

Abstract
The activation of natural killer (NK) cells depends on a change in the balance of signals from inhibitory and activating receptors. The activation threshold values of NK cells are thought to be set by engagement of inhibitory receptors during development. Here, we found that the activating receptor NKG2D specifically set the activation threshold for the activating receptor NCR1 through a process that required the adaptor DAP12. As a result, NKGD2-deficient (Klrk1-/-) mice controlled tumors and cytomegalovirus infection better than wild-type controls through the NCR1-induced production of the cytokine IFN-γ. Expression of NKG2D before the immature NK cell stage increased expression of the adaptor CD3ζ. Reduced expression of CD3ζ in Klrk1-/- mice was associated with enhanced signal transduction through NCR1 and CD3ζ-deficiency resulted in hyper-responsiveness to stimulation via NCR1. Thus, an activating receptor developmentally set the activity of another activating receptor on NK cells and determined NK cell-reactivity to cellular threats.
Keywords: NK cells, NK cell education, NKG2D, NCR1, DAP12, CD3ζ, CMV, cancer immuno-surveillance
NK cells detect and eliminate “stressed” cells, for example following infection or malignant transformation1. Due to their ability to respond without prior sensitization, activation of NK cells needs to be tightly regulated to ensure proper immuno-surveillance whilst avoiding hyperactivity that may lead to inflammatory or autoimmune disorders. Proper responsiveness is mediated through `education` process during NK cell development2. After engagement of inhibitory receptors, NK cells gain full reactivity and develop tolerance toward self. NK cell-responsiveness is further fine-tuned by continuous cues that mature NK cells encounter in the periphery. NK cell activation depends on a shift in the signaling balance between inhibitory and activating receptors. Under homeostatic conditions, inhibitory signals prevail when NK cells interact with peripheral tissues. In response to a cellular threat, host cells downregulate MHCI molecules and/or overexpress stress-induced or non-self-ligands. Upon encounter of `stressed` target cells, a lack of signals through the inhibitory receptors and/or increased stimulation of activating receptors shifts the balance towards NK cell activation3. The factors that control the bandwidth of the equilibrium between inhibitory and activating cues are not completely characterized.
NKG2D and NCR1 are activating receptors expressed on all NK cells. They primarily mediate tissue stress-surveillance by recognizing stress-induced self-ligands on target cells4. NCR1 (NKp46) is the only member of the NCR family expressed in mice. The role of NCR1 and NKG2D in the control of tumors and infection is well documented4, 5. Beyond its effector functions6, NKG2D is expressed from the earliest precursors onwards during NK cell development7,8. NKG2D-deficient mice have bone marrow (BM) NK cell progenitors with enhanced proliferation, faster maturation and augmented sensitivity to apoptosis9, indicating a role for NKG2D signaling in NK cell development. Moreover, as expected, NKG2D-deficiency results in reduced NK cell-responsiveness to target cells expressing NKG2D ligands9, 10. However, in response to specific activating stimuli, NKG2D-deficient NK cells display a hyper-reactive phenotype in terms of production of the cytokine IFN-γ [9, 11] and better control mouse cytomegalovirus (mCMV) infection compared to NKG2D-sufficient NK cells9. How NKG2D-deficiency drives NK cell hyper-reactivity is unclear.
Activating receptors NCR1 and NKG2D require adaptors to convert signals into the cell. NKG2D associates with the adaptors DAP10 or DAP1212 while NCR1 docks the adaptors CD3ζ and FcεRγ13. DAP10 has an YxxM motif through which the PI3K and Grb2-Vav1-SOS1 are engaged14 CD3ζ, FcεRγ and DAP12 possess ITAM motifs, phosphorylation of which activates signaling proteins Syk and ZAP70, resulting in cytotoxicity and/or cytokine production14. It is commonly believed that adaptors with ITAM motifs only transduce activating signals. However, increasing evidence shows that they can also negatively impact signaling cascades14, 15, 16. How adaptors of activating NK cell receptors contribute to negative regulation of signaling is mostly unknown.
Here we demonstrate that NKG2D-deficiency, or blocking of NKG2D signaling early during NK development caused hyper-reactivity of the NCR1 receptor, resulting in enhanced control of mCMV infection and tumors expressing NCR1 ligands. Deficiency of NKG2D or DAP12, resulted in downregulation of CD3ζ and ZAP-70, yet in stronger signaling after NCR1 stimulation. Ablation of NKG2D in immature CD122+NK1.1+NCR1+CD11b-c-Kit- NK cells completely abrogated the hyperactive phenotype of NK cells, indicating that this regulation occurs early during NK development. NKG2D-driven regulation of NCR1 signaling was independent of conventional education mechanisms. Altogether, these results reveal an unknown developmental regulation of one activating NK receptor by another, which controls the sensitivity of immune-surveillance of tumors and viral infections.
Results
Klrk1-/- NK cells efficiently control tumors negative for NKG2D ligands
We addressed whether Klrk1-/- mice, which are NKG2D-deficient, can control tumors which do not express NKG2D ligands using a radiation-induced thymic lymphoma (RITL) model17. In this model, mice receive low-dose irradiation early in life and develop disease after three months. RITL is characterized by expansion of CD4+CD8+ T cells lacking NKG2D ligands (Supplementary Fig.1a, b). When Klrk1-/- and C57BL/6J mice were exposed to this regime, we observed a delay in the onset of disease in Klrk1-/- mice. Notably, whereas penetration of RITL was around 80% seven months after irradiation in C57BL/6J mice, less than half of all Klrk1-/- mice developed disease (Fig. 1a). To substantiate this observation, C57BL/6J and Klrk1-/- mice were injected i.v. with B16-F10 (B16) melanoma cells, which induce lung metastases after i.v. injection or solid tumors when applied subcutaneously18 and do not express NKG2D ligands (Supplementary Fig.1b). We observed a delay in development of disease in Klrk1-/- mice compared to Klrk1+/+ littermates (Fig. 1b and Supplementary Fig.1c). B16 cells applied subcutaneously showed a reduced rate of tumor growth in Klrk1-/- mice than in Klrk1+/+ littermates (Fig. 1c and Supplementary Fig.1d), indicating for better control of cancer cell expansion in Klrk1-/- mice.
Figure 1. Better control of B16 melanoma by Klrk1-/- mice is NK cell and IFNγ mediated.

(a) Survival curve of C57BL/6J and Klrk1-/- mice in model of radiation induced thymic lymphomas. Graphs show survival curves (b,e,g) or tumor sizes (c,f) of Klrk1-/- mice and indicated controls after injection of B16 cells i.v (b,e,g) or s.c. (c,f). One day prior to tumor inoculation, mice were either untreated (b, c) or received depleting antibodies directed against NK cells (e, f) or CD8+ T cells (g). Depletion was repeated every fifth day. (d) Survival curve of Klrk1 fl/fl and CD4CreKlrk1fl/fl mice which received B16 cells i.v.. (h) Killing capacity of NK cells from C57BL/6J or Klrk1-/- mice toward CFSE- labeled B16 cells after 4h of co-cultivation at indicated effector to target (E:T) ratios. (i) Cytokine production by NK cells from C57BL/6J or Klrk1-/- mice 4h after PMA/Ionomycin stimulation. (j) IFN-γ production by NK cells after co-cultivation of splenocytes from Klrk1+/+ and Klrk1-/- mice with B16 cell. (k) Survival curve of Ifng-/-, Klrk1-/-Ifng-/- and Klrk1-/- mice after B16 cells i.v. injection. Graphs for tumor experiments (a-g and k; n=10 mice per group) are representative from 2 independent experiments. Survival curves were analyzed by the Kaplan–Meier model followed by Log-rank (Mantel-Cox) test (two-tailed; **p < 0.01, *** p <0.001). Tumor sizes and difference between groups after stimulations were analyzed using two-tailed unpaired t-test (shown mean ± s.e.m); **P < 0.01. Results in h, i and j (n=5 mice per group) are representative from 2 independent experiments. b-d and j are performed using littermates.
Because NKG2D is expressed on subsets of T cells6 we asked whether NKG2D-deficiency enhanced T cell-mediated control of tumors in Klrk1-/- mice. Klrk1fl/fl mice19 were crossed with Cd4Cre mice to ablate NKG2D on CD4+ T cells, CD8+ T cells and NKT cells (Supplementary Fig. 1e). No differences were observed in the survival of Cd4CreKlrk1fl/fl mice and Klrk1fl/fl littermates in the RITL model (Supplementary Fig. 1f). In addition, i.v. injection of B16 cells did not result in survival differences between Cd4CreKlrk1fl/fl mice and Klrk1fl/fl littermates (Fig. 1d). However, Klrk1-/- mice in which NK cells were depleted by monoclonal antibodies (mAb) one day before i.v. injection of B16 cells no longer had a survival advantage over C57BL/6J control (Fig. 1e). Also, differences in tumor growth between Klrk1-/- mice and C57BL/6J controls after s.c. injection of B16 cells were lost following NK cell depletion(Fig. 1f). NK cells can also limit tumor growth indirectly by controlling T cell priming and expansion20, 21. Klrk1-/- mice in which depletion of CD4+ or CD8+ T cells by mAb started one day before i.v. injection of B16 cells had prolonged survival in comparison to C57BL/6J mice (Fig. 1g and Supplementary Fig. 1g), indicating that NK cells are mediating the survival effect of Klrk1-/- mice independently of T cells. However, splenic Klrk1-/- NK cells killed B16 target cells with equal efficiency as C57BL/6J NK cells in 4 h (Fig. 1h) and 14 h (Supplementary Fig. 1h) in vitro cytotoxicity assays. PMA+Ionomycin stimulated splenic NK cells predominantly produced IFN-γ (Fig. 1i), a cytokine that promotes tumor surveillance22. Klrk1-/- NK cells produced more IFN-γ than C57BL/6J NK cells after 24 hours in co-culture with B16 cells (Fig. 1j). To corroborate this findings in vivo, Ifng mRNA was quantified in tumors isolated from C57BL/6J or Klrk1+/+ mice ten days after s.c. injection of B16 cells. Tumors isolated from Klrk1-/- mice had higher expression of Ifng mRNA than from C57BL/6J mice. (Supplementary Fig. 1i). To confirm the role of IFN-γ in tumor control, we crossed Ifng-/- mice with Klrk1-/- mice and followed the survival of Ifng-/-, Klrk1-/- and Klrk1-/-Ifng-/- mice following i.v. injection of B16 melanoma cells. Klrk1-/-Ifng-/- mice did not have a survival advantage compared with Ifng-/- mice, whereas Klrk1-/- mice did (Fig. 1k), indicating that better control of B16 melanoma was mediated by increased capacity of Klrk1-/- NK cells to produce IFN-γ.
Klrk1-/- NK cells have specific hyper-reactivity through NCR1
To analyze the impact of NKG2D-deficiency on target cell engagement, we performed a conjugation assay with B16 melanoma23. No difference in the amount of NK-target cell complexes was observed between C57BL/6J and Klrk1-/- NK cells (Fig. 2a). To test whether increased IFN-γ production in Klrk1-/- NK cells resulted from enhanced signaling through activating receptors, splenic NK cells from Klrk1-/- mice or Klrk1+/+ littermates were stimulated through different activating receptors. Engagement of receptors NK1.1, DNAM-1, Ly49D or Ly49H by mAb or incubation of NK cells with IL-12 and IL-18 cytokines resulted in similar production of IFN-γ in Klrk1-/- and Klrk1+/+ NK cells (Fig. 2b,c and Supplementary Fig. 1j). In contrast, stimulation with mAb against the NCR1 receptor resulted in higher percentage of IFN-γ+Klrk1-/- NK cells compared to IFNγ+Klrk1+/+ NK cells (Fig. 2b,c). NCR1 expression and stimulation-induced degranulation by mAb (Fig. 2d) were similar in Klrk1-/- and Klrk1+/+ NK cells. Thus, NKG2D-deficient NK cells show hyper-responsiveness to stimulation through the NCR1 receptor.
Figure 2. Klrk1-/- NK cells show specific hyper-reactivity through NCR1 receptor.

(a) Representative graph (left) and dot plots (right) from three independent experiments showing formation of conjugates between labeled NK and B16 cells (mean ± s.e.m; n=5 mice per group). (b) IFN-γ production by NK cells from Klrk1+/+ (n=3) or Klrk1-/- (n=4) mice after stimulation by monoclonal antibodies (mAb) through indicated receptors or with IL-12/IL-18 cytokines. (c) Representative FACS plots gated for CD3-NCR1+ cells (NK1.1 stimulation) or CD3-NK1.1+ cells (other stimuli). (d) Levels of NCR1 expression from untreated (left) and percentage of CD107a+ (right) NK cells, after 4h of NCR1 stimulation using mAb, from Klrk1+/+ (n=3) or Klrk1-/- (n=4) mice. (e) Survival curves for untreated (left) or NK cell depleted (right) indicated groups of mice after B16 i.v. injections (n=10 mice per group). Representative graph from three independent experiments. (f) Viral titers in spleens 4 days after infection of indicated group of mice with 5x 105 PFU Δm157 MCMV. Mice were left untreated (left) or received NK cell depleting antibodies one day prior to infection (right). Graphs show pooled data from two independent experiments. Survival curves were analyzed by the Kaplan–Meier model followed by Log-rank (Mantel-Cox) test (two-tailed; **p < 0.01, *** p <0.001). a, b and d are analyzed using two-tailed unpaired t-test (shown mean ± s.e.m; ns, not significant, *p < 0.05). Viral titers were analyzed using Kruskal-Wallis test (shown mean ± s.e.m; *P< 0.05; ***P< 0.001). b-d show representative data from 2 independent experiments using littermates.
NCR1 is known to have a role in the control of B16 melanoma24, 25. Labeling with NCR1-Ig fusion proteins26 showed high expression of NCR1 ligands on B16 cells (Supplementary Fig. 1k). To investigate whether NCR1 was involved in the enhanced tumor control by Klrk1-/- mice, we used Ncr1GFP/GFP mice, which are deficient in NCR1. Ncr1GFP/GFP mice showed reduced survival in comparison to C57BL/6J mice after i.v. injection of B16 melanoma cells (Fig. 2e). Klrk1-/- mice showed better survival in comparison to C57BL/6J controls, while Klrk1-/- Ncr1GFP/GFP mice had survival comparable to Ncr1GFP/GFP mice (Fig. 2e). Depletion of NK cells by mAb abrogated any difference in survival between all mice (Fig. 2e). These results show that the enhanced tumor control in Klrk1-/- mice is dependent on NCR1 engagement by NK cells.
Klrk1-/- mice have better control of MCMV infection compared to C57BL/6J mice (Supplementary Fig. 2), which is NK cell-dependent9. To test whether this effect was mediated through NCR1, we infected C57BL/6J, Ncr1GFP/GFP, Klrk1-/- and Ncr1GFP/GFPKlrk1-/- mice with Δm157 MCMV, a mutant strain of MCMV lacking ligand for NK cell receptor Ly49H. We used this MCMV strain to avoid the Ly49H-mediated control of viral replication, which may occlude the effects of NCR127. Klrk1-/- mice showed better control of Δm157 MCMV in the spleen compared to all other mice, which was lost after depletion of NK cells by mAb (Fig. 2f). These results show that the enhanced control of MCMV infection by NKG2D-deficient mice is dependent on NCR1 engagement by NK cells.
NKG2D sets NCR1 activation threshold during NK cell development
During NK cell development, NKG2D is expressed from the Lin-CD117dimSca1++Flt3L-CD127+ cells onwards, which represents the earliest NK cell committed precursor (pre-pro NK)7. Because NKG2D-deficiency impacts development of NK cells in the bone marrow (BM)9, as well as NK cells effector responses in the periphery28, 29 we asked whether the hyper-reactivity of Klrk1-/- NK cells to NCR1 stimulation was acquired during development or later on mature NK cells in the periphery. We crossed Klrk1fl/fl mice with Ncr1Cre mice (Supplementary Fig. 3a) to generate Ncr1CreKlrk1fl/fl mice, in which Cre-mediated deletion of Klrk1 occurs in CD122+NK1.1+NCR1+CD11b-c-Kit- NK cells7, 30. Spleen NK cells from Ncr1CreKlrk1fl/fl mice showed comparable production of IFN-γ to Klrk1fl/fl litermates after stimulation of NCR1 by mAb in vitro (Fig. 3a). We did not observe differences in survival between Ncr1CreKlrk1fl/fl mice and Klrk1fl/fl littermates after i.v. injection of B16 cells (Fig. 3b). In contrast, higher percentage of spleen NK cells from Klrk1Δ/Δ mice, which had a germline deletion of Klrk1 and were generated from the cross between deleter (tg-cmvCre) 31 and Klrk1fl/fl mice, produced IFN-γ to NCR1 stimulation by mAb compared to C57BL/6J control (Supplementary Fig. 3b). These results indicate that NKG2D sets the activation threshold for NCR1 developmentaly before CD122+NK1.1+NCR1+CD11b-c-Kit- NK cells.
Figure 3. NKG2D sets an activation threshold for NCR1 early during NK cell development in a process that differs from known mechanisms of NK cell education.

(a) IFNγ production by NK cells from C57BL/6J, Klrk1-/-, Klrk1fl/fl (n=5 mice per group), NCR1Cre (n=6), NCR1Cre Klrk1fl/fl (n=4) after 4h of NCR1 stimulation using mAb (ANOVA, with Bonferroni's post-test correction). (b) Survival curve for Ncr1CreKlrk1fl/fl and Klrk1fl/fl littermates after B16 i.v. injection (Kaplan–Meier model followed by Log-rank (Mantel-Cox) test; n=10 mice per group). (c,f) Analysis of splenic EYFP+ NK cells from RagCreEYFPStop-FloxiDTR mice injected with DT and NKG2D-blocking antibodies or isotype controls (n=6 mice per group). (c) IFN-γ production after 4h of indicated stimulations. (f) Developmental NK cell stages in the BM (see Supplementary Fig. 4a). (d) Hematopoietic precursor populations and development of NK cells (e) were analyzed in BM of Klrk1-/- (n=4) and Klrk1+/+ (n=3) littermates. Analysis of EYFP expression (g) and IFNγ production by NK cells after indicated stimulations in Rag1CreEYFPStop-FloxKlrk1+/+ (n=5 or 6) and Rag1CreEYFPStop-Flox Klrk1-/- (n= 3 or 4) (h). (i) NK cells from Klrk1-/- (n=4) and Klrk1+/+ (n=3) littermates were stimulated through the NCR1 receptor using mAb and IFN-γ production was analyzed after 4h. Cells were untreated and gated based on Ly49I expression (left) or stimulation was performed in presence of SHP1/2 inhibitor (NSC-87877) or solvent only (right). c-g were analyzed using two-tailed unpaired t-test and h-i using ANOVA, with Bonferroni's post-test correction. Shown are means ± s.e.m of representative plots (*P<0.05, ** P<0.01, *** P < 0.001) of two (b,c,d,e,f,i) or four (a,g,h) experiments.
Next we tested whether NKG2D played a role during early NK cell development in a model independent of genetic modification of Klrk1. In Rag1CreEYFPStop-FloxiDTR mice all cells derived from Rag1+ hematopoietic precursors, including T cells, B cells and a large fraction of NK cells, expressed EYFP and dipheteria toxin receptor (DTR) upon Cre-mediated deletion of the transcriptional “stop” sequence and can be eliminated by diphtheria toxin (DT) injection (Supplementary Fig. 3c). Rag1CreEYFPStop-FloxiDTR mice were i.p. injected with DT on the first two consequtive days to deplete all NK cells originating from the Rag1+ precursors. To inhibit NKG2D signaling on all newly generated NK progenitors, the mice were treated from the second day onwards with NKG2D-blocking mAb or isotype control (Supplementary Fig. 3c) and the receptor responsiveness of EYFP+ NK cells was analyzed two weeks later. Spleen NK cells from Rag1CreEYFPStop-FloxiDTR mice that had received i.v. NKG2D-blocking mAb showed increased IFN-γ production after stimulation of NCR1, but not following stimulation of NK1.1 or Ly49H by mAbs (Fig. 3c), indicating that NKG2D sets the activation threshold for NCR1 early in NK cell development.
These observations prompted us to analyze the impact of NKG2D-deficiency on hematopoiesis. NKG2D-deficiency does not affect hematopoietic stem cells or more differentiated precursors, such as the common lymphoid and myeloid precursors19,32 (Fig. 3d). However, there was a reduction in the number of BM Lin-CD117dimSca1++Flt3L-CD127+ NK progenitors in Klrk1-/- mice compared to Klrk1+/+ littermates (Fig. 3d). Further analysis of NK cell development revealed an increase in percentage of CD122+NK1.1+NCR1-CD11b-c-Kit- and decrease of CD122+NK1.1+NCR1+CD11b-c-Kit- NK progenitors in Klrk1-/- mice compared to Klrk1+/+ littermates (Fig. 3e and Supplementary Fig. 4a). To confirm the role of NKG2D on changes of NK progenitors in a second model, we analyzed them in BM of Rag1CreEYFPStop-FloxiDTR mice 15 days after DT injection and treatment with NKG2D-blocking mAb. Similar to the Klrk1-/- mice, we observed an increase in percentage of CD122+NK1.1+NCR1-CD11b-c-Kit- and decrease of CD122+NK1.1+NCR1+CD11b-c-Kit- NK progenitors compared to isotype control-treated Rag1CreEYFPStop-FloxiDTR mice (Fig. 3f). Together, these results indicate a role of NKG2D in NK cell development at the time of NCR1 appearance on NK progenitors.
NKG2D-mediated NK cell-education differs from known mechanisms
NK cells which never expressed Rag1 during development show cell-intrinsic hyper-responsiveness in comparison to NK cells generated from Rag1+ progenitors33. We therefore asked whether NKG2D promoted NK cell development from a Rag1+ progenitor. We generated Klrk1+/+Rag1CreEYFPStop-Flox and Klrk1-/- Rag1CreEYFPStop-Flox mice, in which expression of Rag1 was marked through the expression of EYFP. As shown previously33, we observed that a higher percentage of EYFP- NK cells from Klrk1+/+Rag1CreEYFPStop-Flox were Klrg1+ and CD11b+ compared to EYFP+ NK cells (Supplementary Fig. 4b). We made the same observations in Klrk1-/- Rag1CreEYFPStop-Flox mice (Supplementary Fig. 4c). However, there were no differences in percentages of EYFP+ or EYFP- NK cells between Klrk1+/+Rag1CreEYFPStop-Flox and Klrk1-/- Rag1CreEYFPStop-Flox mice (Fig. 3g). Also, regardless of EYFP expression, NK cells from Klrk1-/-Rag1CreEYFPStop-Flox mice produced more IFN-γ than NK cells from Klrk1+/+Rag1CreEYFPStop-Flox mice upon NCR1 stimulation by mAb, whereas responsiveness to NK1.1 was similar (Fig. 3h), indicating that NKG2D influences NCR1 signaling independently of the Rag-driven developmental pathway.
To investigate the role of Ly49-mediated education in NCR1 signaling we compared the production of IFN-γ in Ly49I+ and Ly49I- Klrk1+/+ and Klrk1-/- NK cells following NCR1 stimulation by mAb. Klrk1-/- Ly49I+ NK cells produced more IFN-γ in comparison to Klrk1+/+ Ly49I- NK cells (Fig. 3i). However, Klrk1-/- NK cells produced more IFN-γ compared to Klrk1+/+ NK cells, regardless of their Ly49I expression (Fig. 3i). Also, there was no difference in the frequency of Ly49I+ NK cells between Klrk1-/- and Klrk1+/+ cells (Supplementary Fig. 4d). These observations indicate that the threshold for the NKG2D-dependent activation of NCR1 is independent of Ly49-mediated education.
SHP-1 plays a role in NK cell education and SHP-1-deficient NK cells are hypo-responsive to MHCI-deficient transplants and tumors34, 35. To test whether SHP-1 played a role in the NKG2D-mediated education, Klrk1-/- and Klrk1+/+ spleen NK cells were treated in vitro with the SHP-1/2 inhibitor NSC-8787736 followed by stimulation through the NCR1 receptor by mAb. SHP-1/2 inhibition resulted in an increase of IFN-γ production in both Klrk1-/- and Klrk1+/+ NK cells. However, production of IFN-γ was higher in Klrk1-/- NK cells compared to Klrk1+/+ (Fig. 3i), indicating that NKG2D sets activation threshold for NCR1 independently of SHP-1/2.
The NKG2D-DAP12 signaling axis regulates NCR1 activity
To investigate the mechanism through which NKG2D regulates the activity of NCR1, we focused on the adaptors DAP10 and DAP1212. We used mice which lack either DAP10 (Hcst-/-) or DAP12 (Tyrobp-/-). There were no differences in the production of IFN-γ between C57BL/6J, Klrk1-/-, Hcst-/- and Tyrobp-/- spleen NK cells after stimulation through NK1.1 by mAb (Fig. 4a). Ly49H and Ly49D use DAP12 for signal transduction14. IFN-γ production from Tyrobp-/-, but not Klrk1-/- or Hcst-/- NK cells was reduced compared to C57BL/6J NK cells after stimulation through these receptors (Fig. 4a). Notably, after stimulation through NCR1, Tyrobp-/-, but not Hcst-/- NK cells showed an increase in IFN-γ production compared to C57BL/6J controls, similar to Klrk1-/- NK cells (Fig. 4a). Similar observations were made after NCR1 stimulation of spleen NK cells from Tyrobp-/- mice and Tyrobp+/+ littermates (Supplementary Fig. 5a). When B16 cells were injected i.v. in Klrk1-/-, Hcst-/-, Tyrobp-/- and C57BL/6J mice, Tyrobp-/- mice showed prolonged survival in comparison to Hcst-/-, C57BL/6J and even Klrk1-/- mice (Fig. 4b), indicating that signaling through DAP12 only was important for NK cell hyper-reactivity to NCR1 stimulation.
Figure 4. The NKG2D-DAP12 signaling axis regulates NCR1 activity.

(a) NK cells from Tyrobp-/-(n=3 or 4 mice), Hcst-/-(n=3 or 5 mice), Klrk1-/- (n=3 or 5 mice) and C57BL/6J (n=3 or 5) mice were stimulated for 4h through the NK1.1, Ly49H, Ly49D or NCR1 receptor by mAb and IFN-γ production was analyzed after 4h. (b) Survival curves of indicated group of mice after i.v. injection of B16 cells (Kaplan–Meier model followed by Log-rank (Mantel-Cox) test; n=10 mice per group). (c) NK cells from Ly49h-/- (n=3 mice), Klrk1-/- (n= 4 mice) and C57BL/6J (n= 4 mice) mice were stimulated through the NK1.1 or NCR1 receptor by mAb or with IL-12 cytokines and IFN-γ production was analyzed after 4h. Shown are representative plots of three (a) or two (b-c) experiments. For analysis of a and c ANOVA, with Bonferroni's post-test correction for multiple comparisons was used. Shown are means ± s.e.m. * P<0.05, ** P<0.01, *** P < 0.001.
We next questioned whether the hyper-responsiveness of DAP12-deficient NK cells was specific for NKG2D, or was observed following deletion of any receptor that signals through this adaptor. When Klrk1-/-, Ly49H-/- or C57BL/6J spleen NK cells were stimulated through NK1.1 or NCR1 by mAbs or with the cytokine IL-12, Ly49H-/- NK cells did not show increased IFN-γ production after any of these stimulations compared to C57BL/6J NK cells (Fig. 4c). In mice, NKG2D has a long (L) and a short (S) isoform, of which only the latter associates with DAP1212. We therefore investigated whether NKG2D-S and DAP12 were expressed during early NK cell development in wild-type mice. qPCR in sorted CD122+NK1.1-NCR1-CD11b-c-Kit- and CD122+NK1.1+NCR1-CD11b-c-Kit- BM NK progenitors detected transcripts for the Tyrobp and short isoform of Klrk1, whose expression increased from CD122+NK1.1-NCR1-CD11b-c-Kit- to CD122+NK1.1+NCR1-CD11b-c-Kit- NK progenitors (Supplementary Fig. 5b,c). Thus, the NKG2D-mediated control of NCR1 signaling occurs through the NKG2D-DAP12 axis early in NK cell development.
CD3ζ and ZAP70 are involved in inhibition of NCR1 signalling
Because NCR1 uses CD3ζ and FcεRγ for signal transduction13, we tested whether the NKG2D-DAP12 axis affects signaling through these adaptors. Flow cytometry analysis indicated that expression of CD3ζ was reduced in both Klrk1-/- and Tyrobp-/- NK cells in comparison to C57BL/6J NK cells, while expression of FcεRγ was comparable in all groups (Fig. 5a,b and Supplementary Fig. 5d,e). In addition, expression of ZAP-70, a signaling component downstream of CD3ζ, was reduced in Klrk1-/- and Tyrobp-/- spleen NK cells in comparison to C57BL/6J NK cells, while expression of the Syk kinase, also downstream of CD3ζ, was comparable in all groups (Fig. 5a,b and Supplementary Fig. 5e). Immunoblot analysis also showed reduced expression of CD3ζ and ZAP-70 in Klrk1-/- spleen NK cells compared to C57BL/6J NK cells (Fig. 5c). In contrast, Ncr1CreKlrk1fl/fl spleen NK cells had no alterations in the expression of CD3ζ or ZAP-70 compared to Klrk1fl/fl NK cells (Fig. 5d). Because CD3ζ-deficient mouse spleen NK cells are hyper-responsive to CD16 stimulation16, we asked whether NKG2D and DAP12 mediated the responsiveness of NK cells to CD16 engagement. Klrk1-/- and Tyrobp-/- spleen NK cells showed enhanced IFN-γ production following CD16 stimulation compared to C57BL/6J NK cells (Supplementary Fig. 5f).
Figure 5. Klrk1-/- and Tyrobp-/- NK cells have reduced levels of CD3ζ and Zap70 signaling molecules.

(a-b) Splenic NK cells from C57BL/6J (n=5 mice per group), Klrk1-/- (n=4-5 mice per group) and Tyrobp-/-(n=4 or 6 mice per group) mice were analyzed for expression of CD3ζ, FcεRγ, Zap-70 and Syk. Shown are graphs of geometric mean values for indicated molecules (a) as well as histograms (b). As a controls for staining, isotype control (for CD3ζ and Zap-70), FMO (for FcεRγ) or secondary antibody only (for Syk) were used. FACS plots were gated for NK cells (CD3-NK1.1+). (c) NK cells from C57BL/6J and Klrk1-/- mice were sorted and expression of indicated proteins was analyzed by western blot. Pan-akt or β-actin were used as controls for equal loading. (d) NK cells from Ncr1Cre (n=6), Ncr1CreKlrk1fl/fl (n=4), Klrk1-/-(n=5) and C57BL/6J (n=5) mice were analyzed for CD3ζ and Zap-70 expression. Figures show graphs of geometric mean. (e) Transcript levels for CD3ζ and FcεRγ of NK cells sorted from spleens from C57BL/6J and Klrk1-/- mice were analyzed by qPCR (n=5 mice per group). Shown are representative plots of three (a and c) or two (b and d) experiments. ANOVA, with Bonferroni's post-test correction for multiple comparisons was used to analyze a and c. Two-tailed unpaired t-test was used to analyze d. Shown are means ± s.e.m. ** P<0.01, *** P < 0.001.
As determined by qPCR, Cd247 or Fcre1g mRNA was similar in Klrk1-/- and C57BL/6J NK cells (Fig. 5e), suggesting altered post-transcriptional regulation. To identify candidates that might impact the expression of CD3ζ and/or ZAP-70, we compared the transcriptome of spleen CD3-NK1.1+NCR1+ NK cells from C57BL/6J, Klrk1 -/- and Tyrobp-/- mice by RNA sequencing. 94 genes were differentially expressed between C57BL/6J and Klrk1-/- NK cells, whereas expression of 543 genes was different between Tyrobp-/- and C57BL/6J NK cells (Figure 6a). We performed qualified cluster analysis of genes differentially expressed in Klrk1-/- NK cells versus C57BL/6J cells, based on data mining of known protein-protein interactions, in order to establish a potential link with CD3ζ and/or ZAP-70 (Supplementary Fig. 6a). Next, we determined which of these genes showed a shared expression pattern between Klrk1 -/- and Tyrobp-/- mice (Fig. 6a,b). Prf, encoding perforin and Sla, encoding adaptor SLAP-1, were identified as potential candidates. Using flow cytometry analysis we did not detect a difference in perforin protein expression between Klrk1-/- and C57BL/6J spleen NK cells (Fig. 6c). SLAP-1 is known to target CD3ζ for degradation in thymocytes37. Sla transcripts were upregulated in Klrk1-/- and Tyrobp-/- NK cells compared to C57BL/6J NK cells (Fig. 6b). Cell surface expression of SLAP-1 on Klrk1-/- NK cells was increased compared to C57BL/6J NK cells (Fig. 6c and Supplementary Fig. 6b). In addition, expression of SLAP-1 in splenic EYFP+ NK cells from Rag1CreEYFPStop-FloxiDTR mice 15 days after DT injection and treatment with NKG2D-blocking Ab was increased compared to EYFP+ NK cells from DT and isotype-treated Rag1CreEYFPStop-FloxiDTR mice (Fig. 6d). Next, we asked whether SLAP-1 downregulates CD3ζ in NK cells. We observed increased expression of CD3ζ protein in spleen NK cells isolated from Sla-/- mice37 compared to wild-type controls, whereas expression of NCR1, FcεRγ, Syk and ZAP-70 were not affected (Fig. 6e and Supplementary Fig. 6c). Thus, NKG2D-deficiency results in an increase of SLAP-1 protein in NK cells, which reduces the expression of CD3ζ.
Figure 6. Hyper-reactivity of Klrk1-/- NK cells in response to NCR1 stimulation is a consequence of reduced expression of CD3ζ.
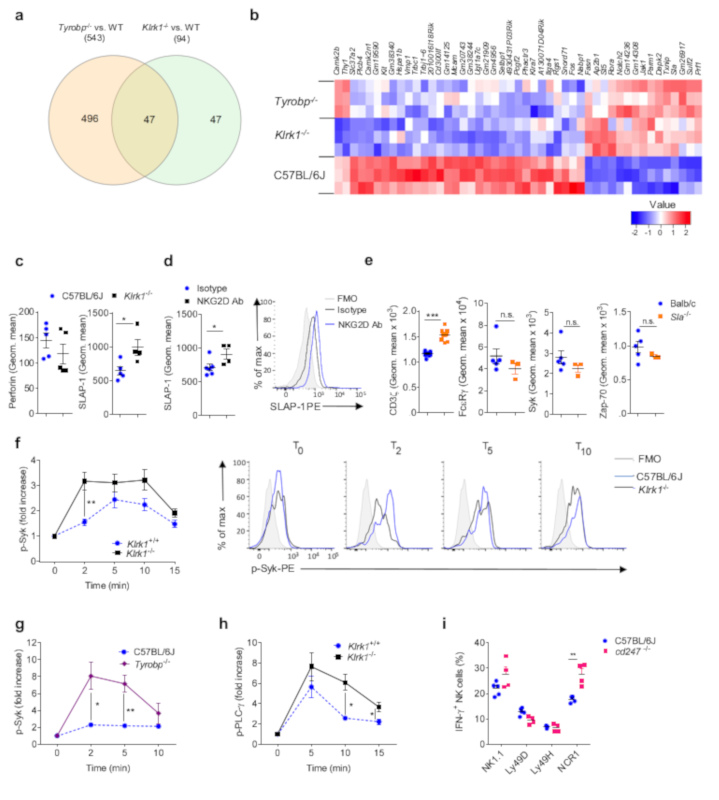
(a-b) RNAseq was performed on sorted NK cells isolated from the spleens of Klrk1-/-, Tyrobp-/- and C57BL/6J mice. (a) Venn diagram and (b) heat map for differentially expressed genes. (c) Analysis of splenic NK cells from C57BL/6J (n=5 mice) and Klrk1-/- (n=4 mice) mice for expression of perforin (left) and SLAP-1 (right). Shown are geometric mean values. (d) Analysis of NK cells from RagCreEYFPStop-FloxiDTR mice injected with DT and in addition with NKG2D-blocking antibodies (n=4 mice) or isotype controls (n=6 mice) splenic EYFP+ NK cells were analyzed for expression of SLAP-1. FACS plot is gated for NK cells (CD3-NK1.1+). (e) Splenic NK cells from Balb/c (n=5 mice) and Sla -/- (n=3 mice) mice were analyzed for expression of Syk, FcεRγ, ZAP-70 and CD3ζ (n= 10 Balb/c and 8 Sla-/- mice). Graphs show geometric mean values. (f-h) NK cells from C57BL/6J (n=4), Tyrobp-/- (n=4) or Klrk1-/- (n=5) mice were stimulated through the NCR1 receptor by mAb and phosphorylation of Syk (f, g) and PLC-γ (h) was analyzed. FACS plots are gated for NK cells (CD3-NK1.1+). (i) NK cells from C57BL/6J and cd247-/- (n=4 mice per group) mice were stimulated through NCR1 by mAb and IFN-γ production was analyzed. Shown are representative plots of one (a,b), two (d,e,i,h) or three (c,f,g) experiments.f and h are performed using littermates. Two-tailed unpaired t-test was used to analyze c-i. Shown are means ± s.e.m. * P<0.05, **P<0.01, *** P < 0.001.
In thymocytes, CD3ζ and ZAP-70 mediate activation upon TCR stimulation, but are also important for the shutdown of signal transduction through recruitment of proteins involved in proximal negative feedback mechanisms16, 38. We therefore asked whether NCR1 stimulation in Klrk1-/- cells results in enhanced proximal signaling. Phosphorylation of Syk, the kinase directly downstream of FcεRγ, was both increased and prolonged in Klrk1-/- NK cells after NCR1 stimulation by mAbs compared to Klrk1+/+ NK cells (Fig. 6f). Similar observations were made in Tyrobp-/- cells (Fig. 6g and Supplementary Fig. 6d). Importantly, phosphorylation of PLC-γ, which is a target of Syk, was prolonged in Klrk1-/- NK cells compared to Klrk1+/+ NK cells, whereas maximal phosphorylation of PLC-γ was only slightly increased (Fig. 6h), indicating a reduced negative feedback loop in proximal NCR1 signaling. Finally, we asked whether CD3ζ-deficiency resulted in hyper-responsive NK cells. CD3ζ-/- spleen NK cells produced more IFN-γ after stimulation through the NCR1 receptor by mAb, but not through NK1.1, Ly49H or Ly49D, compared to C57BL/6J spleen NK cells (Fig. 6i), indicating a role for CD3ζ in the negative regulation of NCR1 signaling. NCR1 expression was similar in all the cells analyzed (Supplementary Fig. 6f). As such, NKG2D sets an activation threshold for the NCR1 receptor by increasing CD3ζ protein and by lowering expression of SLAP-1 during NK cell development.
Discussion
Here we show that NKG2D sets activation threshold for NCR1 early in NK cell development which determines the sensitivity of NK cells to cellular targets expressing NCR1-ligands. This process operates through NKG2D-DAP12 signalling axis which drives downregulation of CD3ζ and ZAP-70, invovlved in negative regulation of NCR1 signalling. Thus, we identified a developmental NK cell regulation, distinct from previously described mechanisms of education, in which one activating receptor regulates the activity of another activating receptor.
Our results indicated that the role of NKG2D in the regulation of NCR1 activation was mediated by DAP12. Although we did not formally show that NKG2D and DAP12 were directly engaged to mediate their regulatory role during NK cell development, the absence of a hyper-responsive phenotype in Ly49H-deficient mice makes it highly unlikely that these two molecules have an identical, yet independent effect. The role of DAP12 in setting activating thresholds does not appear to be unique for NK cells. DAP12-deficient mice have macrophages39 and pDCs40 that produce higher amounts of cytokines after TLR stimulation in comparison to wild-type mice. The mechanism via which DAP12 sets activation thresholds in these cells is unknown, but may be similar to the NKG2D-mediated regulation in NK cells. Because human NKG2D does not bind to DAP12 our findings can not be directly extrapolated. However, several KIRs signal through DAP1241, 42, 43, 44 It has been shown that activating KIRs down-modulate human NK cell-responsiveness in individuals carrying self ligands43. It will therefore be interesting to see whether regulation of NKp46 activation thresholds in humans is regulated through NKG2D or through Dap12-binding KIRs.
Our data indicated the involvement of CD3ζ and ZAP-70 in the negative regulation of NCR1 signaling. NK cells from CD3ζ-deficient mice had higher production of IFN-γ after stimulation through the NCR1 receptor. These observations are in line with reports of an important role for CD3ζ and ZAP-70 in the negative regulation of signaling in T cells45, 46 and NK cells16. Following T cell receptor engagement, CD3ζ-ZAP-70 recruit ubiquitinase Nrdp3 and phosphatases Sts-1 and Sts-2, which dephosphorylate ZAP-70 and cause cessation of the activating signal. Deficiency for Nrdp3, Sts-1 and Sts-2 causes prolonged signal transduction47. We observed that Klrk1-/- mice had delayed signal inhibition following NCR1 stimulation, especially at the level of PLC-γ phosphorylation. Thus, we propose that a reduction in expression of CD3ζ-ZAP-70 causes impaired recruitment of factors that terminate the activating signal directly downstream of the NCR1 receptor. Although a direct interaction between CD16 and CD3ζ has only been shown in human cells, deficiency of CD3ζ or DAP12 causes hyper-reactivity in murine NK cells in response to CD16 stimulation16, 48. We saw increased production of IFN-γ in Klrk1-/- NK cells after stimulation through the CD16 receptor, suggesting that NKG2D may also regulate the responsiveness of CD16.
Chronic exposure to NKG2D ligands can result in cross-tolerance of multiple distinct NK cell activation pathways 28, 29. In addition, weak signaling through NKG2D leads to reduced activity of NK cells, which can be circumvented by administration of soluble high-affinity ligands29. However, peripheral “desensitization” through NKG2D generates a general hypo-responsiveness of NK cells to activating stimuli. Therefore, the developmental and peripheral regulation of activation thresholds by NKG2D appear to use distinct molecular mechanisms.
The NKG2D-mediated ‘education’ of NCR1 is distinct from previously described mechanisms. Education through molecules such as Ly49 receptors2 or via modification of the SHP phosphatases34 results in a general hypo-responsiveness to activating stimuli. The impact of NKG2D-mediated education, in contrast, appears restricted to NCR1. Importantly, we identify a temporal window in which NKG2D permanently controls NCR1 responsiveness. Deletion of Klrk1 from the CD122+NK1.1+NCR1+CD11b-c-Kit- NK progenitors onwards did not cause NK cell hyper-responsivenes to NCR1 as it was the case in mice with germline deletion of Klrk1. Ly49 molecules are expressed from the CD122+NK1.1+NCR1+CD11b-c-Kit- NK progenitors onwards. Indeed, lack of Ly49H, which also signals through DAP12, did not result in NK cell hyper-responsiveness to NCR1. How permanent regulation of CD3ζ expression by NKG2D signaling at a highly-restricted stage of NK cell development is accomplished mechanistically remains unclear. Our data suggests post-transcriptional regulation of CD3ζ. RNA-seq analysis revealed Sla as a candidate involved in this process. Sla encodes SLAP-1, an adaptor that targets CD3ζ for ubiquitin ligase c-Cbl-dependent degradation following receptor activation37. NKG2D-deficiency, or blocking of NKG2D during NK cell development, caused a permanent increase in Sla gene expression, implying increased degradation of CD3ζ and subsequently altered signal transduction through NCR1. Indeed, SLAP-1-deficient NK cells expressed higher amounts of CD3ζ. CD244, another NK activating receptor expressed on multipotent hematopoietic progenitors49, was shown to regulate immune cell function through epigenetic silencing of chromatin regions50. Although methylation analysis of the Sla locus in Klrk1-/- NK cells did not reveal differences compared to wild-type NK cells (unpublished data by T. D. H. and Y. C. B.), this does not exclude another ways of epigenetic regulation of Sla expression.
Materials and Methods
Mice
Mice were strictly age- and sex-matched within experiments and were held in SPF conditions and handled in accordance with institutional, national and/or EU guidelines. Permission for our experiments was given by Ethical Committee of the Faculty of Medicine, University of Rijeka and Croatian Ministry of Agriculture, Veterinary and Food Safety Directorate ((UP/I-322-01/16-01/16, 525-10/0255-16-7). Mice used in experiments were between 6 and 12 weeks of age. Klrk1-/-, Klrk1fl/fl and Klrk1Δ/Δ were generated as described previously 9, 19. Wild-type C57BL/6J (strain 000664), Balb/c (strain 00651) and Ifng-/- (strain 002287) mice were from the Jackson Laboratory. Ncr1GFP/GFP mice were provided by O. Mandelboim (Hebrew University Hadassah Medical School). Hcst-/- and Tyrobp-/- mice were provided by M. Colonna (St. Louis, MO). Rag1cre/+ mice and were provided by M. Busslinger (Vienna, Austria). Rosa26-foxed STOP YFP (EYFPStop-Flox) and iDTR mice were provided by A. Waisman (Mainz, Germany). Ncr1Cre and Cd4Cre mice were provided by V. Sexl (Vienna, Austria) and D. Littman (New York, NY, USA). Deleter-cre mice were kindly provided by K. Rajewsky. Cd247-/- mice were from CNRS, Orleans. Sla-/- mice were provided by J. McGlade (Toronto, Canada). Klrk1-/- or Tyrobp-/- and wild-type littermates were generated by interbreeding heterozygous mice.
Cells
B16 cell line was purchased from the American Type and Culture Collection (ATCC). Cells were cultured in complete DMEM, supplemented with 10mM HEPES (pH 7.2), 2mM L-glutamine, 105 U/L Penicillin, 0.1 g/L Streptomycin, and 10 % FCS. Cells were maintained in incubator at 37°C, 5% CO2.
Tumor models
Radiation induced thymic lymphomas
Young mice (4-6 weeks old) received low doses of γ radiation (1,6 Gy) once a week for 4 weeks as previously described 17. In these experiments we used 10 mice per group and experiments were repeated two times.
B16 melanoma
Mice received 105 B16 clone F10 (B16) cells i.v or s.c and survival (reaching of human end points) or tumor growth was followed respectively. Digital caliper was used to measure tumor size. X-ray pictures of tumors in isolated skin were done using an in vivo imager (MS FX Pro, Carestream) on day 10 after s.c. tumor inoculation. To deplete lymphocyte populations, mice received 250 ug antibodies i.p. one day before tumor inoculation and repeated every 5th day. Antibodies used were αCD4 (YTS 191.1.2), αCD8 (YTS 169.4.2) and αNK1.1 (PK136). In these experiments we used 10 mice per group and experiments were repeated at least two times.
In vitro analysis of NK cells
For NK killer assay B16 cells were labeled with CFSE and co-cultured with C57BL/6J or Klrk1-/- splenocytes, respectively, at an effector to-target ratio adjusted to the number of NK cells. After 4h or 14h co-culture in a 5% CO2 atmosphere at 37°C, specific lysis was determined in triplicate by flow cytometric analysis as measured by To-pro-3 (To-pro-3 iodide, Life technologies) incorporation. Spontaneous death of cells was determined in wells containing targets only 9. In co-cultivation assays for cytokine production analysis, splenocytes from WT or Klrk1-/- mice were co-cultured with B16 cells in a 1:1 ratio overnight at 5% CO2 at 37°C in addition to IL-2 (100U/mL, R&D Systems), IL-12 (50 pg, R&D Systems) and addition of Brefeldin A (eBioscience) for the last 4h. IFN-γ production by NK cells was analyzed by flow cytometry. Formation of conjugates was done as previously described 23. Briefly, B16 cells were CFSE labeled and co-cultured with NK1.1-labeled splenocytes. At indicated time points, the fraction of CFSE+NK1.1+ double positive cells was determined by flow cytometry. For in vitro NK cell stimulations and analysis of cytokine production 5 x105 splenocytes were stimulated for 4h at 5% CO2 atmosphere and 37°C in addition to IL-2 (100U, Preprotech) and Brefeldin A (eBioscience). For stimulation through specific activating receptor 2-5µg/mL of antibody in PBS was pre-coated on an ELISA plate (αLy49H (3D10), αLy49D (4e4), αNK1.1 (PK136), αNCR1 (29A1.4) and IgG2aκ (eBM2a)). Cytokines were added in suspension: IL-12 (10ng/mL, R&D Systems) and IL-18 (20ng/mL, R&D Systems). For SHP-1/2 inhibition we cultured cells in the presence of 50µM SHP-1/2 PTPase inhibitor (NSC-87877, Merck Millipore) dissolved in DMSO, or DMSO only as control. In experiments of in vitro NK cell analysis we used 5 mice per group and experiments were repeated two to three times.
Phosphorylation kinetics
Splenocytes were starved from stimuli in RPMI 1640 for 30 min at 37°C in a humidified incubator with 5% CO2. Next, cells (5 x 105) were stimulated with 2µg/mL αNCR1 (29A1.4) for the indicated times. Cells were fixed with 2% paraformaldehyde, permeabilized vv in 90% methanol, and stained with p-Syk (Y348) (moch1ct, eBioscience) or p-PLC-γ1(Y783) (D6M9S, Cell Signaling). In these experiments 4-5 mice per group were used and experiment were repeated two to three times.
Purification of NK cells and RNA isolation for qPCR
NK cells were enriched from splenocytes using biotinylated DX5 antibodies, streptavidin-coated beads and magnetic cell sorting (Milteny). Next, NK cells (CD3-NK1.1+) were sorted to high (>99% purity) on a FACS Aria II (BD Biosciences). RNA was isolated via the Trizol method, and cDNA was generated with a reverse transcriptase core kit (Eurogentec). The expression of mRNA was examined by quantitative PCR analysis with a 7500 Fast Real Time PCR machine. Taqman assays were used to quantify the expression of Ifng (IFN-γ, Mm00485148_m1). The relative mRNA expression was normalized by quantification of Rn18S (18S, Mm03928990_g1) RNA in each sample.
RNAseq
Sorted cells were stored frozen in RLT buffer (Qiagen). Lysates were thawed and RNA extracted using column-based purification with the inclusion of a DNase I treatment (TurboDNase, Ambion, Thermo Fischer Scientific). Purified RNA was converted to cDNA using Superscript II kit with the addition of 20ng/ml anchored Oligo d (T) (Thermo Fischer Scientific) and 1M Betaine, 12mM MgCl2 (Sigma-Aldrich) to aid conversion. Second strand cDNA synthesis proceeded directly using the NEB second strand cDNA synthesis kit (New England Biolabs Ltd). The completed cDNA reaction was transferred to a Covaris AFA tube and sonicated using 140W peak power at 200 cycles for 3mins to generate average length fragments of 300bp (Covaris S220, Covaris Inc.). DNA was extracted (Micro DNA columns, Qiagen) and used as template in Illuminia compatible sequencing library preparation kit (DNA-seq Thruplex, Rubicon Genomics). Multiplexed libraries were purified twice using AMPure beads (Becton Coulter Inc.) and quantified using Qubit (Thermo Fischer Scientific) and Kapa Library Quantification (Kapa Biosystems). Sequencing was performed on a Hi-Seq 3000 (Illuminia, USA). Sequencing reads were FASTQ quality assessed, trimmed using trim galore and mapped to the mm10 mouse genome assembly using STAR-SEQ. Differential expression analysis was accomplished using limma and edgeR R programmes. For qualified cluster analysis, genes differentially expressed between C57BL/6J and Klrk1 NK cells were analyzed using the String (www.String-db.org) database to identify associations with Zap70 and CD247. Network edges with a confidence interval of 0.4 are included, whereas unconnected nodes were excluded. Clustering was visualized using Cytoscape 3.5.0 software.
Purification of NK cell precursors and RNA isolation for qPCR
NK precursors (stage 1 and stage 2) were sorted according to their expression profiles described in supplementary figure 4a. Total RNA was extracted using RNeasy Micro Kit (Qiagen). The quality of the RNA obtained was evaluated using the Laboratory-Chip technique (Agilent Bioanalyzer) and subsequently preamplified according to the Ambion WTA expression protocol (Thermo Fischer). RNA was reverse transcribed into cDNA using the iScript protocol (Bio-Rad). QT-PCR was performed using the SsoAdvanced™ Universal SYBR® Green Supermix (BIO-RAD) and the MyCycler Thermal cycler system (Bio-Rad).Used primers: mNKG2D-S FW: AGTTGAGTTGAAGGCTTTGACTC, mNKG2D-S REV: ACTTTGCTGGCTTGAGGTC, DAP12 FW: GACTGTGGGAGGATTAAGTCC, DAP12 REV: AACACCAAGTCACCCAGAAC.
Flow cytometry
Cells were pretreated with Fc block (clone 2.4G2, produced in-house). To-pro3 (life Technologies) or Fixable Viability Dye (eBioscience) was used to exclude dead cells. Cells were stained and analyzed in PBS containing 1% BSA and NaN3 with antibodies listed below. For intracellular staining, permeabilization and fixation of cells was done with the Fix/Perm kit (BD Biosciences).The cells were measured on a FACSVerse or FACSAria flow cytometer (BD Biosciences), and data were analyzed using FlowJo v10 software (Tree Star, Ashland, OR). To analyze ligands expression tumor cells were stained with fusion proteins (NKG2D-Fc or NCR1-Fc) or irrelevant fusion protein and secondary FITC labeled anti-human antibody. For flow cytometry, we used monoclonal antibodies to mouse CD3e (145-2C11), NK1.1 (PK136), CD49b (DX5), IFN-γ (XMG1.2), TNFα (MP6-XT22), IL-6 (MP5-32C11), GM-CSF (MP1-22E9), NKG2D (CD314) (CX5), CD247 (CD3ζ) (6B10.2), ZAP-70 (1E7.2), CD122 (TM-b1), CD11b (M1/70) and c-kit (CD117)(ACK2) from eBioscience. FcεR1γ (1D6) from MBL, CD247 (CD3ζ) (H146-968) from Abcam. Ly49H (3D10) and Ly49D (4e4) were kind gift from W. Yokoyama (Washington University in St. Louis, USA). For hematopoietic stem cell staining, bone marrow cells were stained with Abs against CD34 (RAM34), Flt3L (A2F10.1), CD127 (A7R34), CD16/32 (93), Sca1 (D7), and CD117 (2B8). Populations were defined according to previous publication32 as following CLP (CD117dimSca1dimFlt3L+CD127+),PreProNK (CD117dimSca1++Flt3L-CD127+), MPP (CD117+Sca1+CD34+Flt3L+), ST-HSC (CD117+Sca1+CD34+Flt3L-), LT-HSC (CD117+Sca1+CD34-Flt3L-), CMP (CD117+Sca1-CD34+CD16int), GMP (CD117+Sca1-CD34+CD16+), MEP (CD117+Sca1-CD34-CD16-). Biotinylated Abs against CD4 (GK1.5), CD8 (53-6.7), B220 (RA3-6B2), Gr-1 (RB6-8C5), CD11b (M1/70), TER119 (Ter-119), NK.1.1 (PK136) were used to exclude Lin+ cells. In experiment where we analyzed 6 stages of NK cell development NK1.1 (PK136) was excluded from staining for Lin+ cells. Abs were purchased from eBioscience or BD Biosciences. Fusion proteins (NCR1-Fc, NKG2D-Fc and hPVR-Fc) were produced by our in-house facility.
MCMV infection
The tissue culture grown mCMV MW97.01 and Δm157 MCMV was produced in mouse embryonic fibroblasts according to standard protocol. The mice were either treated with αNK1.1 (PK136, 250µg/mouse) or PBS 24h before i.v. injection of 5 x 105 PFU of Δm157 MCMV. Four days after infection mice were sacrificed and viral titers were determined in spleens by standard virus plaque assay. In these experiments 4-5 mice per group were used and experiment were repeated two to three times.
In vivo NKG2D blocking
Rag1CreEYFPStop-FloxiDTR mice were injected i.p. on two consecutive days with 0.8mg diphtheria toxin. From the second day onwards, mice received once every three days 200μg anti-NKG2D (BioXcell, clone HMG2D) or istotype control antibodies. After 15 days, spleens and bone marrow were analyzed.
ImmunoBlot
NK cells were enriched from splenocytes using MACS and then sorted o high (>99% purity) on a FACS Aria II (BD Biosciences). Cell extracts were generated using laemmli sample buffer (Syk, FcR1γ and Zap 70) or RIPA buffer (CD3ζ), in the presence of protease inhibitors (complete Ultra, Roche). Equal amounts of total lysate were analyzed by 12% SDS-polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon-P and incubated with blocking buffer (Tris buffered saline/Tween-20) containing 2% low-fat milk for 1 h before incubating with an antibody against Syk (Cell signaling), FcR1γ (Merckmillipore), Zap-70 (Cell signaling) and CD3ζ (Sigma), Akt (Cell signaling) or β-actin (Santa Cruz). Bands were visualized with ECL Prime Immuno Blotting Detection Reagent (GE Healthcare) using ImageQuant LAS 4000mini (GE Healthcare, Life Science).
Quantitation and Statistical Analysis
To analyze statistical significance, we used Student's t-test, Mann-Whitney, Kruskal-Wallis and ANOVA, with Bonferroni's post-test correction for multiple comparisons. To assess survival rates, the Kaplan–Meier model was used followed by Log-rank (Mantel-Cox) test for pairwise group comparisons. Statistical significance is defined as: * p < 0.05; ** p < 0.01; *** p < 0.001.
Supplementary Material
A Life Science Reporting Summary for this paper is available online.
Acknowledgments
We thank S. Slavić Stupac, M. Samsa, E. Marinović, A. Miše and K. Mikić for technical assistance. We thank S. Jonjić for discussions and overall support to this work. We thank O. Mandelboim (Hebrew University Hadassah Medical School), M. Colonna (Washington University School of Medicine in St. Louis, St. Louis, MO, USA), M. Busslinger (Research Institute of Molecular Pathology, Vienna, Austria), A. Waisman (Institute of Molecular Biology, Mainz, Germany), D.R. Littman (NYU School of Medicine, New York, NY, USA), J. McGlade (University of Toronto, Toronto, Canada) and K. Rajewsky (MDC Center for Molecular Medicine, Berlin, Germany) for providing mouse lines. This work was supported by an EFIS-IL Short-term Fellowship grant to V.J., European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 311335, Swedish Research Council, Norwegian Research Council, Swedish Foundation for Strategic Research, Wallenberg Foundation, Swedish Cancer Foundation, Swedish Childhood Cancer Foundation, as well as the Stockholm County Council and Karolinska Institutet Center for Innovative Medicine to Y.T.B., Netherlands Organization for Scientific Research (91614029), the European Commission (PCIG14-GA-2013-630827), a University of Rijeka Support grant (865.10.2101) and a Croatian Science Foundation Grant (IP-2016-06-8027) to F.M.W., a European Social Fund grant (HR.3.2.01-0263), University of Rijeka Support grant (803.10.1103) and Croatian Science Foundation grant (IP-2016-06-9306) to B.P. and the grant KK.01.1.1.01.0006, awarded to the Scientific Centre of Excellence for Virus Immunology and Vaccines and co-financed by the European Regional Development Fund.
Footnotes
Data availability:
The data that support the findings of this study are available from the corresponding author upon request. The accession codes are: SRX4548789, SRX4548788, SRX4548787, SRX4548786, SRX4548785, SRX4548784, SRX4548783, SRX4548782, SRX4548781
Author Contributions: V.J. carried out most of the experiments and analyzed data. M.Š., I.K., M.L., S.M., B.L. and F.M.W. performed and analyzed experiments. B.P. directed the research. B.P., V.J. and F.M.W. designed experiments and wrote the paper. V.S. and M.P. designed and performed qPCR on NK precursors. T.D.H. and Y.T.B. designed and performed RNAseq experiments.
Competing financial interests:
The authors declare no competing financial interests.
References
- 1.Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nature immunology. 2016;17:758–764. doi: 10.1038/ni.3482. [DOI] [PubMed] [Google Scholar]
- 2.Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011;32:364–372. doi: 10.1016/j.it.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34:182–191. doi: 10.1016/j.it.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Zafirova B, Wensveen FM, Gulin M, Polic B. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell Mol Life Sci. 2011;68:3519–3529. doi: 10.1007/s00018-011-0797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jelencic V, Lenartic M, Wensveen FM, Polic B. NKG2D: A versatile player in the immune system. Immunol Lett. 2017 doi: 10.1016/j.imlet.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Goh W, Huntington ND. Regulation of Murine Natural Killer Cell Development. Front Immunol. 2017;8:130. doi: 10.3389/fimmu.2017.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7:703–714. doi: 10.1038/nri2154. [DOI] [PubMed] [Google Scholar]
- 9.Zafirova B, et al. Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity. 2009;31:270–282. doi: 10.1016/j.immuni.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerra N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–580. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheppard S, et al. Characterization of a novel NKG2D and NKp46 double-mutant mouse reveals subtle variations in the NK cell repertoire. Blood. 2013;121:5025–5033. doi: 10.1182/blood-2012-12-471607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol. 2002;3:1150–1155. doi: 10.1038/ni857. [DOI] [PubMed] [Google Scholar]
- 13.Mandelboim O, Porgador A. NKp46. Int J Biochem Cell Biol. 2001;33:1147–1150. doi: 10.1016/s1357-2725(01)00078-4. [DOI] [PubMed] [Google Scholar]
- 14.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227:150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamerman JA, Lanier LL. Inhibition of immune responses by ITAM-bearing receptors. Sci STKE. 2006;2006:re1. doi: 10.1126/stke.3202006re1. [DOI] [PubMed] [Google Scholar]
- 16.Arase H, et al. Negative regulation of expression and function of Fc gamma RIII by CD3 zeta in murine NK cells. J Immunol. 2001;166:21–25. doi: 10.4049/jimmunol.166.1.21. [DOI] [PubMed] [Google Scholar]
- 17.Utsuyama M, Hirokawa K. Radiation-induced-thymic lymphoma occurs in young, but not in old mice. Exp Mol Pathol. 2003;74:319–325. doi: 10.1016/s0014-4800(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 18.Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2001;Chapter 20:Unit 20 21. doi: 10.1002/0471142735.im2001s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenartic M, et al. NKG2D Promotes B1a Cell Development and Protection against Bacterial Infection. J Immunol. 2017;198:1531–1542. doi: 10.4049/jimmunol.1600461. [DOI] [PubMed] [Google Scholar]
- 20.Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA. “Natural Regulators”: NK Cells as Modulators of T Cell Immunity. Front Immunol. 2016;7:235. doi: 10.3389/fimmu.2016.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soderquest K, et al. Cutting edge: CD8+ T cell priming in the absence of NK cells leads to enhanced memory responses. J Immunol. 2011;186:3304–3308. doi: 10.4049/jimmunol.1004122. [DOI] [PubMed] [Google Scholar]
- 22.Takeda K, et al. IFN-gamma production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J Leukoc Biol. 2011;90:777–785. doi: 10.1189/jlb.0411208. [DOI] [PubMed] [Google Scholar]
- 23.Dong Z, et al. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity. 2012;36:974–985. doi: 10.1016/j.immuni.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Glasner A, et al. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol. 2012;188:2509–2515. doi: 10.4049/jimmunol.1102461. [DOI] [PubMed] [Google Scholar]
- 25.Lakshmikanth T, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119:1251–1263. doi: 10.1172/JCI36022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wensveen FM, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16:376–385. doi: 10.1038/ni.3120. [DOI] [PubMed] [Google Scholar]
- 27.Davis AH, Guseva NV, Ball BL, Heusel JW. Characterization of murine cytomegalovirus m157 from infected cells and identification of critical residues mediating recognition by the NK cell receptor Ly49H. J Immunol. 2008;181:265–275. doi: 10.4049/jimmunol.181.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 2008;111:3571–3578. doi: 10.1182/blood-2007-07-100057. [DOI] [PubMed] [Google Scholar]
- 29.Deng W, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136–139. doi: 10.1126/science.1258867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckelhart E, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. 2011;117:1565–1573. doi: 10.1182/blood-2010-06-291633. [DOI] [PubMed] [Google Scholar]
- 31.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2009;75:14–24. doi: 10.1002/cyto.a.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94–107. doi: 10.1016/j.cell.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viant C, et al. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat Commun. 2014;5 doi: 10.1038/ncomms6108. 5108. [DOI] [PubMed] [Google Scholar]
- 35.Wu N, et al. A hematopoietic cell-driven mechanism involving SLAMF6 receptor, SAP adaptors and SHP-1 phosphatase regulates NK cell education. Nat Immunol. 2016;17:387–396. doi: 10.1038/ni.3369. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–570. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 37.Myers MD, Dragone LL, Weiss A. Src-like adaptor protein down-regulates T cell receptor (TCR)-CD3 expression by targeting TCRzeta for degradation. J Cell Biol. 2005;170:285–294. doi: 10.1083/jcb.200501164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodfellow HS, et al. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci Signal. 2015;8:ra49. doi: 10.1126/scisignal.2005596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sjolin H, et al. DAP12 signaling regulates plasmacytoid dendritic cell homeostasis and down-modulates their function during viral infection. J Immunol. 2006;177:2908–2916. doi: 10.4049/jimmunol.177.5.2908. [DOI] [PubMed] [Google Scholar]
- 41.Mulrooney TJ, Posch PE, Hurley CK. DAP12 impacts trafficking and surface stability of killer immunoglobulin-like receptors on natural killer cells. J Leukoc Biol. 2013;94:301–313. doi: 10.1189/jlb.0213093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Della Chiesa M, et al. Evidence that the KIR2DS5 gene codes for a surface receptor triggering natural killer cell function. Eur J Immunol. 2008;38:2284–2289. doi: 10.1002/eji.200838434. [DOI] [PubMed] [Google Scholar]
- 43.Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaelsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood. 2010;115:1166–1174. doi: 10.1182/blood-2009-09-245746. [DOI] [PubMed] [Google Scholar]
- 44.Carr WH, et al. Cutting Edge: KIR3DS1, a gene implicated in resistance to progression to AIDS, encodes a DAP12-associated receptor expressed on NK cells that triggers NK cell activation. J Immunol. 2007;178:647–651. doi: 10.4049/jimmunol.178.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng GM, Beltran J, Chen C, Terhorst C, Tsokos GC. T cell CD3zeta deficiency enables multiorgan tissue inflammation. J Immunol. 2013;191:3563–3567. doi: 10.4049/jimmunol.1300634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodfellow HS, et al. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Science signaling. 2015;8:ra49. doi: 10.1126/scisignal.2005596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang M, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nat Immunol. 2015;16:1253–1262. doi: 10.1038/ni.3258. [DOI] [PubMed] [Google Scholar]
- 48.Takaki R, Watson SR, Lanier LL. DAP12: an adapter protein with dual functionality. Immunol Rev. 2006;214:118–129. doi: 10.1111/j.1600-065X.2006.00466.x. [DOI] [PubMed] [Google Scholar]
- 49.Kiel MJ, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, et al. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T-cell immune responses in tuberculosis infection. Proc Natl Acad Sci U S A. 2015;112:E3883–3892. doi: 10.1073/pnas.1501662112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.