

Abstract
Control of absolute stereochemistry in radical and ion radical transformations is a major challenge in synthetic chemistry. Herein, we report the design of a photoredox catalyst system comprised of an oxidizing pyrilium salt bearing a chiral N-triflyl phosphoramide anion. This class of chiral organic photoredox catalysts is able to catalyze the formation of cation radical-mediated Diels-Alder transformations in up to 75:25 e.r. in both intramolecular and intermolecular examples.
Keywords: photoredox, catalysis, enantioselective, cycloaddition, chiral anion
Graphical Abstract
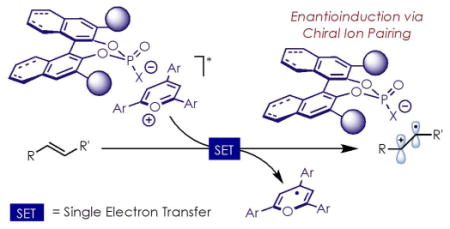
1. Introduction
In recent years, research in the field of photoredox catalysis has resulted in the development of numerous, powerful new transformations for organic synthesis that are complementary to reactions proceeding by two electron processes.1 A central challenge in the field is the development of methods to conduct these reactions in an asymmetric manner, as access to enantiopure products is critical for the development of new bioactive compounds. However, photoredox catalysis is distinct from other common modes of catalysis in a way which makes developing enantioselective reactions especially challenging. Namely, after the key electron (or energy) transfer event occurs between substrate and catalyst, the two are no longer necessarily associated in solution. Subsequent bond-forming steps can therefore take place off-catalyst, which poses a challenge to traditional methods for asymmetric induction, such as introducing chiral ligands to the catalyst. Success in this field therefore requires engineering systems in which the activated substrate is necessarily associated with an element of chirality during the enantiodetermining step of the reaction. Several key advances have recently been made in this area, though reports remain sparse (Scheme 1).
Scheme 1.

Asymmetric photoredox catalysis – prior art
The state of the art in this field includes MacMillan’s use of chiral enamine catalysis, which affords excellent stereocontrol in reactions with electron-deficient radical partners.2 The Yoon group has developed a dual photoredox/Lewis acid catalyst system that enables enantioselective [2+2] cycloaddition reactions, as well as radical conjugate addition reactions.3 The Meggers group has developed a chiral-at-Iridium complex capable of acting both as a Lewis acid and photoredox catalyst for similar conjugate addition reactions.4 The Knowles group has demonstrated absolute stereocontrol in ketyl radical chemistry, enabled by a key proton-coupled electron transfer event to generate the reactive species that is coordinated to a chiral Brønsted acid catalyst.5 Additionally, enzymatic processes have recently been shown to be compatible with photoredox catalysis and allow for enantioinduction.6 These methods have proven to be very powerful, and share many common features. In each case, the reactions proceed by a mechanism in which the starting material is reduced by the photoredox catalyst in order to arrive at the desired reactive intermediate. The substrates for these reactions also all contain functional groups, specifically carbonyls, which serve as sites for association with the chiral co-catalyst, via either covalent or non-covalent interactions.
An archetypal example of a reaction that operates via this manifold is the cation-radical Diels-Alder reaction (Scheme 2). This transformation was initially reported by Bauld7 using ground-state one-electron oxidants, and has been greatly expanded upon through photoredox manifolds in recent years by the Yoon group and others.8 The reaction is powerful due to its ability to enable Diels-Alder reactions between pairs of electron-rich dienes and dienophiles. The transformation begins with the one-electron oxidation of the electron-rich dienophile i (Scheme 2). The dienophile is now rendered electrophilic (ii), and readily undergoes cyclization with diene iii, giving intermediate iv. One-electron reduction of this intermediate furnishes the final product v, and also serves to either turn over the catalyst, or propagate the reaction via a chain mechanism.9
Scheme 2.

The cation radical Diels-Alder reaction and current work
Pyrylium salts are a class of organic photoredox catalysts that are strong one-electron photooxidants and are known catalysts for this transformation.10 Being cationic in nature, they also bear a non-coordinating anion, such as a tetrafluoroborate (BF4) anion. Based on the proposed mechanism for the reaction, if it were to be run in a relatively non-polar solvent, then presumably the BF4 anion would become paired with the cation radical intermediates of the reaction (ii and iv) after electron transfer occurs. This led us to consider whether an ion pairing interaction between a radical cation intermediate and chiral counterion could be used to control the absolute stereochemistry of these types of reactions (Scheme 2). In a single example reported by Schuster, low levels of enantioinduction (15% ee, yield not given) were observed in a cation-radical Diels-Alder reaction promoted by a neutral, axially-chiral cyanoarene photosensitizer, lending support to this hypothesis.11 Moreover, a recent report utilizing an acridinium photooxidant showed enantioinduction in a hydrofunctionalization reaction using a chiral anion.12
We therefore set out to develop a new class of organic photoredox catalysts, in which a pyrylium-based photooxidant is paired with a chiral anion, and ascertain whether these compounds could be used to control the absolute stereochemistry of the cation radical Diels-Alder reaction. The design of this catalyst system is especially appealing to us, as the goal of ion pairing directly with the radical-cation intermediates circumvents the need for chelating functional groups, such as carbonyls, to be present in the substrates.
2. Results and Discussion
At the outset of the project, we hypothesized that the chiral anion would need to be sufficiently non-nucleophilic in nature in order to avoid directly reacting with either the photo-oxidant or charged intermediates. We focused our attention on anions derived from chiral Bronsted acids, due to their tunable steric and electronic properties.13 They have also been successfully employed as chiral anions in a number of previously reported transformations.14 Triaryl pyryliums were chosen as the photooxidant due to their easily tunable high excited state reduction potentials, mono-cationic nature, and prior use as catalysts in Diels-Alder reactions.10
We found that the preparation of the desired oxopyrylium salts could be accomplished in a very straightforward manner. A precursor enedione molecule was found to undergo an acid promoted cyclization to the oxopyrylium when heated in the presence of an appropriate chiral Bronsted acid. This strategy proved general and allowed us to construct our library of chiral catalysts. Furthermore, the salts have proven to be highly stable and can be stored on the bench top for extended periods of time.
A series of catalysts were evaluated bearing different couteranion structures with triene 2a as the substrate (Table 1). In general, anions derived from N-trifylphosphoramides afforded the desired Diels-Alder adduct whereas chiral phosphate anions did not, possibly due to the higher nucleophilicity of the latter species. We also found that sterically demanding groups were necessary at the 3,3′-positions of the BINOL-derived anions, with triphenylsilyl groups ultimately proving most effective. We attribute this success to two factors: in addition to creating a highly demanding chiral environment, the bulky nature of the triphenylsilyl groups may also further decrease the effective nucleophilicity of the anion, thereby enhancing reactivity. Toluene proved uniquely effective as a solvent during an initial screen. Conversion was observed in dichloromethane, but no enantioselectivity. Nonpolar, non-aromatic solvents were not successful, likely due in part to poor catalyst solubility. 1e was explored as well, the conjugate acid of which has recently been reported as a highly effective Brønsted acid catalyst15, gave conversion to the desired product in dichloromethane, but did not give appreciable levels of enantioselectivity under several conditions. Catalyst 1c proved uniquely effective and so was carried forward for further study.
Table 1.
Optimization of enantioselective Diels-Alder reactiona
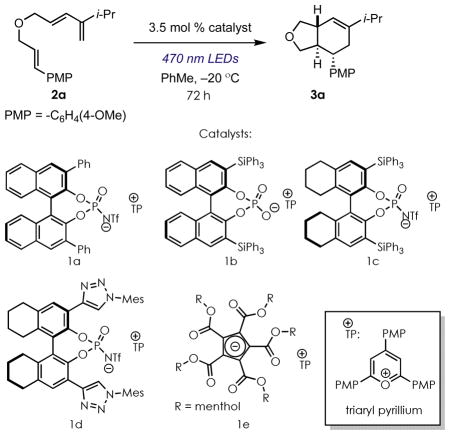
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yield[%]b | d.r.b | e.r.c |
| 1 | 1a | PhMe | <5 | -- | -- |
| 2 | 1b | PhMe | <5 | -- | -- |
| 3 | 1c | PhMe | 72 | 6:1 | 75:25 |
| 4 | 1c | DCM | 44 | 10:1 | 50:50 |
| 5 | 1c | THF | <5 | -- | -- |
| 6 | 1c | Et2O | <5 | -- | -- |
| 7 | 1d | PhMe | <5 | -- | -- |
| 8d | 1e | PhMe | <5 | -- | -- |
| 9d,e | 1e | DCM | 53 | 6:1 | 51:49 |
| 10d | 1e | PhCF3 | 25 | 6:1 | 50:50 |
| 11d | 1e | DCM:PhMe (9:1) | 25 | 6:1 | 53:47 |
Reactions were carried out in dry PhMe [0.2 M] in a cooling bath, with strips of blue LEDs (λmax = 470 nm) coiled around the reactions (see supporting information for details).
Determined by 1H NMR spectroscopic analysis of the crude reaction mixture relative to the internal standard (Me3Si)2O.
Determined by HPLC analysis of purified material using a chiral stationary phase.
Reaction performed using a 2.5 mol % catalyst loading.
Reaction performed at −30 °C.
Given the promising results with toluene, we probed the effect of the aromatic solvent identity further using catalyst 1c and substrate 2a (Table 2). Overall, we observed a general trend that enantioselectivity is inversely correlated with solvent dielectric constant εr. This observation supports our hypothesis that close ion pairing is necessary for the relay of chiral information from anion to substrate.16 The requirement for aromatic solvents to be used is intriguing as well, especially since a modest 61:39 e.r. was observed in trifluorotoluene, despite it being more polar (εr = 9.22) than dichloromethane (εr = 8.93).16 We attribute this to favourable π-stacking interactions helping to organize the transition state of the desired pathway, though further study is required to elucidate the exact nature of the interactions involved.
Table 2.
Solvent effect on enantioselectivity of Diels-Alder reactiona
| Entry | Solvent | Dielectric constant (εr)b | Yield[%] | d.r. | e.r. |
|---|---|---|---|---|---|
| 1 | PhCF3 | 9.22 | 23 | 10:1 | 61:39 |
| 2 | PhBr | 5.45 | 27 | 3:1 | 66:34 |
| 3 | PhMe | 2.38 | 75 | 6:1 | 75:25 |
| 4 | Xylenes | 2.36 | <10 | -- | 72:28 |
We then investigated the scope of the intramolecular cycloaddition reaction with a series of triene substrates under the optimized reaction conditions (Figure 1). Substitution at the 2-position of the diene proved to be critical for obtaining both moderate yields and levels of enantioinduction – replacement with less bulky substituents (i.e. Me, 3d) resulted in complete loss of enantiocontrol. Furthermore, the use of other linkers other than an ether (i.e. NTs, 3e) also resulted in an inexplicable loss of enantioselectivity for the cycloaddition reaction.
Figure 1.

Scope of the intramolecular enantioselective cation radical Diels-Alder reaction
We also tested a number of intermolecular reactions, and the results are summarized in Figure 2. Using the highly reactive cyclopentadiene with PMP-styrene derivatives resulted in the formation of [2.2.1]-bicycloheptenes in good levels of diastereocontrol, with similar levels of enantiocontrol observed in the intramolecular reactions (4a and 4b, eq 1). In the absence of the single electron oxidation catalyst, no product formation was observed. Cryogenic temperatures (−60 °C) were required to observe any enantioselectivity in the [4+2] reaction. Less reactive dienes such as 2,3-dimethyl-1,3-butadiene gave only trace amounts of the desired cyclohexene adduct 4c, but again with similar levels of stereocontrol (75:25 e.r., eq 2).
Figure 2.

Scope of the intermolecular enantioselective on radical Diels-Alder reaction
3. Conclusions
In summary, we have developed a novel class of chiral photoredox catalysts consisting of a cationic oxopyrylium photooxidant bearing a chiral anion. In this report, we have demonstrated that this catalyst system is capable of modest levels of control of the absolute stereochemistry of cation radical Diels-Alder reactions. These results serve as an important proof-of-concept that ion pairing between cation radical intermediates and chiral anions is a viable and potentially general mode of asymmetric induction for these transformations. It is noteworthy that the substrates used in this reaction do not posses any additional functional handles, such as carbonyls, for coordination with the catalyst, which are typically required in other reported enantioselective Diels-Alder reactions, as well as enantioselective photoredox reactions. We are currently interested in using the knowledge gained from this study to further improve the design of our catalysts, as well as apply them to new reaction manifolds.
4. Experimental Data
4.1. General Methods
Infrared (IR) spectra were obtained using a Jasco 260 Plus Fourier transform infrared spectrometer. Proton, carbon, and fluorine magnetic resonance spectra (1H NMR and 13C NMR) were recorded on a Bruker model DRX 400 or 600 (1H NMR at 400 MHz or 600 MHz and 13C NMR at 100 MHz or 150 MHz spectrometer. Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in solvent (1H NMR: CHCl3 at 7.26 ppm). Chemical shifts for carbons are reported in parts per million downfield from tetramethylsilane and are referenced to the carbon resonances of the residual solvent peak (13C NMR: CDCl3 at 77.0 ppm). NMR data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, ddd = doublet of doublet of doublet, dddd = doublet of doublet of doublet of doublet, dtd = doublet of triplet of doublet, t = triplet, q = quartet, qd = quartet of doublet, sept = septuplet, m = multiplet), coupling constants (Hz), and integration. Mass spectra were obtained using either a Micromass Quattro II (triple quad) instrument with nanoelectrospray ionization or an Agilent 6850 series gas chromatograph instrument equipped with a split-mode capillary injection system and Agilent 5973 network mass spec detector (MSD). Thin layer chromatography (TLC) was performed on SiliaPlate 250 μm thick silica gel plates purchased from Silicycle. Visualization was accomplished using fluorescence quenching, KMnO4 stain, or ceric ammonium molybdate (CAM) stain followed by heating. Organic solutions were concentrated under reduced pressure using a Büchi rotary evaporator. Purification of the reaction products was carried out by chromatography using Siliaflash-P60 (40–63 μm) or Siliaflash-T60 (5–20 μm) silica gel purchased from Silicycle.
All reactions were carried out under an inert atmosphere of nitrogen in flame-dried glassware with magnetic stirring unless otherwise noted. Irradiation of photochemical reactions was carried out using a Par38 Royal Blue Aquarium LED lamp (Model # 6851) fabricated with high-power Cree LEDs as purchased from Ecoxotic (www.ecoxotic.com), with standard borosilicate glass vials purchased from Fisher Scientific. Yield refers to isolated yield of analytically pure material unless otherwise noted. NMR yields were determined using hexamethyldisiloxane as an internal standard.
All materials were used as received unless otherwise stated. Starting materials for the synthesis of substrates 2a-e were synthesized according to known procedures and spectral data matched those already reported (see supporting information).
4.2. Procedure for Asymmetric Cation-Radical Diels-Alder Reactions (Figure 1 and 2)
Substrate (0.2 mmol) and 1c (3.5 mol-%) were added to a flame dried vial with stir bar added, covered in aluminum foil to block light, and transferred to a glove box. The material was dissolved in PhMe (1.0 mL) and the vial was sealed. The vial was then removed from the glove box and placed in a cryobath set to the appropriate tempterature after removing the foil cover. The cryobath contained an irradiation setup consisting of blue LED strips wrapped around a small section of a test tube rack (pictured below). After stirring and cooling for 10 minutes in darkness, the sample was irradiated by the LEDs for 72h. The reaction mixture was passed through a plug of silica and eluted with DCM to remove the catalyst. The crude mixture was then concentrated and purified by column chromatography.
4.2.1. 6-isopropyl-4-(4-methoxyphenyl)-1,3,3a,4,5,7a-hexahydroisobenzofuran (3a)
Compound 3a was prepared according to the general procedure and obtained in a 57% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (400 MHz, CDCl3) δ 7.18 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.61 (s, 1H), 4.12 (t, J = 7.2 Hz, 1H), 3.82 (s, 4H), 3.44 (dd, J = 11.6, 7.0 Hz, 1H), 3.34 (dd, J = 11.2, 7.7 Hz, 1H), 2.86 (td, J = 11.2, 6.2 Hz, 1H), 2.67 – 2.54 (m, 1H), 2.54 – 2.42 (m, 1H), 2.34 – 2.08 (m, 3H), 1.05 (dd, J = 6.8, 1.7 Hz, 6H); 13C NMR: (100 MHz, CDCl3) δ 158.0, 146.4, 137.1, 127.6, 115.8, 113.9, 71.4, 71.4, 70.6, 55.2, 55.2, 48.5, 45.3, 42.70, 38.1, 34.4, 21.6, 21.5, 21.4; MS (GC-MS) Calculated m/z = 272.17, Found m/z = 272.20; IR (thin film): 3902, 3870, 3819, 3801, 3749, 3710, 3031, 2959, 2929, 2869, 2003, 1792, 1732, 1584, 1463
4.2.2. 6-cyclohexyl-4-(4-methoxyphenyl)-1,3,3a,4,5,7a-hexahydroisobenzofuran (3b)
Compound 3b was prepared according to the general procedure and obtained in a 63% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (400 MHz, CDCl3) δ 7.14 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.56 (s, 1H), 4.08 (t, J = 7.2 Hz, 1H), 3.79 (s, 3H), 3.76 (t, J = 7.3 Hz, 1H), 3.40 (dd, J = 11.6, 7.0 Hz, 1H), 3.30 (dd, J = 11.2, 7.7 Hz, 1H), 2.83 (td, J = 11.2, 6.2 Hz, 1H), 2.63 – 2.51 (m, 1H), 2.51 – 2.40 (m, 1H), 2.25 – 2.06 (m, 2H), 1.89 – 1.61 (m, 6H), 1.33 – 1.06 (m, 5H); 13C NMR: (101 MHz, CDCl3) δ 158.1, 145.9, 137.2, 127.6, 116.2, 114.0, 71.5, 70.6, 55.2, 48.6, 45.3, 45.0, 42.8, 38.90, 32.2, 32.1, 26.7, 26.3.; MS (GC-MS) Calculated m/z for [M] = 312.21, Found m/z for [M] = 312.22; IR (thin film): 2924, 2851, 1611, 512, 1448, 1302, 1249, 1178, 1110, 1035
4.2.3. 6-benzyl-4-(4-methoxyphenyl)-1,3,3a,4,5,7a-hexahydroisobenzofuran (3c)
Compound 3c was prepared according to the general procedure and obtained in a 43% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (600 MHz, CDCl3) δ 7.28 (dd, J = 15.8, 8.4 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.17 (d, J = 7.5 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.2 Hz, 2H), 5.64 (s, 1H), 4.10 (t, J = 7.3 Hz, 1H), 3.78 (s, 3H), 3.76 (t, J = 7.4 Hz, 1H), 3.44 (dd, J = 11.6, 7.1 Hz, 1H), 3.40 – 3.20 (m, 3H), 2.82 (td, J = 11.2, 6.3 Hz, 1H), 2.61 (q, J = 10.4 Hz, 1H), 2.40 (dd, J = 18.2, 6.4 Hz, 1H), 2.22 (qd, J = 11.2, 6.9 Hz, 1H), 2.12 (ddt, J = 14.5, 10.7, 5.5 Hz, 1H); 13C NMR: (151 MHz, CDCl3) δ 158.1, 140.1, 139.4, 136.7, 128.8, 128.3, 127.6, 126.1, 120.3, 113.9, 77.2, 76.7, 71.28, 70.62, 55.2, 48.6, 45.4, 43.6, 42.6, 39.9; MS (GC-MS) Calculated m/z = 320.43, Found m/z = 320.20; IR (thin film): 3082, 3060, 3027, 3000, 2932, 2908, 2836, 1652, 1607, 1577, 1511, 1495, 1453, 1441, 1420
4.2.4. 6-methyl-4-(4-methoxyphenyl)-1,3,3a,4,5,7a-hexahydroisobenzofuran (3d)
Compound 3d was prepared according to the general procedure and obtained in a 10% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (600 MHz, CDCl3) δ 7.16 – 7.11 (m, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.56 (s, 1H), 4.08 (t, J = 7.2 Hz, 1H), 3.79 (s, 3H), 3.76 (t, J = 7.4 Hz, 1H), 3.41 (dd, J = 11.6, 7.0 Hz, 1H), 3.31 (dd, J = 11.2, 7.7 Hz, 1H), 2.85 (td, J = 11.2, 6.3 Hz, 1H), 2.56 (d, J = 9.9 Hz, 1H), 2.40 (dd, J = 18.1, 6.5 Hz, 1H), 2.19 (m, 2H), 1.70 (s, 3H);13C NMR: (151 MHz, CDCl3) δ 158.1, 137.0, 136.8, 127.7, 118.8, 114.0, 71.4, 70.7, 55.2, 48.6, 45.5, 42.7, 41.9, 22.9; MS (GC-MS) Calculated m/z = 244.15, Found m/z = 244.20; IR (thin film): 3082, 3060, 3027, 3000, 2932, 2908, 2836, 1652, 1607, 1577, 1511, 1495, 1453, 1441, 1420
4.2.5. 6-isopropyl-4-(4-methoxyphenyl)-2-tosyl-2,3,3a,4,5,7a-hexahydro-1H-isoindole (3e)
Compound 3e was prepared according to the general procedure and obtained in a 42% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (600 MHz, CDCl3) δ 7.72 – 7.68 (m, 2H), 7.33 (d, J = 7.9 Hz, 2H), 7.09 – 7.05 (m, 2H), 6.90 – 6.85 (m, 2H), 5.47 (s, 1H), 3.83 (s, 3H), 3.70 (dd, J = 9.1, 7.3 Hz, 1H), 3.33 (dd, J = 9.5, 6.8 Hz, 1H), 2.94 (dd, J = 11.8, 9.1 Hz, 1H), 2.75 (dd, J = 11.3, 9.5 Hz, 1H), 2.68 (td, J = 11.2, 6.2 Hz, 1H), 2.46 (s, 1H), 2.37 (ddt, J = 17.9, 6.2, 1.8 Hz, 1H), 2.30 – 2.22 (m, 1H), 2.17 (p, J = 6.9 Hz, 1H), 2.13 – 2.05 (m, 1H), 1.92 (qd, J = 11.3, 6.8 Hz, 1H), 0.99 (dd, J = 6.9, 3.8 Hz, 6H); 13C NMR: (151 MHz, CDCl3) δ 158.3, 146.7, 143.1, 136.0, 129.6, 127.6, 127.2, 116.1, 114.1, 55.2, 52.3, 51.3, 47.3, 43.8, 43.1, 37.3, 34.4, 21.5, 21.5, 21.3; MS (ESI) Calculated m/z for [M+H] = 426.2103, Found m/z for [M+H] = 426.2082
4.2.2. 5-ethyl-6-(4-methoxyphenyl)bicyclo[2.2.1]hept-2-ene (4b)
Compound 4b was prepared according to the general procedure and obtained in a 43% yield as a colorless solid after purification by column chromatography with silica gel and DCM.
1H NMR: (600 MHz, CDCl3) δ 7.28 (dd, J = 15.8, 8.4 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.17 (d, J = 7.5 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.2 Hz, 2H), 5.64 (s, 1H), 4.10 (t, J = 7.3 Hz, 1H), 3.78 (s, 3H), 3.76 (t, J = 7.4 Hz, 1H), 3.44 (dd, J = 11.6, 7.1 Hz, 1H), 3.40 – 3.20 (m, 3H), 2.82 (td, J = 11.2, 6.3 Hz, 1H), 2.61 (q, J = 10.4 Hz, 1H), 2.40 (dd, J = 18.2, 6.4 Hz, 1H), 2.22 (qd, J = 11.2, 6.9 Hz, 1H), 2.12 (ddt, J = 14.5, 10.7, 5.5 Hz, 1H). 13C NMR: (151 MHz, CDCl3) δ 158.1, 140.1, 139.4, 136.7, 128.8, 128.3, 127.6, 126.1, 120.3, 113.9, 77.2, 76.7, 71.28, 70.62, 55.2, 48.6, 45.4, 43.6, 42.6, 39.9.
MS (GC-MS) Calculated m/z = 320.43, Found m/z = 320.20
IR (thin film): 3082, 3060, 3027, 3000, 2932, 2908, 2836, 1652, 1607, 1577, 1511, 1495, 1453, 1441, 1420
4.3. Procedure for Formation of Chiral Anion Pyrylium Salts (Table 1)
The Brønsted acid (1 equiv.) and 1f (1 equiv.) were added to a vial and suspended in EtOH. Upon heating to approx. 60 °C using a heat gun for 5 minutes, all material dissolved and the solution took on a bright orange color. The crude reaction mixture was allowed to cool and concentrated upon complete consumption of the starting material. The crude product was dissolved in MeOH and shaken with hexanes. After separation, concentration of the MeOH layer and drying under high vacuum overnight afforded pure oxopyrilium salts. These compounds are benchtop stable for several days but should be stored at low temperatures for extended periods.
4.3.1. 2,4,6-tris(4-methoxyphenyl)pyrylium ((11bR)-4-oxido-2,6-diphenyldinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepin-4-yl)((trifluoromethyl)sulfonyl)amide (1a)
Compound 1a was prepared according to the general procedure and obtained in a 74% yield as an orange solid after extraction.; 1H NMR: (600 MHz, CDCl3) δ 8.03 – 7.72 (m, 14H), 7.49 – 7.38 (m, 2H), 7.32 (dd, J = 28.8, 8.5 Hz, 2H), 7.24 (t, J = 7.7 Hz, 4H), 7.09 (d, J = 3.7 Hz, 4H), 6.77 (d, J = 8.5 Hz, 4H), 6.33 (d, J = 8.4 Hz, 2H); 13C NMR: (151 MHz, CDCl3) δ 166.8, 165.4, 164.6, 161.4, 138.0, 137.0, 134.7, 134.1, 132.4, 132.2, 131.2, 131.1, 131.0, 130.4, 130.3, 130.0, 130.0, 128.3, 128.2, 127.7, 127.1, 127.0, 127.0, 126.9, 126.2, 125.9, 125.2, 125.2, 123.4, 120.3, 115.3, 115.2, 109.8, 55.6, 55.2; MS (ESI) Calculated m/z for [M-H] = 862.21, Found m/z for [M-H] = 863.21778, Calculated m/z for [M+] = 399.16, Found m/z for [M+] = 399.15859; IR (thin film): 3432, 2077, 1631, 1606, 1589, 1512, 1484, 1438, 1306, 1261, 1243, 1178, 1096
4.3.2. 2,4,6-tris(4-methoxyphenyl)pyrylium ((11bR)-4-oxido-2,6-bis(triphenylsilyl)dinaphtho[2,1-d:1′,2′ f][1,3,2]dioxaphosphepin-4-yl)((trifluoromethyl)sulfonyl)amide (1b)
Compound 1b was prepared according to the general procedure and obtained in a 83% yield as an orange solid after extraction.; 1H NMR: (600 MHz, CDCl3) δ 8.27 (d, J = 9.0 Hz, 4H), 8.16 (s, 2H), 8.13 (d, J = 8.6 Hz, 2H), 7.97 (s, 2H), 7.69 (d, J = 8.3 Hz, 2H), 7.64 – 7.52 (m, 12H), 7.33 (d, J = 8.5 Hz, 2H), 7.29 (t, J = 7.5 Hz, 2H), 7.22 (t, J = 7.7 Hz, 2H), 7.01 (d, J = 7.8 Hz, 18H), 6.91 (d, J = 8.7 Hz, 4H), 6.48 (d, J = 8.4 Hz, 2H), 3.67 (s, 6H), 3.33 (s, 3H); 13C NMR: (151 MHz, CDCl3) δ 167.0, 165.3, 164.5, 161.9, 141.0, 136.9, 135.4, 134.4, 133.6, 130.7, 129.8, 128.5, 128.5, 127.1, 126.9, 126.3, 124.1, 123.9, 122.4, 121.5, 115.3, 115.1, 111.4, 77.2, 77.0, 76.7, 55.7, 55.5; MS (LTQ FT-ICR MS) Calculated m/z for [M−] = 862.21, Found m/z for [M−] = 863.21778, Calculated m/z for [M+] = 399.16, Found m/z for [M+] = 399.15862; IR (thin film): 3428, 3069, 3049, 2937, 2041, 1628, 1604, 1512, 1486, 1438, 1408, 1262, 1244, 1178, 1104
4.3.3. 2,4,6-tris(4-methoxyphenyl)pyrylium ((11bR)-4-oxido-2,6-bis(triphenylsilyl)-8,9,10,11,12,13,14,15-octahydrodinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepin-4-yl)((trifluoromethyl)sulfonyl)amide (1c)
Compound 1c was prepared according to the general procedure and obtained in a 74% yield as an orange solid after extraction.; 1H NMR: (600 MHz, CDCl3) δ 8.27 (s, 2H), 8.25 – 8.14 (m, 6H), 7.64 – 7.45 (m, 14H), 7.13 – 6.96(m, J = 8.7 Hz, 24H), 6.94 (d, J = 8.4 Hz, 2H), 3.91 (s, 6H), 3.82 (s, 3H), 2.79 – 2.54 (m, 6H), 2.40 (d, J = 16.7 Hz, 1H), 2.30 (d, J = 17.9 Hz, 1H), 1.90 – 1.55 (m, 8H); 13C NMR: (151 MHz, CDCl3) δ 167.6, 166.2, 165.0, 162.2, 136.9, 136.9, 135.5, 134.9, 133.2, 130.5, 128.4, 128.4, 127.0, 126.9, 124.3, 121.3, 116.0, 115.7, 111.0, 56.2, 55.9, 29.2, 29.0, 27.9, 27.8, 22.9, 22.8, 22.7; MS (LTQ FT-ICR MS) Calculated m/z for [M-H] = 1002.25, Found m/z for [M-H] = 1002.26177, Calculated m/z for [M+] = 399.16, Found m/z for [M+] = 399.15867; IR (thin film): 3734. 3648. 3586, 1716, 1588, 1508, 1487, 1243, 1177, 1105
4.3.4. 2,4,6-tris(4-methoxyphenyl)pyrylium ((11bR)-2,6-bis(1-mesityl-1H-1,2,3-triazol-4-yl)-4-oxido-8,9,10,11,12,13,14,15-octahydrodinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepin-4-yl)((trifluoromethyl)sulfonyl)amide (1d)
Compound 1d was prepared according to the general procedure and obtained in a 56% yield as an orange solid after extraction.; 1H NMR: (600 MHz, CDCl3) δ 8.57 (s, 2H), 8.29 (s, 3H), 8.17 (d, J = 8.4 Hz, 6H), 7.92 (s, 2H), 7.03 (d, J = 8.2 Hz, 4H), 6.89 (s, 2H), 6.82 (s, 4H), 3.86 (s, 6H), 3.74 (s, 3H), 2.88 (dd, J = 15.0, 7.7 Hz, 2H), 2.85 – 2.67 (m, 2H), 2.69 – 2.54 (m, 2H), 2.26 (s, 6H), 2.21 (d, J = 16.5 Hz, 2H), 1.88 (s, 12H), 1.82 – 1.64 (m, 6H), 1.52 (dt, J = 10.3, 4.8 Hz, 2H); 13C NMR: (151 MHz, CDCl3) δ 167.9, 165.9, 165.1, 162.3, 142.7, 139.4, 134.9, 133.4, 132.1, 130.2, 128.8, 128.2, 127.9, 125.9, 124.1, 120.9, 115.7, 115.6, 110.4, 77.2, 77.0, 76.7, 55.8, 29.1, 27.8, 22.7, 22.5, 21.0, 17.4; MS (LTQ FT-ICR MS) Calculated m/z for [M−] = 727.31, Found m/z for [M−] = 727.31472, Calculated m/z for [M+] = 399.16, Found m/z for [M+] = 399.15862; IR (thin film): 2938, 2844, 2044, 1630, 1604, 1512, 1485, 1463, 1439, 1307, 1261, 1243, 1178
4.3.5. 2,4,6-tris(4-methoxyphenyl)pyrylium 1,2,3,4,5-pentakis((((1S,2R,5S)-2-isopropyl-5-methylcyclohexyl)oxy)carbonyl)cyclopenta-2,4-dien-1-ide (1e)
Compound 1e was prepared according to the general procedure and obtained in a 86% yield as an orange solid after extraction.; 1H NMR: (600 MHz, CDCl3) δ 8.44 (s, 2H), 8.38 (d, J = 8.5 Hz, 2H), 8.25 (d, J = 8.4 Hz, 4H), 7.05 (d, J = 8.4 Hz, 4H), 6.95 (d, J = 8.5 Hz, 2H), 6.89 (s, 2H), 6.82 (s, 4H), 4.74 (m, 5H) 3.90 (s, 6H), 3.88 (s, 3H), 2.15 – 0.68 (m, 95H); 13C NMR: (151 MHz, CDCl3) δ 169.0, 168.0, 167.1, 166.3, 163.4, 132.9, 131.4, 125.6, 122.0, 117.1, 116.8, 116.6, 111.9, 73.3, 57.3 47.2, 40.0, 34.3, 32.2, 24.5, 23.1, 22.6, 21.8, 16.8; MS (LTQ FT-ICR MS) Calculated m/z for [M−] = 975.6916, Found m/z for [M−] = 975.6930, Calculated m/z for [M+] = 399.1590, Found m/z for [M+] = 399.1590;
4.3.6. (Z)-1,3,5-tris(4-methoxyphenyl)pent-2-ene-1,5-dione (1f)
To a clean dry RBF was added triaryloxopyrylium tetrafluoroborate and sodium acetate. Then the solids were dissolved in a 1:1 mixture of DCM/H2O. Equipped with reflux condenser and heated to 50 °C overnight. Cooled to room temperature then the organic and aqueous layers separated, then the aqueous layer was extracted with DCM 3x, organic layers combined and washed with brine, dried over Na2SO4 and concentrated in vacuo. Chromatographed in 25% EtOAc/Hexanes to give the desired product (40%).
1H NMR: (400 MHz, CDCl3) δ = 8.08 (d, J = 8.8 Hz, 2 H), 8.01 (d, J = 8.8 Hz, 6 H), 7.53 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.01 – 6.90 (m, 6 H), 4.86 (s, 2 H), 3.90 (s, 3 H), 3.89 (s, 3 H), 3.85 (s, 3 H); 13C NMR: (101 MHz, CDCl3) δ = 195.1, 189.4, 163.5, 163.2, 160.6, 151.7, 134.3, 132.3, 130.6, 128.3, 121.8, 114.1, 113.7, 55.5, 42.3; MS (GC-MS) Calculated m/z for [M+H] = 417.17, Found m/z for [M+H] = 417.20; IR (thin film): 3056, 3008, 2961, 2935, 2838, 2571, 2048, 1810, 1677, 1645, 1600, 1509, 1461, 1440, 1423, 1363, 1316
4.4. Procedure for Formation of Diels-Alder Substrates (Figure 1)
Diene alcohol was dissolved in THF (0.2 M) in a flame-dried round bottom flask and kept under positive pressure of N2. The solution was cooled to −78 °C in a dry ice/acetone batch. n-BuLi (2.5 M in hexane, 1.1 equiv.) added dropwise via syringe. After stirring for ~5 minutes, MsCl (1.2 equiv.) was added dropwise via syringe, and solution then stirred for an additional 30 minutes. At the same time, a separate round bottom flask was charged with LiBr (4.5 equiv.), 4 Å molecular sieves, and THF. The solution was stirred for 15 minutes and transferred slowly via cannula into the reaction mixture. Reaction continued stirring at −78 °C for 30 minutes, then the dry ice/acetone bath was removed and the reaction continued to stir at room temperature for 1.5 hours. The mixture was quenched by the addition of saturated aqueous ammonium chloride extracted 3× with Et2O, dried over Na2SO4, and concentrated under reduced pressure. The crude product was dissolved in dry THF (0.2 M) and stirred for 1 hour over 4 Å molecular sieves. Meanwhile, 4-methoxycinnamyl alcohol (1.5 equiv) was added in small portions to a stirred solution of NaH (1.6 equiv, 60% dispersion in mineral oil) in dry THF (0.2 M) at 0 °C. The mixture was stirred for 15 minutes, then the solution containing the alkyl bromide was transferred via cannula over several minutes. The reaction was allowed to warm to room temperature and stirred for 4 hours. The reaction was quenched by the slow addition of saturated aqueous ammonium chloride and extracted 3× with Et2O. Combined organic layers were dried over Na2SO4 and concentrated. Products were purified by column chromatography using EtOAc/Hexanes.
4.4.1. 1-((E)-3-(((E)-4-isopropylpenta-2,4-dien-1-yl)oxy)prop-1-en-1-yl)-4-methoxybenzene n (2a)
Compound 2a was prepared according to the general procedure and obtained in a 57% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (400 MHz, CDCl3) δ 7.18 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.61 (s, 1H), 4.12 (t, J = 7.2 Hz, 1H), 3.82 (s, 4H), 3.44 (dd, J = 11.6, 7.0 Hz, 1H), 3.34 (dd, J = 11.2, 7.7 Hz, 1H), 2.86 (td, J = 11.2, 6.2 Hz, 1H), 2.67 – 2.54 (m, 1H), 2.54 – 2.42 (m, 1H), 2.34 – 2.08 (m, 3H), 1.05 (dd, J = 6.8, 1.7 Hz, 6H); 13C NMR: (100 MHz, CDCl3) δ 158.0, 146.4, 137.1, 127.6, 115.8, 113.9, 71.4, 71.4, 70.6, 55.2, 55.2, 48.5, 45.3, 42.70, 38.1, 34.4, 21.6, 21.5, 21.4; MS (GC-MS) Calculated m/z = 272.17, Found m/z = 272.20; IR (thin film): 3902, 3870, 3819, 3801, 3749, 3710, 3031, 2959, 2929, 2869, 2003, 1792, 1732, 1584, 1463
4.4.2. 1-((E)-3-(((E)-4-cyclohexylpenta-2,4-dien-1-yl)oxy)prop-1-en-1-yl)-4-methoxybenzene (2b)
Compound 2b was prepared according to the general procedure and obtained in a 63% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (400 MHz, CDCl3) δ 7.14 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.56 (s, 1H), 4.08 (t, J = 7.2 Hz, 1H), 3.79 (s, 3H), 3.76 (t, J = 7.3 Hz, 1H), 3.40 (dd, J = 11.6, 7.0 Hz, 1H), 3.30 (dd, J = 11.2, 7.7 Hz, 1H), 2.83 (td, J = 11.2, 6.2 Hz, 1H), 2.63 – 2.51 (m, 1H), 2.51 – 2.40 (m, 1H), 2.25 – 2.06 (m, 2H), 1.89 – 1.61 (m, 6H), 1.33 – 1.06 (m, 5H); 13C NMR: (101 MHz, CDCl3) δ 158.1, 145.9, 137.2, 127.6, 116.2, 114.0, 71.5, 70.6, 55.2, 48.6, 45.3, 45.0, 42.8, 38.90, 32.2, 32.1, 26.7, 26.3.; MS (GC-MS) Calculated m/z for [M] = 312.21, Found m/z for [M] = 312.22; IR (thin film): 2924, 2851, 1611, 512, 1448, 1302, 1249, 1178, 1110, 1035
4.4.3. 1-((E)-3-(((E)-4-benzylpenta-2,4-dien-1-yl)oxy)prop-1-en-1-yl)-4-methoxybenzene (2c)
Compound 2c was prepared according to the general procedure and obtained in a 43% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (600 MHz, CDCl3) δ 7.28 (dd, J = 15.8, 8.4 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.17 (d, J = 7.5 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.2 Hz, 2H), 5.64 (s, 1H), 4.10 (t, J = 7.3 Hz, 1H), 3.78 (s, 3H), 3.76 (t, J = 7.4 Hz, 1H), 3.44 (dd, J = 11.6, 7.1 Hz, 1H), 3.40 – 3.20 (m, 3H), 2.82 (td, J = 11.2, 6.3 Hz, 1H), 2.61 (q, J = 10.4 Hz, 1H), 2.40 (dd, J = 18.2, 6.4 Hz, 1H), 2.22 (qd, J = 11.2, 6.9 Hz, 1H), 2.12 (ddt, J = 14.5, 10.7, 5.5 Hz, 1H); 13C NMR: (151 MHz, CDCl3) δ 158.1, 140.1, 139.4, 136.7, 128.8, 128.3, 127.6, 126.1, 120.3, 113.9, 77.2, 76.7, 71.28, 70.62, 55.2, 48.6, 45.4, 43.6, 42.6, 39.9; MS (GC-MS) Calculated m/z = 320.43, Found m/z = 320.20; IR (thin film): 3082, 3060, 3027, 3000, 2932, 2908, 2836, 1652, 1607, 1577, 1511, 1495, 1453, 1441, 1420
4.4.4. 1-((E)-3-(((E)-4-methylpenta-2,4-dien-1-yl)oxy)prop-1-en-1-yl)-4-methoxybenzene (2d)
Compound 2d was synthesized according to the following alteration to the general procedure: The 4-OMe cinnamyl alcohol was dissolved in THF (0.2 M) in a flame-dried round bottom flask and kept under positive pressure of N2. The solution was cooled to 0 °C in a dry ice/acetone batch. n-BuLi (2.5 M in hexane, 1.1 equiv.) added dropwise via syringe. After stirring for ~5 minutes, MsCl (1.2 equiv.) was added dropwise via syringe, and solution then stirred for an additional 30 minutes. At the same time, a separate round bottom flask was charged with LiBr (4.5 equiv.), 4 Å molecular sieves, and THF. The solution was stirred for 15 minutes and transferred slowly via cannula into the reaction mixture. Reaction continued stirring at 0 °C for 30 minutes, then the bath was removed and the reaction continued to stir at room temperature for 1.5 hours. The mixture was quenched by the addition of saturated aqueous ammonium chloride, extracted 3× with Et2O, dried over Na2SO4, and concentrated under reduced pressure. The crude product was dissolved in dry THF (0.2 M) and stirred for 1 hour over 4 Å molecular sieves. Meanwhile, s15 (1.5 equiv) was added in small portions to a stirred solution of NaH (1.6 equiv, 60% dispersion in mineral oil) in dry THF (0.2 M) at 0 °C. The mixture was stirred for 15 minutes, then the solution containing the alkyl bromide was transferred via cannula over several minutes. The reaction was allowed to warm to room temperature and stirred for 16 hours. The reaction was quenched by the slow addition of saturated aqueous ammonium chloride and extracted 3× with Et2O. Combined organic layers were dried over Na2SO4 and concentrated. Products were purified by column chromatography using 10% EtOAc/Hexanes, affording the final product as a viscous yellow oil (34%).
; 1H NMR: (600 MHz, CDCl3) δ 7.16 – 7.11 (m, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.56 (s, 1H), 4.08 (t, J = 7.2 Hz, 1H), 3.79 (s, 3H), 3.76 (t, J = 7.4 Hz, 1H), 3.41 (dd, J = 11.6, 7.0 Hz, 1H), 3.31 (dd, J = 11.2, 7.7 Hz, 1H), 2.85 (td, J = 11.2, 6.3 Hz, 1H), 2.56 (d, J = 9.9 Hz, 1H), 2.40 (dd, J = 18.1, 6.5 Hz, 1H), 2.19 (m, 2H), 1.70 (s, 3H);13C NMR: (151 MHz, CDCl3) δ 158.1, 137.0, 136.8, 127.7, 118.8, 114.0, 71.4, 70.7, 55.2, 48.6, 45.5, 42.7, 41.9, 22.9; MS (GC-MS) Calculated m/z = 244.15, Found m/z = 244.20; IR (thin film): 3082, 3060, 3027, 3000, 2932, 2908, 2836, 1652, 1607, 1577, 1511, 1495, 1453, 1441, 1420
4.4.5. N-((E)-3-(4-methoxyphenyl)allyl)-4-methyl-N-((E)-5-methyl-4-methylenehex-2-en-1-yl)benzenesulfonamide (2e)
Compound 2e was prepared according to the general procedure and obtained in a 42% yield as a colorless solid after purification by column chromatography with silica gel and DCM; 1H NMR: (600 MHz, CDCl3) δ 7.72 – 7.68 (m, 2H), 7.33 (d, J = 7.9 Hz, 2H), 7.09 – 7.05 (m, 2H), 6.90 – 6.85 (m, 2H), 5.47 (s, 1H), 3.83 (s, 3H), 3.70 (dd, J = 9.1, 7.3 Hz, 1H), 3.33 (dd, J = 9.5, 6.8 Hz, 1H), 2.94 (dd, J = 11.8, 9.1 Hz, 1H), 2.75 (dd, J = 11.3, 9.5 Hz, 1H), 2.68 (td, J = 11.2, 6.2 Hz, 1H), 2.46 (s, 1H), 2.37 (ddt, J = 17.9, 6.2, 1.8 Hz, 1H), 2.30 – 2.22 (m, 1H), 2.17 (p, J = 6.9 Hz, 1H), 2.13 – 2.05 (m, 1H), 1.92 (qd, J = 11.3, 6.8 Hz, 1H), 0.99 (dd, J = 6.9, 3.8 Hz, 6H); 13C NMR: (151 MHz, CDCl3) δ 158.3, 146.7, 143.1, 136.0, 129.6, 127.6, 127.2, 116.1, 114.1, 55.2, 52.3, 51.3, 47.3, 43.8, 43.1, 37.3, 34.4, 21.5, 21.5, 21.3; MS (ESI) Calculated m/z for [M+H] = 426.2103, Found m/z for [M+H] = 426.2082
Supplementary Material
Acknowledgments
This project was supported by Award No. R01 GM098340 from the National Institute of General Medical Sciences and an Eli Lilly Grantee Award (DAN). We are grateful to the Lambert group for providing chiral acid precursor for the synthesis of catalyst 1e.
Footnotes
Supplementary material related to this article can be found at ….
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For recent reviews on photoredox catalysis see: Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r.Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449.
- 2.a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shih HW, Vander Wal MN, Grange RL, MacMillan DWC. J Am Chem Soc. 2010;132:13600–13603. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Welin ER, Warkentin AA, Conrad JC, MacMillan DWC. Angew Chem Int Ed. 2015;54:9668–9672. doi: 10.1002/anie.201503789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Du J, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392–397. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amador AG, Sherbrook EM, Yoon TP. J Am Chem Soc. 2016;138:4722–4725. doi: 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Huo H, Shen X, Wang C, Zhang L, Röse P, Chen LA, Harms K, Marsch M, Hilt G, Meggers E. Nature. 2014;515:100–103. doi: 10.1038/nature13892. [DOI] [PubMed] [Google Scholar]; b) Huo H, Huang X, Shen X, Harms K, Meggers E. Synlett. 2016;27:749–753. [Google Scholar]; c) Huang X, Webster RD, Harms K, Meggers E. J Am Chem Soc. 2016;138:12636–12642. doi: 10.1021/jacs.6b07692. [DOI] [PubMed] [Google Scholar]
- 5.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. J Am Chem Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- 6.Emmanuel M, Greenberg N, Oblinsky D, Hyster T. Nature. 2016;540:414–417. doi: 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]
- 7.Bauld NL, Bellville DJ, Harirchian B, Lorenz KT, Pabon RA, Reynolds DW, Wirth DD, Chiou HS, Marsh BK. Acc Chem Res. 1987;20:371–378. [Google Scholar]
- 8.a) Lin J, Padilla CE, Ischay MA, Yoon TP. Tetrahedron Lett. 2012;53:3073–3076. doi: 10.1016/j.tetlet.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stevenson SM, Higgins RF, Shores MP, Ferreira EM. Chem Sci. 2017;8:654–660. doi: 10.1039/c6sc03303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenz KT, Bauld NL. J Am Chem Soc. 1987;109:1157–1160. [Google Scholar]
- 10.Gieseler A, Steckhan E, Wiest O, Knock F. J Org Chem. 1991;56:1405–1411. [Google Scholar]
- 11.Kim J-I, Schuster GB. J Am Chem Soc. 1990;112:9635–9637. [Google Scholar]
- 12.Yang Z, Li H, Li S, Zhang M-T, Luo S. Org Chem Front. 2017;4:1037–1041. [Google Scholar]
- 13.Parmar D, Sugiono E, Raja S, Rueping M. Chem Rev. 2014;114:9047–9153. doi: 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]
- 14.Mahlau M, List B. Angew Chem Int Ed. 2013;52:518–533. doi: 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- 15.Gheewala CD, Collins BE, Lambert TH. Science. 2016;351:961–965. doi: 10.1126/science.aad0591. [DOI] [PubMed] [Google Scholar]
- 16.Macchioni A. Chem Rev. 2005;105:2039–2074. doi: 10.1021/cr0300439. [DOI] [PubMed] [Google Scholar]
- 17.Lide DR, editor. CRC Handbook of Chemistry and Physics. 90. CRC Press; Boca Raton, FL: 2009. Section 6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.