

Abstract
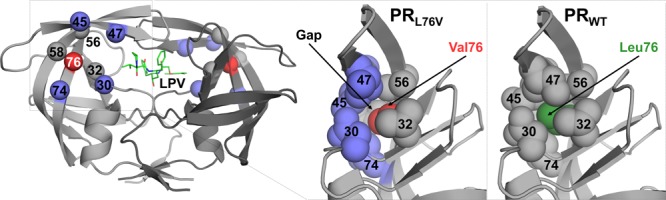
Four HIV-1 protease (PR) inhibitors, clinical inhibitors lopinavir and tipranavir, and two investigational compounds 4 and 5, were studied for their effect on the structure and activity of PR with drug-resistant mutation L76V (PRL76V). Compound 5 exhibited the best Ki value of 1.9 nM for PRL76V, whereas the other three inhibitors had Ki values of 4.5–7.6 nM, 2–3 orders of magnitude worse than for wild-type enzymes. Crystal structures showed only minor differences in interactions of inhibitors with PRL76V compared to wild-type complexes. The shorter side chain of Val76 in the mutant lost hydrophobic interactions with Lys45 and Ile47 in the flap, and with Asp30 and Thr74 in the protein core, consistent with decreased stability. Inhibitors forming additional polar interactions with the flaps or dimer interface of PRL76V were unable to compensate for the decrease in internal hydrophobic contacts. These structures provide insights for inhibitor design.
Introduction
The protease (PR) encoded by the human immunodeficiency virus (HIV) is an important drug target for treatment of the pandemic disease HIV/AIDS. A decrease in AIDS-associated deaths was observed in the mid-90s because of the inclusion of both PR inhibitors (PIs) with reverse transcriptase inhibitors in therapy.1 Despite this notable success, the rapid evolution of drug-resistant viral strains poses a critical challenge, and drug-resistant mutations have been observed for all classes of antiviral drugs.2 HIV PR performs an essential role in viral replication by processing the viral precursor polyproteins into mature viral proteins. Inhibitors bind in the active-site cavity of dimeric HIV PR and block its catalytic activity. More than 100 mutations in the PR gene have been associated with drug resistance.3
Second-generation inhibitors, such as darunavir (1), lopinavir (2), and tipranavir (3) (Figure 1), were designed to target resistant variants of HIV-1 PR. The peptidomimetic inhibitor 2 resembles the natural PR substrate with P2–P3′ groups.4,5 Inhibitor 3 is a highly potent, nonpeptidic PI with a unique oxygen that displaces conserved active-site water and forms direct hydrogen bonds with the main chain amides of Ile50/Ile50′.6,7 The most-recently approved inhibitor, 1, exhibits low toxicity and is equipped with P2/P2′ groups that form strong hydrogen bonds with conserved, main chain atoms, resulting in high binding affinity for PR.8−10 Because of these favorable factors, resistance mutations rarely develop during treatment with 1.11
Figure 1.

Structures of HIV-1 PIs 1–6.
The evolution of resistance toward second-generation inhibitors has fueled the design of novel investigational inhibitors. Compounds GRL-0519 (4) and GRL-5010 (5), derived from the scaffold of 1, are highly potent against several drug-resistant variants (Figure 1). Inhibitor 4 has an enlarged tristetrahydrofuran P2 group, which fits better in the S2 pocket of PR and reinforces a water-mediated network at the dimer interface.12−14 Inhibitor 5 differs from 1 by addition of a gem-difluoro moiety on the P2 bis-THF group, which improves lipophilicity and forms halogen bond interactions with the carbonyl oxygen of flap residue Gly48.15−17
Mutation L76V is associated with clinical resistance to 1, fosamprenavir, 2, and indinavir;2,18 however, it is also linked with increased susceptibility to first-generation inhibitor saquinavir (6), atazanavir, and 3.19,20 This mutation occurs with a frequency of around 3% in PI-experienced patients20,21 and can be transmitted to treatment-naive patients.22,23 Inclusion of L76V in mutants bearing three other resistance mutations is associated with two- and eightfold increase in resistance to 1 and 2, respectively, and an eightfold increase in susceptibility to 6.20
Previous studies of PR with the single mutation of L76V (PRL76V) showed about 100-fold worse inhibition by 1 compared to wild-type PR (PRWT) as assessed by isothermal titration calorimetry,24 although another group using an enzyme inhibition assay reported only 1.5-fold loss in potency.25 Investigational inhibitor GRL-02031 showed a twofold increase in inhibition constant (Ki) for PRL76V in comparison to PRWT.26 Crystal structures of PRL76V in complexes with 1 and 6 showed loss of interactions with 1 and gain of a water-mediated interaction with 6 relative to PRWT,24,25 consistent with the effects on resistance.20 These effects on inhibitors must be indirect as the side chain of Leu76 lies in the interior of the PR dimer and has no van der Waals contacts with these antiviral inhibitors. In the mutant structure, the smaller Val76 side chain has lost hydrophobic interactions with neighboring side chains of Asp30, Lys45, Ile47, and Thr74 compared with those of the wild-type Leu76, consistent with decreased stability and slower autoprocessing observed for the mutant and its precursor.24,26
Here, we have assessed the effect of four antiviral inhibitors, clinical inhibitors 2 and 3 and investigational inhibitors 4 and 5, on the structure and activity of the PRL76V mutant. Clinical inhibitor 2 was selected because L76V is associated with an eightfold increase in resistance to this PI as inferred from genotype–phenotype data.20 Fluorine-containing inhibitors, 3 and 5, form direct interactions with flap residues of the PR,7,16 potentially stabilizing the PR dimer. The larger P2 group and reinforced dimer interface interactions of compound 4 also might overcome the decreased dimer stability observed for PRL76V.12,24 The results show that these chemically diverse inhibitors lose potency against PRL76V, and suggest that local rearrangements in the hydrophobic core because of mutation L76V act to decrease the effectiveness of the inhibitors.
Results
Tested Inhibitors Have Higher Ki Values for PRL76V Relative to PRWT
Inhibition constants (Ki) of the compounds for PRL76V were determined using a fluorescent substrate analog of the HIV-1 p2/NC cleavage site. Table 1 lists Ki values for the mutant in comparison with values reported previously for wild-type enzymes.7,12,16 As reported previously, inhibitor 1 has the best inhibition of this mutant,24 whereas inhibitor 6 retains a similar inhibition of mutant relative to wild-type enzymes.27,28 Compound 5 is the most potent of the new inhibitors for PRL76V with a Ki of 1.9 ± 0.7 nM, whereas 3 and 4 are the worst of the tested inhibitors with Ki of 7.6 ± 0.3 and 7.2 ± 1.4 nM, respectively. Therefore, the tested inhibitors are within a 10-fold difference in potency from each other for PRL76V inhibition. With the exception of 6, all Ki values measured for inhibition of PRL76V were significantly higher than the picomolar Ki values reported for wild-type PR with changes of 1200-fold for 4, 400-fold for 3, 300-fold for 5, 150-fold for 2, and 80-fold for 1.7,12,16 These higher Ki values imply that L76V confers resistance toward the four tested inhibitors.
Table 1. Inhibition Constants for PRL76V and Wild Type PR.
Structures of the PRL76V Dimer with Inhibitor Resemble Each Other as Well as Their PRWT Counterparts
Crystal structures of PRWT complexed with PIs 2 and 3, and of PRL76V complexed with each of the four inhibitors, were determined at high resolution to identify any structural changes because of the single mutation (Table 2). The dimer of PRL76V with 2 and the location of residue 76 are illustrated in Figure 2A. All PRL76V structures were solved in the P21212 space group with one dimer in the asymmetric unit, as were PRWT complexes with inhibitors 2 and 3 in the present study and in previous studies [PRWT complexes with 4 and 5 at 1.27 Å (PDB ID 3OK9) and 1.30 Å (PDB ID 4U8W), respectively].12,16
Table 2. Crystallographic Data Collection and Refinement Statisticsa.
| PRWT–2 | PRL76V–2 | PRWT–3 | PRL76V–3 | PRL76V–4 | PRL76V–5 | |
|---|---|---|---|---|---|---|
| space group | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 |
| Cell Dimensions | ||||||
| a (Å) | 59.73 | 60.01 | 58.30 | 58.60 | 58.67 | 58.26 |
| b (Å) | 85.20 | 85.38 | 86.26 | 86.40 | 86.34 | 86.05 |
| c (Å) | 46.27 | 46.43 | 46.01 | 46.16 | 46.00 | 46.25 |
| resolution range (Å) | 50.00–1.26 | 50.00–1.36 | 50.00–1.20 | 50.00–1.47 | 50.00–1.75 | 50.00–1.31 |
| unique reflections | 64 844 | 47 855 | 71 617 | 38 910 | 24 343 | 55 212 |
| redundancy | 3.9 (3.2) | 6.1 (2.4) | 5.2 (3.2) | 4.0 (3.7) | 4.8 (4.9) | 4.4 (1.8) |
| completeness | 98.9 (92.0) | 91.9 (53.9) | 97.6 (85.4) | 99.6 (98.9) | 100.0 (100.0) | 96.0 (77.6) |
| I/sigma | 13.7 (2.1) | 30.8 (3.3) | 15.9 (2.0) | 11.5 (3.4) | 21.7 (4.2) | 13.3 (2.1) |
| Rsym (%) | 6.7 (49.5) | 4.4 (31.0) | 9.7 (44.4) | 8.8 (49.9) | 6.6 (40.5) | 8.7 (45.4) |
| refinement resolution range (Å) | 50.00–1.26 | 50.00–1.36 | 50.00–1.20 | 50.00–1.47 | 50.00–1.75 | 50.00–1.31 |
| R (%) | 17.3 | 15.1 | 15.4 | 16.3 | 19.8 | 15.8 |
| Rfree (%) | 20.8 | 19.6 | 18.7 | 21.4 | 24.3 | 20.5 |
| number of solvent molecules | 195 | 280 | 217 | 153 | 161 | 197 |
| Average B-Factor (Å2) | ||||||
| main chain | 15.6 | 14.2 | 12.5 | 14.5 | 16.1 | 21.2 |
| side chain | 21.9 | 19.7 | 17.2 | 20.7 | 22.3 | 26.7 |
| inhibitor | 12.9 | 15.8 | 13.4 | 16.9 | 11.0 | 18.1 |
| solvent | 26.0 | 26.5 | 25.6 | 32.5 | 26.6 | 32.3 |
| rms Deviation from Ideality | ||||||
| bond length (Å) | 0.012 | 0.011 | 0.014 | 0.010 | 0.006 | 0.012 |
| angles (Å) | 0.030 | 0.028 | 0.034 | 0.029 | 0.023 | 0.032 |
Values in parentheses are given for the highest resolution shell.
Figure 2.
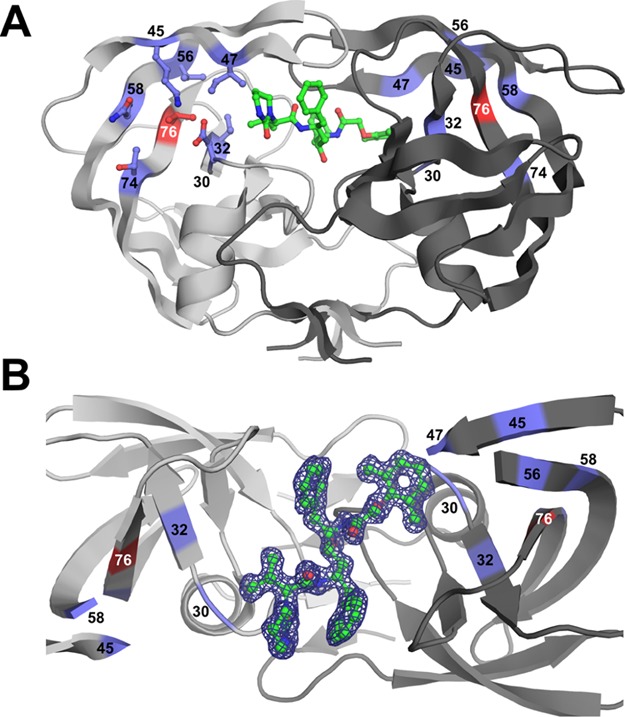
Structure of the PRL76V dimer with inhibitor 2. (A) Structure of the PRL76V dimer in complex with inhibitor 2. The two subunits are shown in light and dark gray ribbons. Inhibitor 2 is shown with green bonds. The side chain of Val76 is in red sticks; side chains of residues that interact with the Leu76 side chain are shown as purple sticks. Side chains were omitted on second subunit for clarity. (B) Electron density map for single conformation of inhibitor 2 (green sticks) bound in the PRL76V dimer. 2Fo – Fc electron density map contoured at the 1.0σ level is represented by the purple mesh. The view is rotated about 90° from (A). Flap residues 46–57 in subunit (A) and 48–54 in subunit (B) have been removed for clarity.
The unit cell dimensions were almost identical for all structures, although 2 complexes had ∼1 Å longer a axis and ∼1 Å shorter b axis compared to the other structures. The six structures were refined to R-factors of 15.1–19.8% with diffraction data at a 1.20–1.75 Å resolution. Atoms were unambiguously modeled, as shown by the electron density map in Figure 2B for the single conformation of 2 in complex with PRL76V. Coordinate errors estimated from Luzzati plots ranged from 0.14 to 0.18 Å for the highest to lowest resolution structures.
The new PRWT complexes with 2 and 3 were refined at near-atomic resolutions of 1.26 and 1.20 Å, respectively, which is a significant improvement compared to previously reported structures determined at 1.54 and 1.80 Å resolutions, respectively.7 The previous PRWT structures in complex with 2 (PDB ID 2O4S) or 3 (PDB ID 2O4P) were solved in the same space group, and the Cα atoms superimposed with the new higher resolution structures with root-mean-square deviation (rmsd) values of 0.19 Å for complexes with 3 and 0.20 Å for complexes with 2. Therefore, the overall folds are very similar.
The four new PRL76V inhibitor structures were superimposed with their corresponding PRWT inhibitor complexes. The overall backbone structures were essentially identical, with low rmsd values ranging from 0.12 to 0.17 Å for all Cα atoms. Therefore, mutation L76V does not produce major alterations in the overall structure of the PR dimer. Furthermore, all PRL76V structures were also very similar to each other, regardless of the inhibitor, with pairwise rmsd values ranging from 0.25 to 0.42 Å.
Polar Interactions are Conserved between Active-Site Residues and Inhibitors
Hydrogen bonds between PR and inhibitor, both direct and water-mediated, were compared for wild-type and mutant PR complexes. Halogen bonds were also observed for two inhibitors, 3 and 5, containing fluorine atoms. The interactions are described for the major conformations of inhibitors in the complexes. Similar interactions were observed for the minor conformations except where noted.
Hydrogen bond interactions in X-ray crystal structures of proteins must be interpreted with caution as hydrogen atoms are poorly scattered by X-rays. Neutron crystallography, however, provides direct evidence for the position of protons. The neutron crystal structure of HIV PR with amprenavir showed nonideal hydrogen bond geometry for inhibitor interactions with the carbonyl oxygen of Gly27, the amide of Asp29, and a water-mediated interaction with the amide of Ile50′.29 Similar effects were observed for the neutron structure of amprenavir complexed with PR mutant V32I/I47V/V82I.30 No neutron crystal structures have been reported for the inhibitor complexes in this study; hence, hydrogen bonds for L76V complexes are described by the same criteria as for previously published wild-type complexes (Figure 3).
Figure 3.

Polar interactions of PRL76V with inhibitors. (A) Inhibitor 2 (green); (B) inhibitor 3 (yellow); (C) inhibitor 4 (cyan); and (D) inhibitor 5 (salmon). PR residues are shown in gray sticks with alpha-carbons as spheres; nitrogen (blue), oxygen (red), fluorine (pale cyan), water (red spheres). Side chains without polar interactions with inhibitors are omitted. Hydrogen bond interactions conserved in wild-type and mutant PR are shown as black dashed lines. Interactions that do not form in the mutant are shown as red dashed lines. Halogen bonds are in green dotted lines. Interatomic distances are given in Å for PRL76V (black) and PRWT (red in parenthesis) if values differ by 0.3 Å or more. In (D), Asp30 and Asp30′ are shown in two alternate conformations, and the interactions of Asp30′ with P2′ group of inhibitors differ for the alternate conformations. Distances for the (B) conformation of Asp30′ are shown after a forward slash.
All clinical inhibitors contain a central hydroxyl group that interacts with the catalytic aspartates, residues 25 and 25′, of PR. The protonation state and hydrogen bond interactions of the catalytic residues, Asp25 and 25′, have been examined in several neutron structures. The side chain carboxylate oxygens of Asp25 and 25′ are almost coplanar with about 2.7 Å between the closest “inner” oxygens of the two side chains. Our neutron structure of wild-type HIV PR with amprenavir showed two protons located on the inner carboxylate oxygen of Asp25 and the outer carboxylate oxygen of Asp25′, which formed hydrogen bonds with the inhibitor hydroxyl.29 Subsequent neutron studies of 1 and amprenavir complexes with a mutant PR demonstrated that the location of the two protons varies, depending on the pH, inhibitor, and mutated residues.30,31 In our X-ray structures of inhibitor-bound PRL76V, we cannot distinguish which hydrogen bond interactions occur with the inhibitor hydroxyl group. In the absence of neutron structures corresponding to the PR complexes with the inhibitors described here, we have indicated distances for four possible interactions between the hydroxyl oxygen of the inhibitor and carboxylate oxygens of Asp25 and 25′ in Figure 3. These interactions are excluded from the description of inhibitor–PR hydrogen bonds in the following sections.
Although inhibitor 2 occurs in two alternative conformations with relative occupancy of 75:25 in the wild-type complex, only a single conformation was observed for 2 bound to PRL76V. Excluding interactions with the catalytic Asp25 and 25′, compound 2 shows only three direct hydrogen bonds with PRL76V and three water-mediated interactions with main chain atoms (Figure 3A). The interactions are in agreement with those reported previously.7 Compound 2 has a pseudosymmetric structure for the central P1–P1′ region. The water-mediated interactions with the amides of Ile50 and 50′ in the flaps are conserved in the majority of PR–inhibitor complexes. The pyrimidine acetamide group of 2 forms hydrogen bonds with the main chain amide and carboxylate side chain of Asp29 and a water-mediated interaction with the carbonyl oxygen of Gly27. These hydrogen bonds contribute to a network of interactions at the dimer interface connecting Gly27 and Asp29 in one subunit with Arg8′ in the second subunit. The hydrophobic dimethylphenoxy group on the opposite end of 2 has van der Waals interactions with the side chains of Ala28′, Asp29′, Asp30′, Val32′, Ile47′ and Ile84′. Inhibitor 2 shows highly conserved hydrogen bond interactions in the complexes with PRWT and PRL76V with differences in hydrogen bond length of no more than 0.2 Å. Therefore, the decreased potency of 2 toward PRL76V cannot be explained by changes in PR interactions with the inhibitor.
The distinctive features of the interactions of compound 3 with PR are the presence of direct hydrogen bonds of the inhibitor with the amides of flap residues Ile50 and Ile50′, as well as fluoride halogen bonds with the guanidinium side chain group of Arg8′. None of the other clinical inhibitors can form these interactions. Furthermore, 3 adopts a bent conformation at the sulfonamide group, and does not bind in the same pockets as more peptidic inhibitors. As shown in Figure 3B, inhibitor 3 has five direct hydrogen bonds with the PR, two halogen interactions with the guanidinium side chain of Arg8′, and a water-mediated interaction with the amide of Gly48. The carboxylate groups of the catalytic Asp25 and Asp25′ are rotated in the 3 complex relative to their orientation in the complexes with other inhibitors and show altered distances to the hydroxyl of 3. Inhibitor 3 retains wild-type hydrogen-bonding interactions with the PRL76V mutant, with 0.1 Å difference in distance observed for most interactions. The largest increases of 0.2 and 0.3 Å in the mutant are seen for the hydrogen bond to the carbonyl oxygen of Gly48 and the water-mediated interaction with the amide of Gly48, respectively. Thus, 3 forms direct hydrogen bonds with the main chain atoms of flap residues, Gly48, Ile50, and Ile50′ in both wild-type and mutant PRs. Inhibitor 3 shows two alternative conformations in both complexes with 70:30 relative occupancy; the minor conformation with 30% occupancy loses a hydrogen bond to the amide of Asp29 in the mutant structure. The conservation of 3 interactions with PRL76V and wild-type enzyme implies that the poor Ki value for the mutant does not arise from altered binding interactions.
In both the PRWT and mutant complexes, compound 4 binds in two alternative conformations with 50:50 relative occupancy. Inhibitor 4 forms five direct hydrogen bonds with PR and three water-mediated interactions in both mutant and wild-type complexes (Figure 3C). In comparison to the wild-type complex, both conformations of 4 in the mutant show a slight shift of the water interacting with the amides of Ile50 and Ile50′, yet this change maintains the interactions with the flaps. A second water, which is highly conserved in many PR structures and was mentioned earlier in the description for complexes with 2, is integrated into a dimer-stabilizing network of interactions that coordinates the tris-THF rings of 4, the carbonyl oxygen of Gly27, and the side chains of Asp29 and Arg8′. Compared to the wild-type complex, most hydrogen bond interactions between inhibitors and PRL76V have insignificant changes of less than 0.1 Å. Greater variation is observed for the interactions with the flap water with differences of up to 0.4 Å in length. One alternate conformation in both wild-type and mutant structures shows longer 3.4–3.5 Å hydrogen bonds between the tris-THF group of 4 and the amides of Asp29 and Asp30. Once again, the lack of significant differences in PR active-site interactions with 4 indicates that the resistance mechanism induced by L76V relies on an alternate strategy.
Compound 5 crystallized in two alternate conformations in PRWT as well as in PRL76V with 55:45 relative occupancy. Overall, PRWT shows seven direct hydrogen bond interactions, two halogen bond interactions, and three water-mediated interactions with the major conformation of 5 (Figure 3D). The majority of the direct hydrogen bond interactions of PRL76V with inhibitors are identical in length or within a 0.1 Å range from those observed in the wild-type complex. Asp30 and Asp30′ occur in two alternate conformations, resulting in differences in the interactions of the P2 aniline group of inhibitors with Asp30′ in the PRL76V structure. In the wild-type complex, the amino group of P2 forms hydrogen bond interactions with the side chain carboxylate of Asp30′ and with the main chain amide and carbonyl oxygen. In the PRL76V structure, the P2 aniline has shifted slightly relative to its position in the wild-type complex. In the major conformation of Asp30′ in the mutant, the carboxylate side chain is positioned to form a shorter hydrogen bond interaction with the aniline amino group relative to the wild-type complex, whereas the hydrogen bond of the main chain amide of Asp30′ with the inhibitor NH2 is elongated to 3.5 Å, and the interaction of the main chain carbonyl oxygen with inhibitor is lost (interatomic distance of 4.4 Å). Similar shifts in P2 aniline and altered interactions with Asp30′ were reported for the PRL76V complex with 1.24 This major conformation of Asp30′ is stabilized by an ionic interaction with the side chain of Lys45′. The minor conformation of the Asp30′ side chain has rotated away from the inhibitor; however, the main chain amide and carbonyl oxygen form hydrogen bonds of 3.0 and 3.3 Å, respectively, with NH2 of inhibitors. Although 5 is the most effective of the four tested inhibitors for PRL76V, it did not retain the picomolar inhibition reported for wild-type enzymes. Overall, the interactions of the major conformation of 5 with Asp30′ are altered in the mutant; however, the rest of the hydrogen-bonding network is maintained. Therefore, the loss in potency against the mutant is not completely explained by interactions between inhibitors and PR.
Polar and hydrophobic interactions of inhibitors 1–6 with mutant PRL76V and wild-type enzyme are summarized in Table 3. Hydrogen bond interactions include direct inhibitor–protein interactions and water-mediated interactions and showed little change for alternate conformations of inhibitors. The count of van der Waals contacts is complicated by the existence of alternate conformations of inhibitors, and frequently for adjacent amino acid side chains or main chains. The exact contacts may be impossible to determine when alternate conformations show 50:50 relative occupancy and, in fact, multiple conformations will contribute to the ensemble present in solution. The binding affinities of clinical inhibitors for wild-type PR have been divided into enthalpic and entropic components using isothermal scanning calorimetry.7,28 The binding of inhibitors 2, 3, and 6 is dominated by the large entropic component, and only inhibitor 1 showed enthalpically driven binding to wild-type PR. This thermodynamic analysis implies that in most cases inhibition cannot easily be assessed by summing inhibitor–PR interactions. In fact, thermodynamic dissection of inhibitor affinity for mutant PRs showed unfavorable changes in both entropic and enthalpic components.7 Inhibitor 1 loses hydrogen bond interactions with mutant PRL76V, countered by a small increase in hydrophobic contacts, and worse inhibition of the mutant. Inhibitor 6 showed gains in hydrogen bond and hydrophobic interactions with this mutant in agreement with insignificant differences in inhibition compared to wild-type enzymes. Apart from inhibitor 2, the other inhibitors showed fewer hydrophobic contacts with mutants as well as worse inhibition values.
Table 3. Summary of Inhibitor–PR Interactionsa.
| inhibitor | MW | PRWT H-bondb | PRL76V H-bondb | PRWT vdWc | PRL76V vdWc |
|---|---|---|---|---|---|
| 1 | 548 | 7 + 2 | 5 + 2 | 136(140) | 152(157) |
| 6 | 671 | 4 + 3 | 5 + 4 | 141(138) | 173(173) |
| 5 | 584 | 7 + 3 | 6 + 3 | 171(146) | 150(137) |
| 2 | 629 | 3 + 3 | 3 + 3 | 184(151) | 191 |
| 4 | 605 | 5 + 3 | 5 + 3 | 170(173) | 155(163) |
| 3 | 603 | 5 + 1 | 5 + 1 | 161(161) | 157(147) |
Compounds are listed in order of best to worst inhibition of mutant.
Hydrogen bond interactions are indicated as direct + water-mediated.
van der Waals contacts are shown for major inhibitor conformation with number for minor conformation in parentheses.
Hydrophobic Interactions of Leu76 are Decreased in the PRL76V Mutant
Residue 76 occupies a region critical for internal hydrophobic contacts between the flap and the core of the protein. Residue 76 lies in the central strand of a three-stranded β-sheet forming one flank of the substrate binding cavity near the base of the flap (Figure 2). The mobility of the flaps is essential for substrate binding and catalysis as the flaps act as lids over the active-site cavity and must open to allow substrate entry and release of products.32 Consequently, altered flap dynamics has been reported for drug-resistant mutants.25,27,33,34
In all the structures of PRWT and PRL76V with various inhibitors, the main chain of residue 76 forms conserved hydrogen bond interactions with adjacent strands of the β-sheet comprising residues 31–33 and 57–59. In addition, the side chain of residue 76 forms hydrophobic interactions with the side chains of Val32, Val56, and Gln58 (Figure 4). Furthermore, the wild-type residue, Leu76, forms van der Waals interactions with Asp30, Thr74, and with the side chains of Lys45 and Ile47 in the first β-strand of the two-stranded flap (Figure 4A). These interactions of Leu76 with residues 30–33 and both strands of the flap are conserved in the open conformation of the PR dimer (PDB ID 2PC0).35 However, in the PRL76V mutant, the shorter side chain of Val76 loses hydrophobic contacts with the first β-strand of the flaps, and additionally loses interactions with Asp30 and Thr74 (Figure 4B). These changes agree with those reported previously for PRL76V complexes with 1 and 6.24
Figure 4.
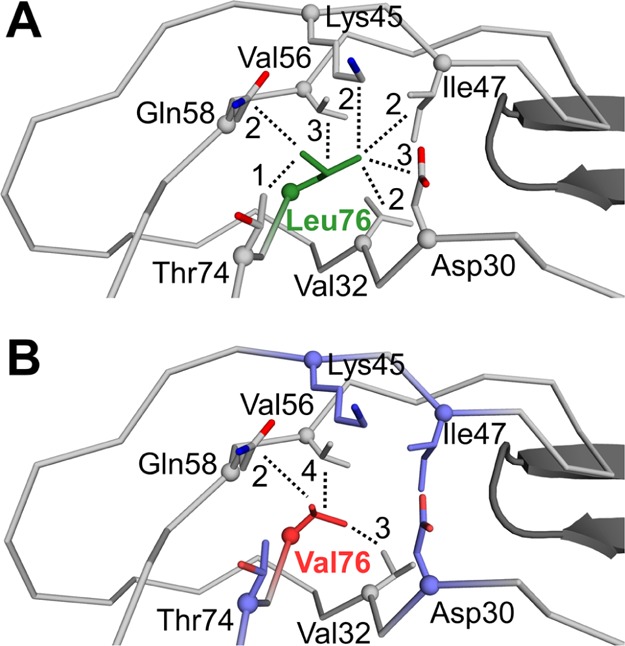
Hydrophobic interactions of residue 76. Hydrophobic interactions are shown for inhibitor 3 complexes with (A) PRWT and (B) PRL76V. The main chain of PR is shown as gray ribbons. The side chains are represented by sticks with Cα atoms as spheres. The tip of the flap in the second subunit of the dimer is shown as black cartoons. Leu76 is green, Val76 is red, and side chains of residues that lose interactions in the mutant are shown in purple in (B). The number of van der Waals contacts of the side chain of residue 76 with neighboring residues is indicated next to each dotted line.
The flap–core interface is composed of residues Lys45, Ile47, Ile54, Val56, and Gln58 in the flap, residues Asp30 and Val32 in the inhibitor binding site, and Thr74 and Leu76 in the protein core. The side chains of these residues form hydrophobic interactions and shield the interface from the solvent. Leu76 is a central component of the flap–core interface and provides extensive hydrophobic interactions connecting the flaps to the active site. The loss of interactions across the flap–core interface in the PRL76V mutant is expected to influence flap mobility and promote inhibitor dissociation from the dimer. This effect is consistent with the observations of increased dimer dissociation and decreased dimer stability.24 Increased conformational flexibility of the flaps in the PRL76V mutant compared to the wild-type enzyme was reported in 1 ns molecular dynamics simulations of the complexes with inhibitor 2 consistent with increased calculated interaction free energy and clinical resistance.36
Discussion
Drug-resistant mutation L76V is rare in clinical isolates of HIV; however, this mutation alone acts to decrease the stability of the PR dimer, alters precursor processing, and reduces viral fitness.20,24,37 In this study, we examined the effect of four antiviral inhibitors, two clinical drugs and two investigational inhibitors, on the single mutant PRL76V. These inhibitors exhibit different chemical structures and binding interactions with the PR.
The tested inhibitors have Ki values of ∼2–8 nM for PRL76V, which are 2–3 orders of magnitude worse compared to values reported for the wild-type enzyme.7,12,16 None of these inhibitors were more effective than 1 for the mutant PRL76V. Inhibition of PRL76V by 1 has been reported as 0.79 nM24 or 1.5-fold worse25 than for wild-type enzymes in different assays with two distinct substrates. GRL-02031, a different antiviral inhibitor based on the 1 scaffold, also gave an inhibition constant of 0.8 nM for PRL76V, consistent with decreased interactions of the P1′ pyrrolidinone group with PR atoms.26 In contrast to other tested inhibitors, 6 exhibited similar inhibition of the PRL76V mutant compared to wild-type enzymes, consistent with retaining antiviral effectiveness.19,20,24 In the current study, 5 was the better of the two investigational inhibitors with Ki of 1.9 nM for PRL76V, possibly due to the two halide interactions with the carbonyl oxygen of Gly48 in the flap. Compounds 3 and 4 showed the worst inhibition, which suggests neither the larger tris-THF group at P2 in 4 nor the direct interactions of 3 with the flaps are beneficial for binding to this mutant.
The structures of PRL76V–inhibitor complexes are very similar to the corresponding wild-type enzyme structures. Only inhibitor 5, which was the best inhibitor of the four for PRL76V, showed distinct changes in the interactions of the aniline amine with alternate conformations of Asp30′. Elongations of water-mediated bonds by up to 0.3 and 0.4 Å, respectively, were observed in complexes 3 and 4. The active-site interactions with 2 were essentially identical in both the wild-type and the mutant. Fewer van der Waals contacts occurred with the mutant compared to wild-type enzymes for inhibitors 3, 4, and 5, whereas inhibitors 1, 2, and 6 showed the opposite effect. Therefore, the mechanism of resistance does not seem to rely solely on the loss of active-site interactions with inhibitors. Mutation L76V is not unusual in this respect; mutations L90M and N88D/S also have no direct interactions with inhibitors, yet are strongly associated with resistance to one or more clinical inhibitors.2,38
The rarity of L76V as a single mutation in clinical samples, at 0.4%,20 may be due to its debilitating effects on precursor processing. Furthermore, the slower turnover of substrate and poor inhibition may arise from alterations in the dynamics of flap opening and closing rather than altered interactions with inhibitors. The mutation is likely to confer resistance by a mechanism that is independent of the inhibitor interactions in the active site. Instead, the loss of interactions of Val76 with residues at the base of the flap could increase flap mobility. If the virus accumulates additional mutations, as observed in 3.2% of clinical samples,20 these mutations might compensate for the loss of stability because of L76V, while retaining resistance to a specific drug.
Interestingly, five of the seven residues adjacent to Leu76 in the PR structure are sites of major resistance mutations, D30N, V32I, I47V, Q58E, and T74P2. Of these, only the resistance mutation Q58E is strongly associated with L76V in resistant mutants.2,39 Experimental studies corroborate findings from statistical analyses. Mutation L76V, which is associated with resistance to 1, is selected during viral passage with increasing concentrations of 1.40 In contrast, L76V is associated with increased susceptibility to 6 and atazanavir, and experiments suggest that L76V re-sensitizes multi-drug resistant viruses to therapy with those two inhibitors.19 Flap mutation M46I is strongly associated with L76V as shown by a large proportion of coprevalence in L76V-containing sequences.20,39,41 Impaired autoprocessing of precursor PR-containing L76V is partly rescued by addition of a second mutation of M46I24 and, although L76V reduces viral replication, viral fitness is partly rescued by combination with this mutation.37 The effects of combining L76V with other mutations on the structure and dynamics of the PR dimer have not yet been explored.
An inhibitor capable of forming strong interactions with residues 45–47 of the flaps might assist in retaining a closed conformation dimer, and be effective against resistant mutants with defects such as those observed for L76V. Therefore, this hydrophobic region between the flaps and the outer edge of the active site in each monomer is a potential target site that should be considered during the design of next-generation inhibitors for resistant viruses.
Experimental Section
Enzymes and Inhibitors
PRWT and PRL76V proteins contain optimizing mutations Q7K/L33I/L63I to reduce autoproteolysis and C67A/C95A to prevent thiol bond formation.42 Recombinant PR was expressed in Escherichia coli BL21 DE(3) and purified by size-exclusion chromatography followed by reverse phase chromatography as described previously.43 PR was refolded via buffer-exchange dialysis in 25 mM formic acid and 1 mM dithiothreitol. PR was activated via buffer-exchange dialysis in 50 mM sodium acetate, pH 5.0. PR was concentrated to 3.5–5.0 mg/mL for crystallization or further diluted for inhibition assays as needed. Inhibitors 2 and 3 with HPLC purity of 99.3 and 100%, respectively, were obtained from the AIDS Reagent Program, Division of AIDS, NIAID, NIH. Inhibitors 4 and 5 (>95% purity by HPLC) were provided by Dr. Arun Ghosh at Purdue University.
Enzyme Inhibition Assays
A continuous kinetic assay employing a Foster resonance energy transfer substrate analog of the HIV-1 p2/NC cleavage site (Abz–Thr–Ile–Nle–p-nitro-Phe–Gln–Arg–NH2 where Abz is anthranilic acid, Nle is norleucine, and p-nitro-Phe is p-nitrophenylalanine) was performed as previously described.26 Microtiter plates (96-well) were loaded with 10 μL PR stock (final well concentration 12–26 nM determined via active-site titration with tight binding inhibitor), 98 μL reaction buffer (0.1 M 2-morpholinoethanesulfonate (MES), pH 5.6, 0.4 M NaCl, 1 mM ethylenediaminetetraacetic acid, and 5% glycerol), and 2 μL inhibitor in dimethyl sulfoxide (DMSO) (final well concentration of 0–100 nM). Samples were equilibrated at 25 °C for 5 min and the reactions were measured at the same temperature. The reactions were initiated by addition of 90 μL substrate at a final well concentration of 81 μM. Reactions were measured under steady-state conditions using a PolarStar Optima (BMG Labtech) with emission wavelength at 340 nm and excitation wavelength at 420 nm. Fluorescence resulting from substrate hydrolysis at each inhibitor concentration [I] was measured and plotted against time. Initial velocities (V0), corresponding to the slope per minute of each reaction, were determined using MARS software (BMG Labtech). A dose–response plot of V0 versus [I] was constructed using SigmaPlot (Systat Software) by nonlinear regression curve fitting to determine IC50. Reactions were performed in triplicate, and Ki values were calculated from IC50 using the tight-binding inhibitor equation Ki = (IC50 – 0.5[E])/(1 + [S]/Km).44 The Km for PRL76V at 37 μM was determined previously under similar conditions.24
Protein Crystallization
Each inhibitor suspended in DMSO was complexed with PR on ice at a molar ratio of at least 5:1 and crystallized using vapor diffusion hanging drop method. Each drop contained equal volumes of protein and reservoir solution. Crystals grew in 0.8–1.5 M sodium chloride as precipitant. Crystals for most complexes grew in 0.1 M sodium acetate, pH 4.0–5.4, as buffer. Buffers used for crystallization for the following were exceptions: 0.1 M MES, pH 5.6, for PRWT in complex with 2, and 0.1 M citrate phosphate, pH 6.0, for PRL76V in complex with 2. Moreover, 5% DMSO was used for crystallization conditions of PRL76V complexes with 2 and 3. Crystals were soaked in reservoir solution with 30% glycerol (v/v) as cryoprotectant and then flash-frozen in liquid nitrogen.
X-ray Diffraction, Processing, and Refinement
X-ray diffraction data were collected remotely using Southeastern Regional Collaborative Access Team ID-22 and BM-22 beamlines of the Advanced Photon Source in Argonne National Laboratory (Argonne, IL). Diffraction data were indexed, integrated, and scaled using HKL-2000.45 Molecular replacement was performed using CCP4 Phaser(46,47) with PRWT in complex with amprenavir (PDB 3NU3) as the initial model. Structures were refined iteratively with Coot(48) and SHELX.49 The inhibitor and any side chains with incomplete 2Fo – Fc density were removed during the initial rounds of refinement to avoid bias, and atoms were added according to density in Fo – Fc omit maps. Alternate conformations were modeled if visible in the electron density maps for the inhibitor and protein residues. Anisotropic B factors were included in the last stages of refinement for all structures, except for the lowest resolution structure of PRL76V in complex with 4. Structures were analyzed using Coot, CCP4 Superpose, CCP4 Baverage, CCP4 Contact, and CCP4 Sfcheck. Illustrations were created using PyMol (Schrödinger, LLC.).
Crystallographic coordinate and structure factors have been deposited in the Protein Data Bank with accession codes 6DJ1 for PRWT–2, 6DJ2 for PRL76V–2, 6DIF for PRWT–3, 6DIL for PRL76V–3, 6DJ5 for PRL76V–4, and 6DJ7 for PRL76V–5. The authors will release the atomic coordinates and experimental data upon article publication.
Acknowledgments
We are grateful to Johnson Agniswamy and Daniel Kneller for valuable discussions. This research was supported in part by the National Institute of Health grants GM062920 (I.T.W., R.W.H.) and GM053386 (A.K.G.), an NIH diversity supplement (A.W.-S.), and a fellowship from the Molecular Basis of Disease Program of Georgia State University (A.W.-S.). We thank the staff at the Southeast Regional-Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38.
The authors declare no competing financial interest.
References
- Konvalinka J.; Kräusslich H.-G.; Müller B. Retroviral Proteases and Their Roles in Virion Maturation. Virology 2015, 479–480, 403–417. 10.1016/j.virol.2015.03.021. [DOI] [PubMed] [Google Scholar]
- Wensing A. M.; Calvez V.; Günthard H. F.; Johnson V. A.; Paredes R.; Pillay D.; Shafer R. W.; Richman D. D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2017, 24, 132–133. [PMC free article] [PubMed] [Google Scholar]
- Rhee S.-Y.; Sankaran K.; Varghese V.; Winters M. A.; Hurt C. B.; Eron J. J.; Parkin N.; Holmes S. P.; Holodniy M.; Shafer R. W. HIV-1 Protease, Reverse Transcriptase, and Integrase Variation. J. Virol. 2016, 90, 6058–6070. 10.1128/jvi.00495-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham H. L.; Kempf D. J.; Molla A.; Marsh K. C.; Kumar G. N.; Chen C. M.; Kati W.; Stewart K.; Lal R.; Hsu A.; et al. ABT-378, a Highly Potent Inhibitor of the Human Immunodeficiency Virus Protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll V.; Qin W.; Stewart K. D.; Jakob C.; Park C.; Walter K.; Simmer R. L.; Helfrich R.; Bussiere D.; Kao J.; et al. X-Ray Crystallographic Structure of ABT-378 (Lopinavir) Bound to HIV-1 Protease. Bioorg. Med. Chem. 2002, 10, 2803–2806. 10.1016/s0968-0896(02)00051-2. [DOI] [PubMed] [Google Scholar]
- Turner S. R.; Strohbach J. W.; Tommasi R. A.; Aristoff P. A.; Johnson P. D.; Skulnick H. I.; Dolak L. A.; Seest E. P.; Tomich P. K.; Bohanon M. J.; et al. Tipranavir (PNU-140690): A Potent, Orally Bioavailable Nonpeptidic HIV Protease Inhibitor of the 5,6-Dihydro-4-Hydroxy-2-Pyrone Sulfonamide Class. J. Med. Chem. 1998, 41, 3467–3476. 10.1021/jm9802158. [DOI] [PubMed] [Google Scholar]
- Muzammil S.; Armstrong A. A.; Kang L. W.; Jakalian A.; Bonneau P. R.; Schmelmer V.; Amzel L. M.; Freire E. Unique Thermodynamic Response of Tipranavir to Human Immunodeficiency Virus Type 1 Protease Drug Resistance Mutations. J. Virol. 2007, 81, 5144–5154. 10.1128/jvi.02706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin C.; Dejesus E.; Khanlou H.; Stoehr A.; Supparatpinyo K.; Lathouwers E.; Lefebvre E.; Opsomer M.; Van de Casteele T.; Tomaka F. Final 192-Week Efficacy and Safety of Once-Daily Darunavir/Ritonavir Compared with Lopinavir/Ritonavir in HIV-1-Infected Treatment-Naïve Patients in the ARTEMIS Trial. HIV Med. 2013, 14, 49–59. 10.1111/j.1468-1293.2012.01060.x. [DOI] [PubMed] [Google Scholar]
- Koh Y.; Nakata H.; Maeda K.; Ogata H.; Bilcer G.; Devasamudram T.; Kincaid J. F.; Boross P.; Wang Y.-W.; Tie Y.; et al. Novel Bis -Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor ( PI ) UIC-94017 ( TMC114 ) with Potent Activity against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob. Agents Chemother. 2003, 47, 3123–3129. 10.1128/aac.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Y.; Boross P. I.; Wang Y.-F.; Gaddis L.; Hussain A. K.; Leshchenko S.; Ghosh A. K.; Louis J. M.; Harrison R. W.; Weber I. T. High Resolution Crystal Structures of HIV-1 Protease with a Potent Non-Peptide Inhibitor (UIC-94017) Active against Multi-Drug-Resistant Clinical Strains. J. Mol. Biol. 2004, 338, 341–352. 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- Lathouwers E.; Wong E. Y.; Luo D.; Seyedkazemi S.; De Meyer S.; Brown K. HIV-1 Resistance Rarely Observed in Patients Using Darunavir Once-Daily Regimens across Clinical Studies. HIV Clin. Trials 2017, 18, 196–204. 10.1080/15284336.2017.1387690. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Xu C.-X.; Rao K. V.; Baldridge A.; Agniswamy J.; Wang Y.-F.; Weber I. T.; Aoki M.; Miguel S. G. P.; Amano M.; et al. Probing Multidrug-Resistance and Protein-Ligand Interactions with Oxatricyclic Designed Ligands in HIV-1 Protease Inhibitors. ChemMedChem 2010, 5, 1850–1854. 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Wang Y.-F.; Shen C.-H.; Agniswamy J.; Rao K. V.; Xu C.-X.; Ghosh A. K.; Harrison R. W.; Weber I. T. Novel P2 Tris-Tetrahydrofuran Group in Antiviral Compound 1 (GRL-0519) Fills the S2 Binding Pocket of Selected Mutants of HIV-1 Protease. J. Med. Chem. 2013, 56, 1074–1083. 10.1021/jm301519z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M.; Tojo Y.; Salcedo-Gómez P. M.; Campbell J. R.; Das D.; Aoki M.; Xu C.-X.; Rao K. V.; Ghosh A. K.; Mitsuya H. GRL-0519, a Novel Oxatricyclic Ligand-Containing Nonpeptidic HIV-1 Protease Inhibitor (PI), Potently Suppresses Replication of a Wide Spectrum of Multi-PI-Resistant HIV-1 Variants in Vitro. Antimicrob. Agents Chemother. 2013, 57, 2036–2046. 10.1128/aac.02189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez P. M. S.; Amano M.; Yashchuk S.; Mizuno A.; Das D.; Ghosh A. K.; Mitsuya H. GRL-04810 and GRL-05010, Difluoride-Containing Nonpeptidic HIV-1 Protease Inhibitors (PIs) That Inhibit the Replication of Multi-PI-Resistant HIV-1 in Vitro and Possess Favorable Lipophilicity That May Allow Blood-Brain Barrier Penetration. Antimicrob. Agents Chemother. 2013, 57, 6110–6121. 10.1128/aac.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Yashchuk S.; Mizuno A.; Chakraborty N.; Agniswamy J.; Wang Y.-F.; Aoki M.; Gomez P. M. S.; Amano M.; Weber I. T.; et al. Design of Gem-Difluoro-Bis-Tetrahydrofuran as P2 Ligand for HIV-1 Protease Inhibitors to Improve Brain Penetration: Synthesis, X-Ray Studies, and Biological Evaluation. ChemMedChem 2015, 10, 107–115. 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agniswamy J.; Louis J. M.; Shen C.-H.; Yashchuk S.; Ghosh A. K.; Weber I. T. Substituted Bis-THF Protease Inhibitors with Improved Potency against Highly Resistant Mature HIV-1 Protease PR20. J. Med. Chem. 2015, 58, 5088–5095. 10.1021/acs.jmedchem.5b00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S.-Y.; Taylor J.; Fessel W. J.; Kaufman D.; Towner W.; Troia P.; Ruane P.; Hellinger J.; Shirvani V.; Zolopa A.; et al. HIV-1 Protease Mutations and Protease Inhibitor Cross-Resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. 10.1128/aac.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann F.; Vachta J.; Ehret R.; Walter H.; Kaiser R.; Stürmer M.; Tappe A.; Däumer M.; Berg T.; Naeth G.; et al. The L76V Mutation in HIV-1 Protease Is Potentially Associated with Hypersusceptibility to Protease Inhibitors Atazanavir and Saquinavir: Is There a Clinical Advantage?. AIDS Res. Ther. 2011, 8, 7. 10.1186/1742-6405-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T. P.; Parkin N. T.; Stawiski E.; Pilot-Matias T.; Trinh R.; Kempf D. J.; Norton M. Prevalence, Mutation Patterns, and Effects on Protease Inhibitor Susceptibility of the L76V Mutation in HIV-1 Protease. Antimicrob. Agents Chemother. 2010, 54, 4903–4906. 10.1128/aac.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda E.; de Mendoza C.; Martin-Carbonero L.; Corral A.; Briz V.; Gonzalez-Lahoz J.; Soriano V. Prevalence of Darunavir Resistance Mutations in HIV-1-Infected Patients Failing Other Protease Inhibitors. J. Antimicrob. Chemother. 2007, 60, 885–888. 10.1093/jac/dkm276. [DOI] [PubMed] [Google Scholar]
- Sayan M.; Sargin F.; Inan D.; Sevgi D. Y.; Celikbas A. K.; Yasar K.; Kaptan F.; Kutlu S.; Fisgin N. T.; Inci A.; et al. HIV-1 Transmitted Drug Resistance Mutations in Newly Diagnosed Antiretroviral-Naive Patients in Turkey. AIDS Res. Hum. Retroviruses 2016, 32, 26–31. 10.1089/aid.2015.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Qian F.; Yuan T.; Xu W.; Zhu L.; Huang J.; Wang H.; Zhu Y.; Wang Y.; Li X.; et al. Drug Resistance Mutation Profiles of the Drug-Naïve and First-Line Regimen-Treated HIV-1-Infected Population of Suzhou, China. Virol. Sin. 2017, 32, 271–279. 10.1007/s12250-017-4002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis J. M.; Zhang Y.; Sayer J. M.; Wang Y.-F.; Harrison R. W.; Weber I. T. The L76V Drug Resistance Mutation Decreases the Dimer Stability and Rate of Autoprocessing of HIV-1 Protease by Reducing Internal Hydrophobic Contacts. Biochemistry 2011, 50, 4786–4795. 10.1021/bi200033z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragland D. A.; Nalivaika E. A.; Nalam M. N. L.; Prachanronarong K. L.; Cao H.; Bandaranayake R. M.; Cai Y.; Kurt-Yilmaz N.; Schiffer C. A. Drug Resistance Conferred by Mutations Outside the Active Site through Alterations in the Dynamic and Structural Ensemble of HIV-1 Protease. J. Am. Chem. Soc. 2014, 136, 11956–11963. 10.1021/ja504096m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-C. E.; Yu X.; Zhang Y.; Tie Y.; Wang Y.-F.; Yashchuk S.; Ghosh A. K.; Harrison R. W.; Weber I. T. Potent Antiviral HIV-1 Protease Inhibitor GRL-02031 Adapts to the Structures of Drug Resistant Mutants with Its P1′-Pyrrolidinone Ring. J. Med. Chem. 2012, 55, 3387–3397. 10.1021/jm300072d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agniswamy J.; Shen C.-H.; Aniana A.; Sayer J. M.; Louis J. M.; Weber I. T. HIV-1 Protease with 20 Mutations Exhibits Extreme Resistance to Clinical Inhibitors through Coordinated Structural Rearrangements. Biochemistry 2012, 51, 2819–2828. 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd M. J.; Luque I.; Velázquez-Campoy A.; Freire E. Thermodynamic Basis of Resistance to HIV-1 Protease Inhibition: Calorimetric Analysis of the V82F/I84V Active Site Resistant Mutant. Biochemistry 2000, 39, 11876–11883. 10.1021/bi001013s. [DOI] [PubMed] [Google Scholar]
- Weber I. T.; Waltman M. J.; Mustyakimov M.; Blakeley M. P.; Keen D. A.; Ghosh A. K.; Langan P.; Kovalevsky A. Y. Joint X-Ray/Neutron Crystallographic Study of HIV-1 Protease with Clinical Inhibitor Amprenavir: Insights for Drug Design. J. Med. Chem. 2013, 56, 5631–5635. 10.1021/jm400684f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O.; Keen D. A.; Blakeley M. P.; Louis J. M.; Weber I. T.; Kovalevsky A. Room Temperature Neutron Crystallography of Drug Resistant HIV-1 Protease Uncovers Limitations of X-Ray Structural Analysis at 100 K. J. Med. Chem. 2017, 60, 2018–2025. 10.1021/acs.jmedchem.6b01767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O.; Wymore T.; Das A.; Shen C.-H.; Parks J. M.; Smith J. C.; Weiss K. L.; Keen D. A.; Blakeley M. P.; Louis J. M.; et al. Long-Range Electrostatics-Induced Two-Proton Transfer Captured by Neutron Crystallography in an Enzyme Catalytic Site. Angew. Chem., Int. Ed. 2016, 55, 4924–4927. 10.1002/anie.201509989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh E.; Louis J. M.; Yamazaki T.; Gronenborn A. M.; Torchia D. A.; Ishima R. A Solution NMR Study of the Binding Kinetics and the Internal Dynamics of an HIV-1 Protease-Substrate Complex. Protein Sci. 2003, 12, 1376–1385. 10.1110/ps.0300703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agniswamy J.; Louis J. M.; Roche J.; Harrison R. W.; Weber I. T. Structural Studies of a Rationally Selected Multi-Drug Resistant HIV-1 Protease Reveal Synergistic Effect of Distal Mutations on Flap Dynamics. PLoS One 2016, 11, e0168616 10.1371/journal.pone.0168616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vera I. M. S.; Blackburn M. E.; Fanucci G. E. Correlating Conformational Shift Induction with Altered Inhibitor Potency in a Multidrug Resistant HIV-1 Protease Variant. Biochemistry 2012, 51, 7813–7815. 10.1021/bi301010z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslet H.; Rosenfeld R.; Giffin M.; Lin Y.-C.; Tam K.; Torbett B. E.; Elder J. H.; McRee D. E.; Stout C. D. Conformational Flexibility in the Flap Domains of Ligand-Free HIV Protease. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 866–875. 10.1107/s0907444907029125. [DOI] [PubMed] [Google Scholar]
- Alcaro S.; Artese A.; Ceccherini-Silberstein F.; Ortuso F.; Perno C. F.; Sing T.; Svicher V. Molecular Dynamics and Free Energy Studies on the Wild-Type and Mutated HIV-1 Protease Complexed with Four Approved Drugs: Mechanism of Binding and Drug Resistance. J. Chem. Inf. Model. 2009, 49, 1751–1761. 10.1021/ci900012k. [DOI] [PubMed] [Google Scholar]
- Nijhuis M.; Wensing A. M. J.; Bierman W. F. W.; de Jong D.; Kagan R.; Fun A.; Jaspers C. A. J. J.; Schurink K. A. M.; van Agtmael M. A.; Boucher C. A. B. Failure of Treatment with First-Line Lopinavir Boosted with Ritonavir Can Be Explained by Novel Resistance Pathways with Protease Mutation 76V. J. Infect. Dis. 2009, 200, 698–709. 10.1086/605329. [DOI] [PubMed] [Google Scholar]
- Weber I.; Agniswamy J. HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses 2009, 1, 1110–1136. 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champenois K.; Baras A.; Choisy P.; Ajana F.; Melliez H.; Bocket L.; Yazdanpanah Y. Lopinavir/Ritonavir Resistance in Patients Infected with HIV-1: Two Divergent Resistance Pathways?. J. Med. Virol. 2011, 83, 1677–1681. 10.1002/jmv.22161. [DOI] [PubMed] [Google Scholar]
- Dierynck I.; Van Marck H.; Van Ginderen M.; Jonckers T. H. M.; Nalam M. N. L.; Schiffer C. A.; Raoof A.; Kraus G.; Picchio G. TMC310911, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor, Shows in Vitro an Improved Resistance Profile and Higher Genetic Barrier to Resistance Compared with Current Protease Inhibitors. Antimicrob. Agents Chemother. 2011, 55, 5723–5731. 10.1128/aac.00748-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier C.; Lambert-Niclot S.; Alteri C.; Storto A.; Flandre P.; Svicher V.; Perno C.-F.; Brun-Vézinet F.; Calvez V.; Marcelin A.-G.; et al. Description of the L76V Resistance Protease Mutation in HIV-1 B and “Non-B” Subtypes. PLoS One 2013, 8, e54381 10.1371/journal.pone.0054381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondrak E. M.; Louis J. M. Influence of Flanking Sequences on the Dimer Stability of Human Immunodeficiency Virus Type 1 Protease. Biochemistry 1996, 35, 12957–12962. 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- Sayer J. M.; Agniswamy J.; Weber I. T.; Louis J. M. Autocatalytic Maturation, Physical/Chemical Properties, and Crystal Structure of Group N HIV-1 Protease: Relevance to Drug Resistance. Protein Sci. 2010, 19, 2055–2072. 10.1002/pro.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Lombardo D.; Giannaras J.; Decicco C. P. Estimating Ki Values for Tight Binding Inhibitors from Dose-Response Curves. Bioorg. Med. Chem. Lett. 1995, 5, 1947–1952. 10.1016/0960-894x(95)00330-v. [DOI] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/s0076-6879(97)76066-x. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2007, 64, 112–122. 10.1107/s0108767307043930. [DOI] [PubMed] [Google Scholar]