium/potassium-transporting ATPase subunit beta-1 | ATP1B1 | 8.65 | 0.03 | 3.91E+06 | 3.38E+07 |
| Basigin | BSG | 3.64 | 0.0052 | 7.66E+07 | 2.79E+08 |
| CD151 antigen | CD151 | 3.93 | 0.0025 | 6.82E+06 | 2.68E+07 |
| CD44 antigen | CD44 | 2.70 | 0.026 | 1.34E+08 | 3.60E+08 |
| Integrin alpha-6 | ITGA6 | 5.05 | 0.025 | 1.49E+07 | 7.50E+07 |
| Integrin beta-4 | ITGB4 | 4.15 | 0.032 | 2.68E+07 | 1.11E+08 |
| Junction plakoglobin | JUP | 2.64 | 0.039 | 4.50E+06 | 1.19E+07 |
| Cell surface glycoprotein MUC18 | MCAM | 14.19 | 0.033 | 1.45E+06 | 2.05E+07 |
| Multiple epidermal growth factor-like domains protein 10 | MEGF10 | 3.63 | 0.015 | 7.75E+05 | 2.81E+06 |
| Lactadherin | MFGE8 | 2.43 | 0.0091 | 4.97E+08 | 1.21E+09 |
| Neuroplastin | NPTN | 34.48 | 0.013 | 1.71E+05 | 5.88E+06 |
| Protocadherin-10 | PCDH10 | U373vIII EV only | 0.022 | 0.00E+00 | 1.52E+06 |
| Plakophilin-2 | PKP2 | 85.66 | 0.027 | 1.28E+05 | 1.10E+07 |
| Thrombospondin-1 | THBS1 | 3.59 | 0.05 | 1.01E+07 | 3.64E+07 |
Impact of EGFRvIII on the Cellular Uptake of Glioma EVs
Interestingly, a subset of EV proteins that are deregulated by oncogenic EGFRvIII have been implicated in homophilic interaction between cells. This includes CD151 tetraspanin (54), BSG (known also as EMPIRIN or CD147) (55), NPTN (56), and PCDH10 (57) (Table I). These changes may suggest interactive preferences of the respective EVs for specific recipient cell populations. In agreement with this prediction U373 and U373vIII cells exhibit different uptake efficiencies of their homologous or heterologous EVs (supplemental Fig. S6). For example, fluorescently (PKH67) labeled U373vIII-derived EVs were preferentially taken up by U373vIII cells, whereas U373 EVs interacted somewhat more readily with surfaces of U373 cell, as demonstrated by flow cytometry (supplemental Fig. S6). Nonetheless, the cumulative ability to uptake EVs was greater in the case of EGFRvIII-transformed glioma cells, which is consistent with the role of oncogeneses in regulating EV uptake (58, 59). Thus, EGFRvIII-mediated oncogenic transformation alters the magnitude and specificity of the EV internalization by changing the surface properties of both EVs and cellular recipients.
Oncogenic EGFRvIII Influences the Heterogeneity of Glioma EVs
Although heterogeneity of EVs is widely acknowledged and linked to their subsets and diverse biogenetic pathways (17), the impact of oncogenic transformation in this regard is poorly understood. To glean some related insights, we applied nano-flow cytometry to analyze the distribution of previously identified proteins among individual EVs emanating from either EGFRvIII-expressing or -non-expressing glioma cells. For this purpose, EVs were labeled using antibodies against the aforementioned selected EV-markers (CD9, CD81) and surface proteins, such as BSG, CD44, and CD151. This choice also included proteins with the high total TIC and a known involvement in invasive characteristics of EVs as indicated in Table I. EV populations single- or double-labeled for these proteins were subsequently resolved by nano-flow cytometry revealing several levels of EV heterogeneity. For example, according to MS analysis CD9 was slightly upregulated (1.5-fold), whereas CD81 was downregulated (0.3-fold) in U373vIII EVs. As expected, according to nano-flow cytometry, the population of CD9+ EVs was a little higher among U373vIII EVs (43.49%) as compared with U373 EVs (35.10%) (Fig. 7A). In contrast, CD81+ EV population was more abundant in the case of U373 cells (14.34%) than for their U373vIII counterparts (7.68%) (Fig. 7B, also compare Figs. 1D and supplemental Fig. S4). Moreover, invasion-related proteins including CD44, BSG, and CD151 were represented by larger numbers of EVs positive for these markers in the U373vIII EV population (Fig. 7C–7E). Similarly, CD151 expression was also higher in the case of U373vIII cells versus U373 (Fig. 2C) and this difference was reflected in the cargo of the corresponding EVs. Thus, oncogenic EGFRvIII alters not only the total level of EV-associated proteins per EV, but also the distribution of several proteins among EV subsets with potentially different biological activities.
Fig. 7.

Nano-flow cytometry analyses reveal heterogeneity of glioma EVs. Dot plots of U373 EVs and U373vIII EVs were acquired following staining with: FITC-conjugated-anti-CD9 (A) and APC-conjugated-anti-CD81 (B). Isotype controls were used to establish the nonspecific binding of antibodies (left panel). Nano-flow cytometry shows the distribution of single EVs labeled for invasion-related proteins including: BSG (C), CD44 (D), and CD151 (E), which are significantly overexpressed in U373vIII EVs. F, Heterogeneity of EGFR content in glioma EVs. Dot plots shows the double immunofluorescent labeling with FITC-conjugated-anti-CD9 and APC-conjugated-anti-EGFR on U373 EVs and U373vIII EVs. Fold change by quantitative proteomics is indicated in parentheses.
To further explore the interrelationships among different EV subpopulation we applied double immunofluorescent labeling. Interestingly, when EVs derived from U373vIII cells were labeled for EGFR (to highlight the predominant EGFRvIII oncoprotein) and CD9, the common EV marker, only 10.2% EVs were shown to be double positive, whereas 31.3% of all EVs stained for CD9 only. The latter is comparable to the EV staining pattern for parental U373 cells (Fig. 7F). This observation suggests that only a fraction of the EGFRvIII driven EV output carries the oncoprotein itself, mostly in the context of CD9, even though all cells express the oncogene and produce EVs. The mechanisms of EGFRvIII inclusion/exclusion into EV subsets remain to be investigated.
Double staining of EVs for BSG/CD44 and BSG/CD81 was also carried out and analyzed by nano-flow cytometry (Fig. 8A). We reasoned that because CD81 is closely linked to the exosomal pathway (24) and BSG was described primarily in the context of cellular microvesicles/blebs (60) this comparison may shed new light on a possible shift in the vesiculation mechanisms (61) following EGFRvIII-dependent transformation. However, this possibility remains to be examined more directly, as the expression of these markers does prove their use by the respective vesiculation pathways. Nonetheless, in the case of U373vIII EVs, BSG and CD44 showed strong colocalization, in which the majority of CD44+ EVs were also BSG+ suggesting a cosorting of these proteins during EV biogenesis. Interestingly, the population of CD81+ EV was markedly reduced among U373vIII EVs, which were instead highly positive for BSG, relative to EVs derived from U373 parental cells (Fig. 8A). These results are not only consistent with the global protein analysis (supplemental Fig. S2), but also suggest a reduced generation of CD81+ (exosome-like) EVs by cells harboring EGFRvIII oncogene with a possible contribution of microvesicle-like EVs positive for BSG.
Fig. 8.

BSG, CD44, and CD81 levels in EVs and glioma cells following EGFRvIII-dependent transformation. A, Nano flow cytometry following double staining of indicated EVs with FITC-conjugated-anti-BSG/APC-conjugated-anti-CD44 antibodies, and FITC-conjugated-anti-BSG/APC-conjugated-anti-CD81 antibodies. Dot plots indicate preferential co-expression of CD44 and BSG in EVs of U373vIII cells but not in EVs of U373 parental cells. B-C, Confocal images showing notable differences of cellular co-expression and distribution of BSG/CD44 (B) and BSG/CD81 (C), between EGFRvIII-expressing and -non-expressing glioma cells (see text).
To examine whether the profiles of BSG, CD44, and CD81 in glioma-derived EV represent altered protein packaging mechanisms into the EV cargo or reflect cellular expression and distribution profiles of these respective proteins we employed immunofluorescence and confocal microscopy of EV donor cells (Fig. 8B and 8C). Indeed, as in the case of the corresponding EVs, U373vIII cells exhibited a strong colocalization of BSG and CD44 throughout the whole plasma membrane and in filopodia (Fig. 8B). In contrast, in U373 cells BSG and CD44 colocalized in specific domains of membrane ruffles, and were generally less abundant except for weak, but continuous staining of cellular plasma membrane for CD44 (Fig. 8B). CD81 was mainly expressed along the plasma membrane, but overall the levels of this signal were markedly lower for U373vIII cells (as in EVs). Consequently, the colocalization of BSG and CD81 in these cells was virtually undetectable (Fig. 8C). Overall these observations suggest that oncogenic EGFRvIII reprograms the landscape of the cancer cell membrane resulting in altered vesiculation pathways, as well as features and possibly subsets of the resulting EVs.
DISCUSSION
In this study, we present a comprehensive analysis of the impact exerted by oncogenic EGFRvIII on the protein profiles of EVs released by human GBM cells. We used an isogenic GBM model system where EGFRvIII possesses a strong and well characterized transforming ability and the changes that occur downstream are not obscured by intercellular variability. In this setting, we made several novel observations, which suggest a multifaceted effect of the oncogenic transformation on glioma vesiculation processes, including the molecular regulation of EV biogenesis, as well as their protein composition, molecular heterogeneity, uptake by recipient cells and possible changes in biological activity (17).
Genetic and epigenetic driver events have already been implicated by us and others as regulators of the EV release and intercellular communication in cancer, including intercellular trafficking of transforming mutant macromolecules, such as HRAS, EGFR, and EGFRvIII (12, 17, 18, 58, 62). In this regard, our present study contains the first detailed description of the landscape of proteins that accompany EV-mediated release of the EGFRvIII oncoprotein from glioma cells. These observations may guide further explorations of the complexities associated with this emission, including the composition of EGFRvIII containing oncosomes (63), associated biomarkers (64, 65), as well as potential for a broader biological effect resulting from interactions between EGFRvIII-driven EVs, tumor microenvironment and recipient cells (12, 66).
Among biological responses elicited by glioma EVs, such as angiogenesis, altered growth, survival and therapy responses (12, 15, 18, 67) invasion is relatively less studied. This is paradoxical given the crucial involvement of the infiltrative growth pattern in the biology of GBM (1), and the emerging evidence for the role of tumor-derived EVs in invasion related processes, such as directional cell motility (14, 30, 68), proteolysis (46), pre-metastatic niche formation (16) and frank tumor dissemination (69). Although GBM is normally confined to the brain some of the cellular phenotypes involved could be relevant to the regional and systemic pathogenesis of this aggressive disease.
In this regard, our study suggests that EGFRvIII mutation may lead to EV-mediated release of several proteins with a documented role in tumor dissemination and interaction with the ECM, including CD44, MCAM, THBS1, and integrin α6β4. Among them CD44 is known to interact with hyaluronic acid, whereas MCAM interacts with laminin, which is also a ligand for exosomal integrin α6β4 implicated in organ specific metastasis (16, 70). Hyaluronic acid is highly overexpressed in patients with malignant glioma (71) and laminins are major constituents of blood vessel basement membranes in brain tumor microenvironment (72). Thus, EGFRvIII may reprogram glioma related EVs to interact with ECM and structures within the brain microenvironment and thereby modulate tumor invasiveness. Because GBM-related EVs are also detected systemically, changes in their surface properties may contribute to distant paraneoplastic effects of these intracranial tumors (3).
Different activating EGFR mutations may impact EV mediated processes in cancer. For example constitutively active EGFR mutant (E746 - A750 deletion in tyrosine kinase domain) expressed in HCC827 lung cancer cells, was reported to stimulate protein sorting and the resulting enrichment of the EV compartment in EGFR, SRC, GRB2, RALA, RAC1, and KRAS, relative to EVs produced by immortalized bronchial epithelial cell HBE4 (73). Notably, BSG and CD151 were upregulated whereas CD81 was downregulated in HCC827-derived EVs (73), which is similar to our observations involving EGFRvIII.
Other oncogenic events within the EGFR signaling pathway have also been implicated in proinvasive reprogramming of the EV cargo. For example, integrins and metalloproteases associated with EVs are upregulated by HRAS (74) EGFR, KRAS, and SRC (75). Overexpression of the EGFR-related proto-oncogene, ERBB2/HER2 in C5.2 breast cancer cells leads to increased content of CD44 in tumor EVs (76). Such changes may be influenced by both oncogenic mutations and cellular contexts in which they occur. For example, integrin beta-1 in EVs is upregulated in the case of EGFR-driven HCC827 cells (73) and mutant HRAS overexpressing MDCK cells (74), but not in U373vIII cells. Thus, a better definition of signaling cues and cellular modifiers (77) operative in different tumor subtypes (78) may be needed to fully understand the regulation of EV biogenesis and cargo assembly in cancer.
Cancer-related EVs have been implicated as carriers of proteolytic activity. Indeed, we observed a trend for enrichment in U373vIII EVs (as compared with their U373-derived counterparts) of several proteases, including ADAM9, ADAM10, cathepsin Z (CTSZ), and MMP14 involved in the degradation of ECM components, including laminins and collagens (supplemental Table S2). Moreover, two elements of the fibrinolytic system (PLAT and PLAU) are significantly overexpressed in U373vIII EVs. This is consistent with the notion that activated EGFR upregulates PLAU and its receptor (PLAUR) resulting in an increase of cellular invasiveness (79), as well as signaling interactions (80) in which the role of EVs remains to be studied. Collectively, our observations suggest that EGFRvIII expression may enable GBM cells to emit EVs endowed with the enhanced capacity to selectively bind and degrade specific ECM proteins (81), thereby possibly contributing to migratory, signaling and stromal responses (82).
Another interesting finding is the role of EGFRvIII in differential EV uptake. Quantitative proteomics and GO term analysis indicated that EVs released by glioma cells expressing this potent oncogene are enriched in adhesion molecules of which some are known for homophilic interactions, including CD151 (54), BSG (55), NPTN (56), and PCDH10 (57). We have also observed a greater homophilic EV uptake by U373vIII cells relative to their U373 counterparts, suggesting the existence of an autocrine or a homotypic ('homocrine') interaction between GBM cell subsets and their own EVs. Such interactions have been implicated in directional cell migration (14) and may represent programs linked to oncogenic transformation, as documented in cells harboring mutant KRAS or HRAS (58, 59). Further studies are needed to understand the role of such processes in GBM.
Our study also revealed novel aspects of EV heterogeneity in the context of EGFRvIII-dependent oncogenic transformation. Although traditionally EVs were analyzed in bulk, or according to their physical properties, recent technologies enabled greater insights into the properties of EV subsets or even single EVs. This includes Raman spectroscopy (83), microfluidic platforms (84) and high-resolution flow cytometry (85–87). Indeed, EVs originating from the same parental cell population may exhibit a remarkable spectrum of characteristics (83, 88, 89), which may change dynamically with the state of donor cells (90, 91). Our analysis revealed several important characteristics associated with EV subsets generated by EGFRvIII-transformed glioma cells. First, we observed that only a fraction of EVs carries the EGFRvIII oncoprotein, mostly in the context of CD9 tetraspanin. However, only subsets of these EVs carried traditional exosomal markers such as CD9 and CD81. Notably, EGFRvIII expression suppressed some of the exosomal features (24) in glioma EVs, including downregulation of CD81 and a complete loss of CD82, as reported earlier (92). This may reflect antagonistic interactions between the activated EGFR pathway and CD82 (93), worthy of further exploration in GBM. EGFRvIII transformation also resulted in the enrichment for BSG+/CD44+ double positive EVs, a pattern reminiscent of the colocalization these proteins near active filopodia of U373vIII cells. Interestingly, EGFR-Ras-ERK signaling cascade may promote the assembly of BSG, CD44, and EGFR to form invadopodia (94). The pro-invasive role of this complex is underscored by the recruitment of MMP14 (MT1-MMP) by BSG (55), all of which may contribute to the pathogenetic effects of glioma EVs.
Overall, our results suggest that EV mediated communication between glioma cells and their microenvironment are regulated by oncogenic transformation. Consequently, the impact of EGFRvIII and other transforming events associated with GBM (IDH1, PTEN) (1) on the proteome of tumor EVs may have meaningful and underappreciated implications for EV-mediated biological effects, biomarker development and therapeutic applications.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE (27) partner repository with the dataset identifier PXD008311 and 10.6019/PXD008311. Summary of ProteomeXchange accession: PXD008311; PubMed ID: 30006486; Project Webpage: http://www.ebi.ac.uk/pride/archive/projects/PXD008311; FTP Download: ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2018/07/PXD008311.
Supplementary Material
Acknowledgments
We are grateful to the Montreal Children's Hospital Foundation and Donors for supporting our purchase of Nano-Flow Cytometry Instrument. We thank the Clinical Proteomics Platform for mass spectrometry analyses, Immunophenotyping Platform for nano-flow cytometry, and Molecular Imaging Platform for confocal analyses at RI-MUHC. We would also like to thank our families for their patient and unwavering support for our efforts.
Footnotes
* This work was supported by the operating grants from Canadian Institutes for Health Research (CIHR Foundation Grant, MOP 102736, MOP 111119) and Innovation to Impact Grant from the Canadian Cancer Society Research Institute to J.R., who is also a recipient of the Jack Cole Chair in Pediatric Hematology/Oncology. DSC was supported by a Fellowship from McGill Integrated Cancer Research Training Program (MICRTP, CIHR/FRSQ-FRN53888). DKK was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) through Banting fellowship. F.P.R. was supported by the Canada Excellence Research Chairs Program, and by NIH grant HG001715. Infrastructure funds were provided by the Fonds de Recherche en Santé du Quebec (FRSQ), Research Institute of the McGill University Health Centre (RI-MUHC), Montreal Children's Hospital, and McGill University.
 This article contains supplemental Figures and Tables. We declare no conflicts of interest.
This article contains supplemental Figures and Tables. We declare no conflicts of interest.
1 The abbreviations used are:
- GBM
- glioblastoma multiforme
- EGFR
- epidermal growth factor receptor
- EGFRvIII
- EGFR variant III
- EVs
- extracellular vesicles
- ECM
- extracellular matrix
- FBS
- fetal bovine serum
- CM
- conditioned medium
- NTA
- nanoparticle tracking analysis
- TIC
- total ion chromatogram
- GO
- gene ontology
- V-SSC
- violet SSC.
REFERENCES
- 1. Reifenberger G., Wirsching H. G., Knobbe-Thomsen C. B., and Weller M. (2017) Advances in the molecular genetics of gliomas - implications for classification and therapy. Nature reviews. Clin. Oncol. 14, 434–452 [DOI] [PubMed] [Google Scholar]
- 2. Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., Chin L., DePinho R. A., and Cavenee W. K. (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710 [DOI] [PubMed] [Google Scholar]
- 3. Magnus N., Garnier D., Meehan B., McGraw S., Lee T. H., Caron M., Bourque G., Milsom C., Jabado N., Trasler J., Pawlinski R., Mackman N., and Rak J. (2014) Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc. Natl. Acad. Sci. U.S.A. 111, 3544–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Talasila K. M., Soentgerath A., Euskirchen P., Rosland G. V., Wang J., Huszthy P. C., Prestegarden L., Skaftnesmo K. O., Sakariassen P. O., Eskilsson E., Stieber D., Keunen O., Brekka N., Moen I., Nigro J. M., Vintermyr O. K., Lund-Johansen M., Niclou S., Mork S. J., Enger P. O., Bjerkvig R., and Miletic H. (2013) EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 125, 683–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avraham R., and Yarden Y. (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 6. Ohgaki H., Dessen P., Jourde B., Horstmann S., Nishikawa T., Di Patre P. L., Burkhard C., Schuler D., Probst-Hensch N. M., Maiorka P. C., Baeza N., Pisani P., Yonekawa Y., Yasargil M. G., Lutolf U. M., and Kleihues P. (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res. 64, 6892–6899 [DOI] [PubMed] [Google Scholar]
- 7. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., Nikolsky Y., Busam D. A., Tekleab H., Diaz L. A. Jr, Hartigan J., Smith D. R., Strausberg R. L., Marie S. K., Shinjo S. M., Yan H., Riggins G. J., Bigner D. D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., and Kinzler K. W. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research, N. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frattini V., Trifonov V., Chan J. M., Castano A., Lia M., Abate F., Keir S. T., Ji A. X., Zoppoli P., Niola F., Danussi C., Dolgalev I., Porrati P., Pellegatta S., Heguy A., Gupta G., Pisapia D. J., Canoll P., Bruce J. N., McLendon R. E., Yan H., Aldape K., Finocchiaro G., Mikkelsen T., Prive G. G., Bigner D. D., Lasorella A., Rabadan R., and Iavarone A. (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 45, 1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brennan C. W., Verhaak R. G., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H., Beroukhim R., Bernard B., Wu C. J., Genovese G., Shmulevich I., Barnholtz-Sloan J., Zou L., Vegesna R., Shukla S. A., Ciriello G., Yung W. K., Zhang W., Sougnez C., Mikkelsen T., Aldape K., Bigner D. D., Van Meir E. G., Prados M., Sloan A., Black K. L., Eschbacher J., Finocchiaro G., Friedman W., Andrews D. W., Guha A., Iacocca M., O'Neill B. P., Foltz G., Myers J., Weisenberger D. J., Penny R., Kucherlapati R., Perou C. M., Hayes D. N., Gibbs R., Marra M., Mills G. B., Lander E., Spellman P., Wilson R., Sander C., Weinstein J., Meyerson M., Gabriel S., Laird P. W., Haussler D., Getz G., Chin L., and Network T. R. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Magnus N., Garnier D., and Rak J. (2010) Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 116, 815–818 [DOI] [PubMed] [Google Scholar]
- 12. Al-Nedawi K., Meehan B., Micallef J., Lhotak V., May L., Guha A., and Rak J. (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 10, 619–624 [DOI] [PubMed] [Google Scholar]
- 13. Colombo M., Raposo G., and Thery C. (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 [DOI] [PubMed] [Google Scholar]
- 14. Sung B. H., Ketova T., Hoshino D., Zijlstra A., and Weaver A. M. (2015) Directional cell movement through tissues is controlled by exosome secretion. Nature Commun. 6, 7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Al-Nedawi K., Meehan B., Kerbel R. S., Allison A. C., and Rak J. (2009) Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. U.S.A. 106, 3794–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoshino A., Costa-Silva B., Shen T. L., Rodrigues G., Hashimoto A., Tesic M. M., Molina H., Kohsaka S., Di G. A., Ceder S., Singh S., Williams C., Soplop N., Uryu K., Pharmer L., King T., Bojmar L., Davies A. E., Ararso Y., Zhang T., Zhang H., Hernandez J., Weiss J. M., Dumont-Cole V. D., Kramer K., Wexler L. H., Narendran A., Schwartz G. K., Healey J. H., Sandstrom P., Jorgen L. K., Kure E. H., Grandgenett P. M., Hollingsworth M. A., de S. M., Kaur S., Jain M., Mallya K., Batra S. K., Jarnagin W. R., Brady M. S., Fodstad O., Muller V., Pantel K., Minn A. J., Bissell M. J., Garcia B. A., Kang Y., Rajasekhar V. K., Ghajar C. M., Matei I., Peinado H., Bromberg J., and Lyden D. (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi D., Lee T. H., Spinelli C., Chennakrishnaiah S., D'Asti E., and Rak J. (2017) Extracellular vesicle communication pathways as regulatory targets of oncogenic transformation. Sem. Cell Develop. Biol. 67, 11–22 [DOI] [PubMed] [Google Scholar]
- 18. Skog J., Wurdinger T., van Rijn S., Meijer D. H., Gainche L., Sena-Esteves M., Curry W. T. Jr, Carter B. S., Krichevsky A. M., and Breakefield X. O. (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kucharzewska P., Christianson H. C., Welch J. E., Svensson K. J., Fredlund E., Ringner M., Morgelin M., Bourseau-Guilmain E., Bengzon J., and Belting M. (2013) Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. U.S.A. 110, 7312–7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mallawaaratchy D. M., Hallal S., Russell B., Ly L., Ebrahimkhani S., Wei H., Christopherson R. I., Buckland M. E., and Kaufman K. L. (2017) Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J. Neurooncol. 131, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujii T., Sakata A., Nishimura S., Eto K., and Nagata S. (2015) TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. U.S.A. 112, 12800–12805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi D. S., Choi D. Y., Hong B. S., Jang S. C., Kim D. K., Lee J., Kim Y. K., Kim K. P., and Gho Y. S. (2012) Quantitative proteomics of extracellular vesicles derived from human primary and metastatic colorectal cancer cells. J. Extracell. Vesicles 1, 1–15, http://journalofextracellularvesicles.net/index.php/jev/article/view/18704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee H. M., Choi E. J., Kim J. H., Kim T. D., Kim Y. K., Kang C., and Gho Y. S. (2010) A membranous form of ICAM-1 on exosomes efficiently blocks leukocyte adhesion to activated endothelial cells. Biochem. Biophys. Res. Commun. 397, 251–256 [DOI] [PubMed] [Google Scholar]
- 24. Kowal J., Arras G., Colombo M., Jouve M., Morath J. P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., and Thery C. (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U.S.A. 113, E968–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi D. S., and Gho Y. S. (2015) Isolation of extracellular vesicles for proteomic profiling. Methods Mol. Biol. 1295, 167–177 [DOI] [PubMed] [Google Scholar]
- 26. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 27. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 29. Nie F., Yang J., Wen S., An Y. L., Ding J., Ju S. H., Zhao Z., Chen H. J., Peng X. G., Wong S. T., Zhao H., and Teng G. J. (2012) Involvement of epidermal growth factor receptor overexpression in the promotion of breast cancer brain metastasis. Cancer 118, 5198–5209 [DOI] [PubMed] [Google Scholar]
- 30. Di Vizio D., Kim J., Hager M. H., Morello M., Yang W., Lafargue C. J., True L. D., Rubin M. A., Adam R. M., Beroukhim R., Demichelis F., and Freeman M. R. (2009) Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 69, 5601–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Montermini L., Meehan B., Garnier D., Lee W. J., Lee T. H., Guha A., Al-Nedawi K., and Rak J. (2015) Inhibition of oncogenic epidermal growth factor receptor kinase triggers release of exosome-like extracellular vesicles and impacts their phosphoprotein and DNA content. J. Biol. Chem. 290, 24534–24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lotvall J., Hill A. F., Hochberg F., Buzas E. I., Di V. D., Gardiner C., Gho Y. S., Kurochkin I. V., Mathivanan S., Quesenberry P., Sahoo S., tahara H., Wauben M. H., Witwer K. W., and Thery C. (2014) Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 3:26913. doi: 10.3402/jev.v3.26913 eCollection 2014, 26913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yue S., Mu W., Erb U., and Zoller M. (2015) The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget 6, 2366–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luga V., Zhang L., Viloria-Petit A. M., Ogunjimi A. A., Inanlou M. R., Chiu E., Buchanan M., Hosein A. N., Basik M., and Wrana J. L. (2012) Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 151, 1542–1556 [DOI] [PubMed] [Google Scholar]
- 35. Chairoungdua A., Smith D. L., Pochard P., Hull M., and Caplan M. J. (2010) Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalra H., Simpson R. J., Ji H., Aikawa E., Altevogt P., Askenase P., Bond V. C., Borras F. E., Breakefield X., Budnik V., Buzas E., Camussi G., Clayton A., Cocucci E., Falcon-Perez J. M., Gabrielsson S., Gho Y. S., Gupta D., Harsha H. C., Hendrix A., Hill A. F., Inal J. M., Jenster G., Kramer-Albers E. M., Lim S. K., Llorente A., Lotvall J., Marcilla A., Mincheva-Nilsson L., Nazarenko I., Nieuwland R., Nolte-'t Hoen E. N., Pandey A., Patel T., Piper M. G., Pluchino S., Prasad T. S., Rajendran L., Raposo G., Record M., Reid G. E., Sanchez-Madrid F., Schiffelers R. M., Siljander P., Stensballe A., Stoorvogel W., Taylor D., Thery C., Valadi H., van Balkom B. W., Vazquez J., Vidal M., Wauben M. H., Yanez-Mo M., Zoeller M., and Mathivanan S. (2012) Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 10, e1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim D. K., Kang B., Kim O. Y., Choi D. S., Lee J., Kim S. R., Go G., Yoon Y. J., Kim J. H., Jang S. C., Park K. S., Choi E. J., Kim K. P., Desiderio D. M., Kim Y. K., Lotvall J., Hwang D., and Gho Y. S. (2013) EVpedia: an integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2, doi: 10.3402/jev.v2i0.20384 eCollection 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rubin B. P., Tucker R. P., Brown-Luedi M., Martin D., and Chiquet-Ehrismann R. (2002) Teneurin 2 is expressed by the neurons of the thalamofugal visual system in situ and promotes homophilic cell-cell adhesion in vitro. Development 129, 4697–4705 [DOI] [PubMed] [Google Scholar]
- 39. Struyk A. F., Canoll P. D., Wolfgang M. J., Rosen C. L., D'Eustachio P., and Salzer J. L. (1995) Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 15, 2141–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Werner H., Dimou L., Klugmann M., Pfeiffer S., and Nave K. A. (2001) Multiple splice isoforms of proteolipid M6B in neurons and oligodendrocytes. Mol. Cell Neurosci. 18, 593–605 [DOI] [PubMed] [Google Scholar]
- 41. Choi D. S., Kim D. K., Kim Y. K., and Gho Y. S. (2015) Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 34, 474–490 [DOI] [PubMed] [Google Scholar]
- 42. Huang D. W., Sherman B. T., Tan Q., Collins J. R., Alvord W. G., Roayaei J., Stephens R., Baseler M. W., Lane H. C., and Lempicki R. A. (2007) The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Furnari F. B., Cloughesy T. F., Cavenee W. K., and Mischel P. S. (2015) Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sangar V., Funk C. C., Kusebauch U., Campbell D. S., Moritz R. L., and Price N. D. (2014) Quantitative proteomic analysis reveals effects of epidermal growth factor receptor (EGFR) on invasion-promoting proteins secreted by glioblastoma cells. Mol. Cell. Proteomics 13, 2618–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mallawaaratchy D. M., Buckland M. E., McDonald K. L., Li C. C., Ly L., Sykes E. K., Christopherson R. I., and Kaufman K. L. (2015) Membrane proteome analysis of glioblastoma cell invasion. J. Neuropathol. Exp. Neurol. 74, 425–441 [DOI] [PubMed] [Google Scholar]
- 46. Hendrix A., Westbroek W., Bracke M., and De W. O. (2010) An ex(o)citing machinery for invasive tumor growth. Cancer Res. 70, 9533–9537 [DOI] [PubMed] [Google Scholar]
- 47. Friedl P., and Alexander S. (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 [DOI] [PubMed] [Google Scholar]
- 48. Wood C. R., Huang K., Diener D. R., and Rosenbaum J. L. (2013) The cilium secretes bioactive ectosomes. Curr. Biol. 23, 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoshino D., Kirkbride K. C., Costello K., Clark E. S., Sinha S., Grega-Larson N., Tyska M. J., and Weaver A. M. (2013) Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 5, 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yokobori T., and Nishiyama M. (2017) TGF-beta signaling in gastrointestinal cancers: progress in basic and clinical research. J. Clin. Med. 6, 11; doi: 10.3390/jcm6010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ma C., Rong Y., Radiloff D. R., Datto M. B., Centeno B., Bao S., Cheng A. W., Lin F., Jiang S., Yeatman T. J., and Wang X. F. (2008) Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 22, 308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xia S., Lal B., Tung B., Wang S., Goodwin C. R., and Laterra J. (2016) Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro-oncology 18, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amos S., Redpath G. T., Dipierro C. G., Carpenter J. E., and Hussaini I. M. (2010) Epidermal growth factor receptor-mediated regulation of urokinase plasminogen activator expression and glioblastoma invasion via C-SRC/MAPK/AP-1 signaling pathways. J. Neuropathol. Exp. Neurol. 69, 582–592 [DOI] [PubMed] [Google Scholar]
- 54. Hong I. K., Jin Y. J., Byun H. J., Jeoung D. I., Kim Y. M., and Lee H. (2006) Homophilic interactions of Tetraspanin CD151 up-regulate motility and matrix metalloproteinase-9 expression of human melanoma cells through adhesion-dependent c-Jun activation signaling pathways. J. Biol. Chem. 281, 24279–24292 [DOI] [PubMed] [Google Scholar]
- 55. Grass G. D., and Toole B. P. (2015) How, with whom and when: an overview of CD147-mediated regulatory networks influencing matrix metalloproteinase activity. Biosci. Rep. 36, e00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Owczarek S., and Berezin V. (2012) Neuroplastin: cell adhesion molecule and signaling receptor. Int. J. Biochem. Cell Biol. 44, 1–5 [DOI] [PubMed] [Google Scholar]
- 57. Kim S. Y., Yasuda S., Tanaka H., Yamagata K., and Kim H. (2011) Non-clustered protocadherin. Cell Adh. Migr. 5, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee T. H., Chennakrishnaiah S., Meehan B., Montermini L., Garnier D., D'Asti E., Hou W., Magnus N., Gayden T., Jabado N., Eppert K., Majewska L., and Rak J. (2016) Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 7, 51991–52002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kamerkar S., LeBleu V. S., Sugimoto H., Yang S., Ruivo C. F., Melo S. A., Lee J. J., and Kalluri R. (2017) Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alcazar O., Hawkridge A. M., Collier T. S., Cousins S. W., Bhattacharya S. K., Muddiman D. C., and Marin-Castano M. E. (2009) Proteomics characterization of cell membrane blebs in human retinal pigment epithelium cells. Mol. Cell. Proteomics 8, 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Menck K., Sonmezer C., Worst T. S., Schulz M., Dihazi G. H., Streit F., Erdmann G., Kling S., Boutros M., Binder C., and Gross J. C. (2017) Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracellular Vesicles 6, 1378056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Garnier D., Magnus N., Lee T. H., Bentley V., Meehan B., Milsom C., Montermini L., Kislinger T., and Rak J. (2012) Cancer Cells Induced to Express Mesenchymal Phenotype Release Exosome-like Extracellular Vesicles Carrying Tissue Factor. J. Biol. Chem. 287, 43565–43572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meehan B., Rak J., and Di Vizio D. (2016) Oncosomes - large and small: what are they, where they came from? J. Extracellular Vesicles 5, 33109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jayaram S., Gupta M. K., Polisetty R. V., Cho W. C., and Sirdeshmukh R. (2014) Towards developing biomarkers for glioblastoma multiforme: a proteomics view. Expert Rev. Proteomics 11, 621–639 [DOI] [PubMed] [Google Scholar]
- 65. Garnier D., Jabado N., and Rak J. (2013) Extracellular vesicles as prospective carriers of oncogenic protein signatures in adult and paediatric brain tumours. Proteomics 13, 1595–1607 [DOI] [PubMed] [Google Scholar]
- 66. Godlewski J., Krichevsky A. M., Johnson M. D., Chiocca E. A., and Bronisz A. (2015) Belonging to a network–microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro-oncology 17, 652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arscott W. T., Tandle A. T., Zhao S., Shabason J. E., Gordon I. K., Schlaff C. D., Zhang G., Tofilon P. J., and Camphausen K. A. (2013) Ionizing radiation and glioblastoma exosomes: implications in tumor biology and cell migration. Transl. Oncol. 6, 638–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu D., Xu Y., Ding T., Zu Y., Yang C., and Yu L. (2017) Pairing of integrins with ECM proteins determines migrasome formation. Cell Res. 27, 1397–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hood J. L., San R. S., and Wickline S. A. (2011) Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 71, 3792–3801 [DOI] [PubMed] [Google Scholar]
- 70. Mu W., Rana S., and Zoller M. (2013) Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 15, 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cha J., Kang S. G., and Kim P. (2016) Strategies of Mesenchymal Invasion of Patient-derived Brain Tumors: Microenvironmental Adaptation. Sci. Rep. 6, 24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ljubimova J. Y., Fujita M., Khazenzon N. M., Ljubimov A. V., and Black K. L. (2006) Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front Biosci. 11, 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clark D. J., Fondrie W. E., Yang A., and Mao L. (2016) Triple SILAC quantitative proteomic analysis reveals differential abundance of cell signaling proteins between normal and lung cancer-derived exosomes. J. Proteomics 133, 161–169 [DOI] [PubMed] [Google Scholar]
- 74. Tauro B. J., Mathias R. A., Greening D. W., Gopal S. K., Ji H., Kapp E. A., Coleman B. M., Hill A. F., Kusebauch U., Hallows J. L., Shteynberg D., Moritz R. L., Zhu H. J., and Simpson R. J. (2013) Oncogenic H-ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol. Cell. Proteomics 12, 2148–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Demory Beckler M., Higginbotham J. N., Franklin J. L., Ham A. J., Halvey P. J., Imasuen I. E., Whitwell C., Li M., Liebler D. C., and Coffey R. J. (2013) Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteomics 12, 343–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Amorim M., Fernandes G., Oliveira P., Martins-de-Souza D., Dias-Neto E., and Nunes D. (2014) The overexpression of a single oncogene (ERBB2/HER2) alters the proteomic landscape of extracellular vesicles. Proteomics 14, 1472–1479 [DOI] [PubMed] [Google Scholar]
- 77. Bobrie A., Krumeich S., Reyal F., Recchi C., Moita L. F., Seabra M. C., Ostrowski M., and Thery C. (2012) Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 72, 4920–4930 [DOI] [PubMed] [Google Scholar]
- 78. Nakano I., Garnier D., Minata M., and Rak J. (2015) Extracellular vesicles in the biology of brain tumour stem cells - Implications for inter-cellular communication, therapy and biomarker development. Semin. Cell Dev. Biol., 10. [DOI] [PubMed] [Google Scholar]
- 79. Mori T., Abe T., Wakabayashi Y., Hikawa T., Matsuo K., Yamada Y., Kuwano M., and Hori S. (2000) Upregulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-alpha or basic fibroblast growth factor. J. Neurooncol. 46, 115–123 [DOI] [PubMed] [Google Scholar]
- 80. Hu J., Jo M., Cavenee W. K., Furnari F., VandenBerg S. R., and Gonias S. L. (2011) Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 108, 15984–15989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McCready J., Sims J. D., Chan D., and Jay D. G. (2010) Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Charles N. A., Holland E. C., Gilbertson R., Glass R., and Kettenmann H. (2011) The brain tumor microenvironment. Glia 59, 1169–1180 [DOI] [PubMed] [Google Scholar]
- 83. Smith Z. J., Lee C., Rojalin T., Carney R. P., Hazari S., Knudson A., Lam K., Saari H., Ibanez E. L., Viitala T., Laaksonen T., Yliperttula M., and Wachsmann-Hogiu S. (2015) Single exosome study reveals subpopulations distributed among cell lines with variability related to membrane content. J. Extracell. Vesicles 4, 28533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Olcum S., Cermak N., Wasserman S. C., Christine K. S., Atsumi H., Payer K. R., Shen W., Lee J., Belcher A. M., Bhatia S. N., and Manalis S. R. (2014) Weighing nanoparticles in solution at the attogram scale. Proc. Natl. Acad. Sci. U.S.A. 111, 1310–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. van der Vlist E. J., Nolte-'t Hoen E. N., Stoorvogel W., Arkesteijn G. J., and Wauben M. H. (2012) Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 7, 1311–1326 [DOI] [PubMed] [Google Scholar]
- 86. Pospichalova V., Svoboda J., Dave Z., Kotrbova A., Kaiser K., Klemova D., Ilkovics L., Hampl A., Crha I., Jandakova E., Minar L., Weinberger V., and Bryja V. (2015) Simplified protocol for flow cytometry analysis of fluorescently labeled exosomes and microvesicles using dedicated flow cytometer. J. Extracell. Vesicles 4, 25530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Higginbotham J. N., Zhang Q., Jeppesen D. K., Scott A. M., Manning H. C., Ochieng J., Franklin J. L., and Coffey R. J. (2016) Identification and characterization of EGF receptor in individual exosomes by fluorescence-activated vesicle sorting. J. Extracell. Vesicles 5, 29254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Laulagnier K., Vincent-Schneider H., Hamdi S., Subra C., Lankar D., and Record M. (2005) Characterization of exosome subpopulations from RBL-2H3 cells using fluorescent lipids. Blood Cells Mol. Dis. 35, 116–121 [DOI] [PubMed] [Google Scholar]
- 89. Oksvold M. P., Kullmann A., Forfang L., Kierulf B., Li M., Brech A., Vlassov A. V., Smeland E. B., Neurauter A., and Pedersen K. W. (2014) Expression of B-cell surface antigens in subpopulations of exosomes released from B-cell lymphoma cells. Clin. Ther. 36, 847–862 e841 [DOI] [PubMed] [Google Scholar]
- 90. Tauro B. J., Greening D. W., Mathias R. A., Mathivanan S., Ji H., and Simpson R. J. (2012) Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol. Cell. Proteomics 12, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Roucourt B., Meeussen S., Bao J., Zimmermann P., and David G. (2015) Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 25, 412–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yang C. H., Chou H. C., Fu Y. N., Yeh C. L., Cheng H. W., Chang I. C., Liu K. J., Chang G. C., Tsai T. F., Tsai S. F., Liu H. P., Wu Y. C., Chen Y. T., Huang S. F., and Chen Y. R. (2015) EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim. Biophys. Acta 1852, 1540–1549 [DOI] [PubMed] [Google Scholar]
- 93. Wang X. Q., Yan Q., Sun P., Liu J. W., Go L., McDaniel S. M., and Paller A. S. (2007) Suppression of epidermal growth factor receptor signaling by protein kinase C-alpha activation requires CD82, caveolin-1, and ganglioside. Cancer Res. 67, 9986–9995 [DOI] [PubMed] [Google Scholar]
- 94. Grass G. D., Tolliver L. B., Bratoeva M., and Toole B. P. (2013) CD147, CD44, and the epidermal growth factor receptor (EGFR) signaling pathway cooperate to regulate breast epithelial cell invasiveness. J. Biol. Chem. 288, 26089–26104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE (27) partner repository with the dataset identifier PXD008311 and 10.6019/PXD008311. Summary of ProteomeXchange accession: PXD008311; PubMed ID: 30006486; Project Webpage: http://www.ebi.ac.uk/pride/archive/projects/PXD008311; FTP Download: ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2018/07/PXD008311.