

Abstract
The classical targets for antipsychotic and antidepressant drugs are G protein-coupled receptors and neurotransmitter transporters, respectively. Full therapeutic actions of these drugs require several weeks. We show how therapeutic effects may eventually accrue after existing therapeutic ligands bind to these classical targets, not on the plasma membrane but rather within endoplasmic reticulum (ER) and cis-Golgi. Consequences of such binding may include pharmacological chaperoning: the nascent drug targets are stabilized against degradation and can therefore exit the ER more readily. Another effect may be matchmaking: heterodimers and homodimers of the target form and can more readily exit the ER. Summarizing recent data for nicotinic receptors, we explain how such effects could lead to reduced ER stress and to a decreased unfolded protein response, including changes in gene activation and protein synthesis. In effects not directly related to cellular stress, escorting would allow increased ER exit and trafficking of known associated proteins, as well as other proteins such as growth factors and their receptors, producing both cell-autonomous and non-cell-autonomous effects. Axonal transport of relevant proteins may underlie the several weeks required for full therapy. In contrast, the antidepressant effects of ketamine and other N-methyl-D-aspartate receptor ligands, which occur within <2 hours, could arise from dendritically localized intracellular binding, followed by chaperoning, matchmaking, escorting, and reduced ER stress. Thus, the effects of intracellular binding extend beyond proteostasis of the targets themselves and involve pathways distinct from ion channel and G protein activation. We propose experimental tests and note pathophysiological correlates.
Keywords: Antipsychotics, depression, gene activation, nicotine, schizophrenia, serotonin selective reuptake inhibitors
What events take place during the 2 to 3 weeks required for the full therapeutic actions of an antidepressant or antipsychotic drug? Most workers agree that a process is activated long after the few seconds required for the drug-receptor interaction to attain steady state at the plasma membrane. Signal transduction cascades, maintained for several weeks, are thought to be involved. Among the postulated downstream mechanisms are gene activation and neurogenesis. A satisfactory mechanistic picture would also explain the more rapid antidepressant effects of ketamine and other N-methyl-D-aspartate (NMDA) glutamate receptor blockers.
The new therapeutic hypotheses reviewed here continue to focus on the classical targets. For the serotonin selective reuptake inhibitors (SSRIs), the targets are the serotonin transporter (SERT) and, to a lesser extent, the norepinephrine transporter. For antipsychotic drugs, the dopamine D2/D3 and serotonin 2A (5-HT2A) G protein-coupled receptors (GPCRs) comprise the classical targets. Recently described antidepressant effects of ketamine and related compounds occur within 2 hours; the target is the NMDA receptor. But in the new hypotheses, the targets are in a novel location: the endoplasmic reticulum (ER) and cis-Golgi, where they are being synthesized and glycosylated (Figure S1 in Supplement 1).
Intracellular actions of psychiatric drugs are not a novel concept. For decades, we have assumed that valproate and lithium act intracellularly, primarily because no high-affinity plasma membrane binding sites have been identified. The sigma-1 receptor, originally thought to be an opioid receptor, presents another relevant example, because it is an intracellular chaperone protein that participates in ER stress signaling (1).
We will not soon have full pathophysiological information about schizophrenia, bipolar disease, or depression. Many therapeutic drugs have been analyzed in the absence of detailed pathophysiology about their target disease. This essay proceeds similarly, but we comment briefly on pathophysiology in the final section.
Statement of the Hypotheses
Antidepressants and antipsychotic drugs are able to bind intracellularly, in the ER and cis-Golgi, to their nascent receptors. There are several possible sequelae.
The target protein achieves a stable state that resists ER associated degradation and/or ER retrieval. This is pharmacological chaperoning.
In some cases, the intracellular binding enhances assembly or dimerization of the target, providing further stability. This is matchmaking.
These processes increase the ER exit rate of the receptors.
The increased ER exit suppresses one or more arms of ER stress and the unfolded protein response (UPR), improving neuronal function in a cell-autonomous fashion.
The increased ER exit allows the targets to co-traffic more effectively with candidate beneficial secreted proteins, such as neurotrophins; these can then act in a non-cell-autonomous fashion. This is escorting.
The binding of ligand within the ER can disrupt endogenous trafficking of either the target or a co-trafficked protein. This could be termed abduction.
Note that items 5 and 6 emphasize the molecules escorted or abducted by the drug-target complex, not the drug targets themselves.
Thus, the hypothesis states that the therapeutic effects of psychiatric drugs occur via inside-out signal transduction beginning in the ER. This contrasts with the usual assumption that therapeutic drugs act outside-in, via the membrane-localized drug-receptor interaction. Importantly, we will show how downstream effects can extend beyond proteostasis (2) of the targets themselves.
Recent Work on Nicotinic Receptors Reveals the Principles
Pharmacological Chaperoning and Matchmaking
Our laboratory’s work focuses on explaining the molecular, cellular, and circuit-based instantiation of such terms as plasticity and adaptation during chronic exposure to nicotine. At the subcellular level, nicotine acts as a pharmacological chaperone for α4β2 nicotinic acetylcholine receptors (nAChRs) (3–5) (Figure 1). A pharmacological chaperone is a small molecule that stabilizes a protein by binding, as either a substrate, agonist, antagonist, or allosteric modulator, at a physiologically relevant site on the target protein, but the binding primarily occurs within an organelle and usually during biosynthesis and trafficking of the target protein. A pharmacological chaperone is not a chaperone protein, although in some cases the effects might be similar. We are beginning to understand how pharmacological chaperoning by nicotine underlies both some initial events of nicotine addiction and some apparent neuroprotective actions of nicotine in Parkinson’s disease (6).
Figure 1.
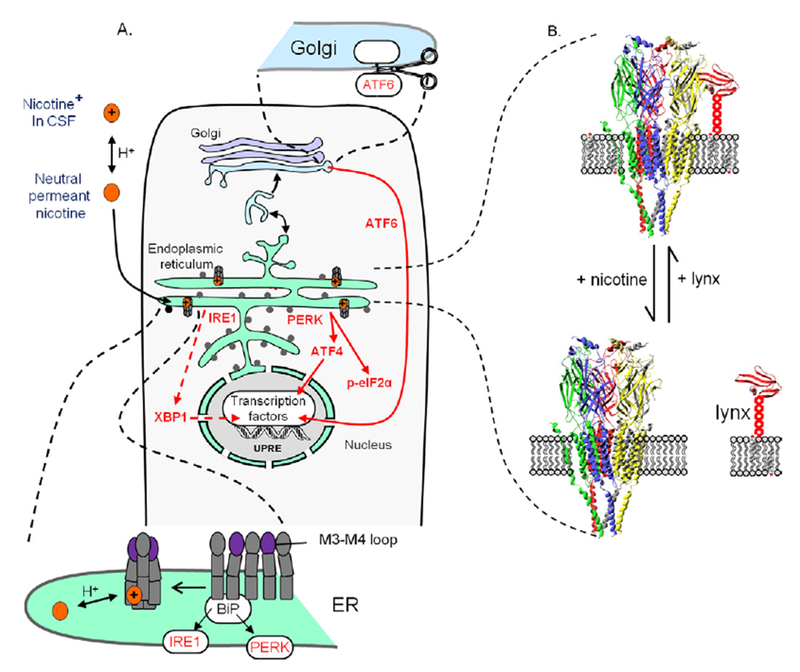
Insights from intracellular nicotine actions on the α4β2 nicotinic acetylcholine receptors (nAChR). (A) Chaperoning, matchmaking, reduction of endoplasmic reticulum (ER) stress, and the unfolded protein response (UPR) (5,16,88). Nicotine enters the neuron, permeates into the ER, and serves as a chaperone that favors assembly and stabilization of α4β2 nAChRs (shown in the insert at bottom). This decreases interactions with immunoglobulin binding protein (BiP), modulating protein kinase R-like ER-localized eukaryotic initiation factor 2α kinase (PERK)-activating transcription factor 4 (ATF4) and inositol-requiring enzyme 1 (IRE1)-X-box binding protein 1 (XBP1) (also shown in the insert at bottom). The insert at top shows that during ER stress, activating transcription factor 6 (ATF6) leaves the ER and enters the Golgi, where a fragment is cleaved; this then translocates to the nucleus and becomes a transcription factor. The UPR influences gene activation, via transcription factor binding to at least three unfolded protein response elements (UPRE). Experiments in our lab have not yet explored the IRE1 branch of the UPR, and it is shown as a dashed line. The M3-M4 loop of some nAChR subunits (purple) mediates ER retention and export via interactions with vesicle coat protein I and II complex proteins (COPI and COPII) (Figure S1 in Supplement 1). Ribosomes bound to the ER membrane are shown as gray dots. (B) An escort mechanism. The prototoxin lynx is synthesized, then transported to the ER lumen, guided by the usual signal sequence. Lynx resembles nAChR toxins from snake venom and is thought to bind like these toxins, at the interface between nAChR subunits (6,93). Unlike the snake venom toxins, lynx has a Glycophosphatidylinositol (GPI) anchor in the membrane. Lynx can therefore guide nAChRs toward cholesterol-rich regions of intracellular membranes (cholesterol molecules are shown in the membranes that anchor lynx). Nicotine binds at the same interface. One postulated consequence of nicotine binding would be displacement of lynx, abducting nAChRs from cholesterol-rich regions. This mechanism has not been tested. CSF, cerebrospinal fluid; p-eIF2α, phosphorylated eukaryotic initiation factor 2α.
Nicotinic Ligands, Acting on nAChR, Modify the Unfolded Protein Response
The unfolded protein response is a homeostatic mechanism that fine-tunes the cell in response to the demands of newly synthesized proteins that enter the ER. In mammalian cells (Figure 1), the unfolded protein response is thought to become activated when an ER-resident chaperone, immunoglobulin binding protein (BiP; also known as 78 kDa glucose-regulated protein GRP-78 or heat shock 70 kDa protein 5 HSPA5), binds to hydrophobic groups on unfolded or partially folded proteins. As a consequence, BiP dissociates from three other proteins in the ER membrane: protein kinase R-like ER-localized eukaryotic initiation factor 2α (eIF2α) kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). Each of these three proteins then activates a pathway resulting in gene activation, protein synthesis, and post-translational modifications. The overall result initially increases the protein processing capacity in the ER, but the PERK-eIF2α pathway eventually decreases the level of translation by membrane-bound ribosomes (7–10).
Many details of the UPR are emerging. During the UPR, some genes are activated, while others are repressed. Of particular interest to those studying receptors and transporters, membrane proteins may produce a signal that triggers a UPR, even if they have no ER lumenal domain (which would eventually become extracellular), and the three pathways may be activated differentially during the lumenal versus transmembrane triggers (11).
When ER stress and the UPR continue for long periods, they appear capable of reducing cell function as well. Endoplasmic reticulum stress occurs in certain dystonia subtypes caused by defective ER-associated proteins (12) and in several examples of inflammation (13). The most extreme outcome is apoptosis. Endoplasmic reticulum stress and the UPR are clearly activated during several neurodegenerative diseases (14,15).
We review here, for the first time, our recent work indicating that the intracellular interaction between nicotine and nAChRs is sufficient to modulate ER stress and to decrease the UPR. These effects were monitored by the number of Sec24d molecules in condensed endoplasmic reticulum exit sites, by translocation of ATF6, and by phosphorylation of eIF2α. Three ligands tested—the full agonist nicotine, the partial agonist cytisine, and the competitive antagonist dihydro-β-erythroidine—suppressed the UPR. Interestingly, these ligands had diverse effects on the subunit stoichiometry of the assembly receptors, on the trans-Golgi network, and on the eventual upregulation of plasma membrane nAChRs. These observations provide the first suggestions of a pathway leading from intracellular pharmacological chaperoning to modified gene activation (16).
Psychiatric Drugs Are Candidate Intracellular Ligands
Because they are weak bases, orally available central nervous system drugs have high membrane permeability, allowing them to pass through the blood-brain barrier, at least in their neutral, deprotonated form. No specific membrane transporter is required. An appreciation for this permeability may be gained by inspecting the record for any drug in PubChem Compound, a sister database to PubMed. The logP or clogP entry describes the logarithm of a compound’s partition constant between octanol and water. All drugs described in this review have logP values >2, rendering them even more membrane-permeant than nicotine (logP = 1.1). Thus, they also readily penetrate into neurons and into organelles. The ER has a pH very similar to that of cerebrospinal fluid. The protonation-deprotonation process occurs in milliseconds, and both in the extracellular solution and the ER, it is the protonated forms of the drug that usually bind to its receptor (Figure 1 A).
Central nervous system drugs are designed not to be substrates for various plasma membrane efflux pumps, which would remove them from cerebrospinal fluid. Thus, they also remain intracellular and intraorganellar. They bind very tightly to their targets. They are resistant to enzymatic metabolism, thus allowing them to interact with their targets for hours to days. Thermodynamics dominates: within the ER, the drug-receptor interaction spends several minutes, hours, or days finding its lowest free-energy state (which is also the tightest-binding state, explaining how therapeutic effects of psychiatric drugs occur at surprisingly low doses). This tightly bound state is often resistant to ER associated degradation, hiding the hydrophobic domains that would otherwise bind BiP and thus allowing binding with the vesicle coat protein II complex (COPII) and exit from the ER (Figure S1 in Supplement 1). The COPII complex comprises five distinct proteins first identified by yeast genetics (10). For this essay, a key protein is Sec24, which binds the transported cargo protein and has four isoforms (a through d) in higher eukaryotes (17).
Recent Work on GPCRs Relevant to Psychiatric Drugs
Some concepts in this section are illustrated in Figure 2. Pharmacological chaperoning of psychiatric drug targets would be most important for cases where these GPCRs are substantially retained in the early secretory pathway. Early experiments showed that GPCRs have a marked intracellular component (18), for instance, in microsomal fractions enriched with markers for endoplasmic reticulum and Golgi (19,20).
Figure 2.
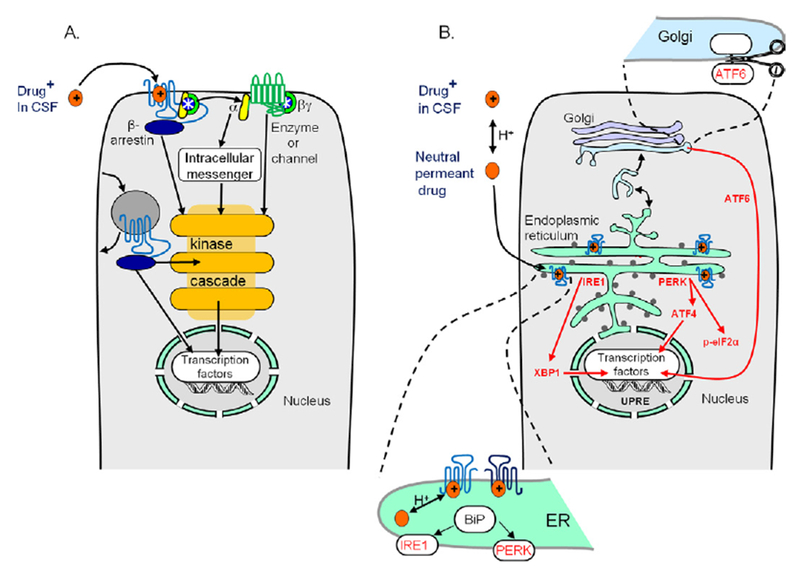
(A, B) Intracellular pharmacological chaperoning and matchmaking of G protein-coupled receptors (GPCRs) and downstream effects. (A) The conventional outside-in assumptions that GPCR antagonists manipulate second messengers and kinase cascades, resulting in gene activation. The diagram includes receptor trafficking in the late exocytotic/endocytotic pathway and signaling by p-arrestin. The diagram omits known facts about homodimerization and heterodimerization of GPCRs. (B) The inside-out view. Intracellular pharmacological chaperoning and matchmaking of GPCR and downstream effects, including reduction of endoplasmic reticulum (ER) stress and suppression of unfolded protein response. See also Figure 1 and Figure SI in Supplement 1. ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; BiP, immunoglobulin binding protein; CSF, cerebrospinal fluid; IRE1, inositol-requiring enzyme 1; p-eIF2α, phosphorylated eukaryotic initiation factor 2α; PERK, protein kinase R-like endoplasmic reticulum-localized eukaryotic initiation factor 2α kinase; UPRE, unfolded protein response elements; XBP1, X-box binding protein 1.
5-HT2A and Dopamine D2/D3 Receptors Localize Partially to the ER
The major accepted targets for antipsychotic drugs are dopamine D2/D3 and serotonin 5-HT2A. Systematic and anecdotal observations from many laboratories suggest that D2/D3 receptors remain to some extent in intracellular membranes (21), and probably in the ER (22), even under normal circumstances. Studies of 5-HT2A receptors also show that they are strongly intracellular (23,24) and are partially localized in ER (25). It cannot yet be claimed that this situation is fully analogous to that for α4β2 nAChRs, but ER and cis-Golgi retention does increase the possibility for ER stress. Dopaminergic neurons may exhibit ER stress even in normal circumstances (26).
Many elegant experiments also show how ligands control the trafficking of 5-HT2A receptors in the late exocytotic/endocytotic pathway (27) (Figure 2A), consequent on their phosphorylation by receptor kinases and interaction with β-arrestins. However, a recent article suggests that the ability of drugs to induce internalization and downregulation of 5-HT2A receptors is unrelated to antipsychotic actions (28).
GPCR Ligands Act as Pharmacological Chaperones
That GPCR ligands, both agonists and antagonists, can act as pharmacological chaperones is an established concept (Table S1 in Supplement 1) (29–39). Previous experiments have concentrated on direct upregulation of the GPCR by pharmacological ligands, with only passing attention (33) to the consequences for ER physiology. Antipsychotic drugs that are GPCR ligands, like other psychiatric drugs, possess pharmacokinetic, binding, and metabolic characteristics that render them highly accessible to the lumen of the ER and roughly as stable within that lumen as in the extracellular solution. Furthermore, the endogenous neurotransmitters are not present within the lumen to compete with the therapeutic ligands. The major classes of GPCR ligands include agonists, antagonists, allosteric modulators, and inverse agonists. As explained above, the ligand-receptor complex can thoroughly explore the energy landscape within intracellular compartments, eventually finding the state of tightest binding. As a result, class A GPCRs display a range of binding states, representing distinct conformations (40,41). These conformations are stabilized by binding of G proteins, β-arrestin (42), ions, and accessory proteins. Inverse agonism may be the mode in which antipsychotic drugs bind most tightly within the ER.
Evidence for involvement of GPCRs in ER stress is found in a recent Caenorhabditis elegans study, showing that an octopamine receptor, an invertebrate homolog of mammalian adrenergic receptors, is associated with an unfolded protein response (43). The identified genes are part of the “activated in blocked UPR” pathway, which differs from the classical mammalian UPR” pathway. An interesting point of the study is that the consequences of the UPR manipulation are read out in non-cell-autonomous fashion, by effects on the animal’s immune system.
Matchmaking in GPCR Homodimers and Heterodimers
Matchmaking is considerably less developed at GPCRs. That the atypical antipsychotics seem to bind weakly to D2 receptors and more strongly to 5HT2A receptors has been interpreted previously in terms of either cell-autonomous or non-cell-autonomous interactions in signaling downstream from inhibition of G protein and/or arrestin signaling. However, in the context of possible matchmaking, one should consider whether direct or indirect physical contact occurs between these two GPCR classes and, if so, whether such interaction is altered by drugs. Many articles show that class A and class C GPCRs can exist as dimers, and in some cases, heterodimerization is required for ER exit (40). Homodimerization or heterodimerization of GPCRs would be favored while the GPCRs are being concentrated by direct binding to the several dozen Sec24 molecules associated with each COPII vesicle. Of specific interest, heterodimers between D2 receptors and 5-HT2A receptors have been reported (44,45), but it is not clear whether ligand binding affects the strength of this binding or indeed any heterodimeric interaction between GPCRs.
Heterodimers between 5-HT2A and a metabotropic glutamate receptor, metabotropic glutamate receptor 2 (mGluR2), occur in cortical neurons (46,47). An experimental mGluR2 prodrug, LY2140023, has shown promise for schizophrenia (48–50). In Supplement 1, we explain how the prodrug strategy (51) yields a ligand that can bind to its target in the early exocytotic pathway. Again, whether the mGluR2 ligand affects the probability of heterodimerization is not known.
Endoplasmic reticulum-resident metabotropic glutamate receptor 5 (mGluR5), and probably other ER-resident GPCRs, participate in signal transduction, including G protein activation (52). Membrane permeant mGluR5 allosteric modulators are active in animal models of depression (53). Whether the apparent benefits arise from ER-based activation, chaperoning, or matchmaking is not known.
Several antipsychotic drugs interact with literally dozens of molecular targets (54). This enhances the possibility for matchmaking within the ER, as explained below for SERT.
Escorting Effects with GPCRs
Escorting is the least developed, but perhaps most powerful, concept for the effects of ER binding to GPCRs, and is not explained in Figure 2. In principle, any protein that 1) interacts with GPCRs within the early exocytotic pathway, 2) interacts with other proteins as well, and 3) exists in rate-limiting quantities might affect cell function if it is differentially escorted or abducted as a result of ligand binding to GPCRs. The GPCR-associated sorting proteins family should be studied in this regard (55). Homer proteins, when overexpressed, may interact with metabotropic glutamate receptors in the ER (56). Receptor-associated membrane proteins (57) and major histocompatibility proteins (58) present other possible families. If the escorted protein becomes secreted, non-cell-autonomous results are possible.
Side Effects of GPCR Drugs
That psychiatric drugs activate off-target receptors has long been suspected as the cause for agranulocytosis, weight gain, and other side effects. These side effects could also arise from intracellular binding.
That both agonists and antagonists of D2 receptors lead to several types of dyskinesias is usually ascribed to circuit-based phenomena. We point out that both agonists and antagonists could act as pharmacological chaperones of GPCRs. A partially analogous situation, in which both agonists and antagonists of nAChRs act as pharmacological chaperones to suppress ER stress, has been described at nAChRs (16). Thus, pharmacological chaperoning, matchmaking, escorting, abduction, or UPR-related changes in gene activation could cause some dyskinesias.
Recent Work on SERT
Raphe neurons are the major neuronal type that express SERT, the primary target of SSRIs. In raphe neurons, most axonal SERT is on the plasma membrane (PM), but most SERT is cytoplasmic in the soma and dendrites of neurons, in platelets, and in astrocytes (59-61). Thus, intracellular events such as chaperoning, matchmaking, and escorting have the potential to alter SERT biology.
Chaperoning is an established concept at SERT. The alternating access model of neurotransmitter transport (62) summarizes the concept that neurotransmitter transporters shuttle between two major conformations, in which a central region of the transporter faces either the cytosolic or cytoplasmic/ER lumenal compartment. Within and between each of these two orientations, several substates occur. For neurotransmitter transporters as for other proteins, “by saturating SERT with a ligand, we shift the equilibrium toward protein conformations optimal for binding that ligand” (63). The most stable state often has the greatest resistance to degradation. Each class of ligands favors distinct conformations, in some cases specifically favoring posttranslational modification, trafficking, and interaction of the transporter with regulatory proteins (63). These conformations are stabilized by binding of substrates such as Na+, Cl−, and K+, which themselves have different concentrations in the cytoplasm versus ER lumen versus extracellular space. Binding of fluoxetine has been best studied, and in its presence the most stable conformation appears to be midway between the extracellular/lumenal-facing and cytosol-facing orientation of the permeation pathway (63). There is evidence that even among the SSRIs, the various ligands interact differentially with the binding of the inorganic co-substrates (for instance fluoxetine vs. paroxetine) (64). These differences add to the possibilities for explaining how subsets of patients are benefited by distinct SSRIs.
Within the ER, neurotransmitter transporters interact with the protein chaperones calnexin, calreticulin, and BiP. This binding facilitates both intramolecular and intermolecular arrangements of hydrophobic segments (65,66). Presumably, like most molecules that exit the ER, the transporter must be correctly folded before binding to Sec24c or other members of the COPII cargo-binding protein Sec24 family (67). As noted, SSRIs may favor this specific form of chaperoning. Ibogaine, a hallucinogenic alkaloid, has complex pharmacology including blockade of SERT and the dopamine transporter. At SERT, ibogaine acts as a pharmacological chaperone to rescue some ER exit mutations (67). Interestingly, SERT appears to be the only neurotransmitter transporter studied to date that specifically requires Sec24c to exit the ER. The structural basis for this difference is not known (68), but that neuronal proteins specifically utilize individual Sec24 isoforms has precedents in the ER exit literature (69,70). This concept also applies to neuronal proteins (68,71).
Matchmaking is less well established at SERT. Serotonin transporter and neurotransmitter transporters dimerize in the ER as a prerequisite for binding to Sec24 (68). Ligands for SERT enhance ER exit, but it is not known whether SSRIs affect this dimerization. Once dimerized, mutations in the C-terminus of SERT impair its ability to bind to Sec24 proteins (67).
What are the escort consequences of enhanced ER exit of SERT? Baudry et al. (72) showed that an SSRI suppresses microRNA16 and enhances S100β exit. Other research suggests a role for p11 (S100A10) (73). Most researchers postulate that these are consequences of excess extracellular serotonin (74). Within the framework of the present review, one would suggest that these actions arise via suppression of ER stress and/or escorting or abduction.
Axonal Transport
The effects of psychiatric drugs have long been assumed to involve gene activation, and the ER stress/UPR pathway has direct influences on gene activation but this process takes just a day or two. The inside-out view, in which therapeutic effects result from action in the ER, leads one to emphasize the role of intracellular trafficking during the several weeks involved in the full action of antipsychotic and antidepressant drugs. Some effects occur within days, perhaps because of effects that occur in the soma; on an intermediate time scale, events might involve transport to dendrites. Slow axonal transport and restructuring occur at a rate of ~1 mm per day, accounting for the most delayed effects, for instance, ~30 days at the axon terminal of a 3 cm human axon. This might be briefer in animals with shorter axons. No retrograde transport need be invoked.
Rapid Antidepressant Effects of Ketamine Occur in Dendrites
Figure 3A shows a previously suggested mechanistic framework (75–77) for the antidepressant effects of ketamine, which become established within 1 to 2 hours. Figure 3B shows mechanisms within the framework of this review, beginning with the binding of ketamine to nascent NMDA receptors in the dendritic ER.
Figure 3.
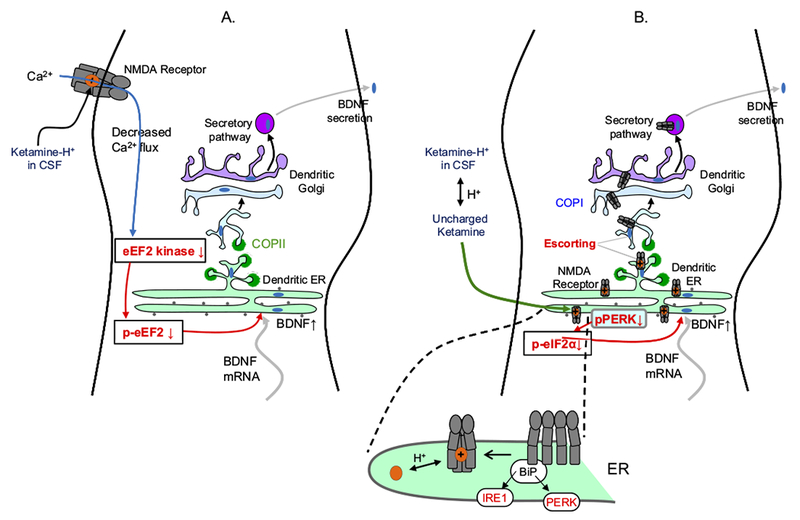
Diagrams of a dendrite, showing possible explanations for the 1- to 2-hour delay before antidepressant actions of ketamine and related N-methyl-D-aspartate (NMDA) receptor blockers. Ketamine exerts at least some of these antidepressant effects via increases in brain-derived neurotrophic factor (BDNF) secretion (76,77,94,95), and we therefore diagram mechanisms leading to BDNF secretion. Brain-derived neurotrophic factor secretion presumably requires depolarization, which may explain the requirement for 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptor activation (82). Note that dendritic endoplasmic reticulum (ER) and Golgi are thought to be simpler than the somatically located organelles shown in previous figures. The selective NMDA receptor 2B antagonist Ro 25-6981 had similar effects to ketamine (77). The transcription of BDNF is not required (as shown by insensitivity to actinomycin D), but its synthesis is required (as shown by sensitivity to anisomycin). In hippocampus, ketamine and NMDA, in the absence of neuronal activity, led to dephosphorylation of eukaryotic elongation factor (eEF2) (also called calcium/calmodulin-dependent eukaryotic elongation factor 2 kinase), but only ketamine produced this effect in cortex. Rottlerin and NH125, which inhibit several kinases including eEF2 kinase, also had BDNF-dependent antidepressant activity. (A) An outside-in mechanism, downstream from NMDA receptor block. Ketamine binds within the NMDA receptor pore but does not enter the neuron. The data have generally been interpreted in light of the knowledge that ketamine blocks spontaneous miniature excitatory postsynaptic currents through NMDA receptors (76). It is implied that decreased Ca2+ influx through NMDA receptors begins the transduction pathway leading to effects on presynaptic or postsynaptic efficiency. (B) Possible inside-out mechanisms, resulting from intracellular binding to nascent NMDA receptors. Existing data show that ligands can act as pharmacological chaperones for glutamate receptors within the ER (96), analogous to experiments in which nicotine acts as a pharmacological chaperone for nicotinic acetylcholine receptors. Two possible sequelae could lead to increased BDNF secretion. First, enhanced ER exit would decrease ER stress, for instance by decreasing phosphorylated protein kinase R-like ER-localized eukaryotic initiation factor 2α kinase (pPERK). This would decrease phosphorylated eukaryotic initiation factor 2α (p-eIF2α), increasing synthesis of ER proteins, including BDNF, thus producing the observed BDNF increase. Activation of NMDA receptors increases ER stress markers such as eIF2α phosphorylation and CCAAT/enhancer-binding protein homologous protein (CHOP) (97). However, it is not known whether blockade of NMDA receptors by ketamine has any effect on eIF2α phosphorylation in the absence of ER stress. This would be a key test of the chaperoning-ER stress hypothesis. Blockade by MK-801 did decrease caspase-12 activation, even in the absence of ER stress; however, caspase-12 activation may occur in a pathway distinct from ER stress (98). Second, an escort effect of intracellular ketamine-NMDA receptor binding arises from the fact that both NMDA receptors and BDNF are trafficked via a nonstandard ER and Golgi vesicle pathway involving synapse-associated protein 97 (SAP97) and calcium/calmodulin-dependent serine protein kinase (CASK) (99,100). Additional knowledge about the synapse-associated protein 97-calcium/calmodulin-dependent serine protein kinase trafficking pathway is crucial for evaluating the escort hypothesis. Chaperoning and/or matchmaking would occur in the ER. Escorting would occur when both the NMDA receptor and BDNF bind to Sec24 or at a later step. BiP, immunoglobulin binding protein; COPI and COPII, vesicle coat protein I and II complex; CSF, cerebrospinal fluid; IRE1, inositol-requiring enzyme 1; mRNA, messenger RNA; p-eEF2, phosphorylated eukaryotic elongation factor.
The intracellular binding explanation for the 1- to 2-hour effects of ketamine seems at first glance inconsistent with the idea that intracellular binding is also the source of the 2- to 3-week time course of SSRI antidepressant action. It is, therefore, a key observation that these antidepressant effects vanish in valine/valine and methionine/methionine brain-derived neurotrophic factor (BDNF) knockin mice, which cannot transport BDNF message to dendrites (77). One straightforward interpretation: whereas other psychiatric drugs require protein synthesis at the somatic compartment followed by transport to axons, the ketamine actions are local to dendrites, so that the newly synthesized or escorted BDNF can be secreted near dendrites within minutes. Dendrites have ribosomes, ER, endoplasmic reticulum exit sites, and Golgi and are fully competent to translate and secrete BDNF, without requiring the soma (78–80). Supplement 1 discusses local ketamine concentrations (76,81–85).
Testing and Applying Inside-Out Hypotheses
We must test hypotheses that intracellular binding in the early exocytotic pathway explains actions of antipsychotic and antidepressant drugs. The compartmentalization of drug actions must be studied, presumably with isolated neurons. The extent to which drugs bind within the ER could be tested most generally with stable isotope-labeled drugs and nanometer scale secondary ion mass spectrometry. The compartment in which drugs act could be studied by quaternizing the amines to reduce membrane permeation (86). Compartmented cultures also seem well suited for such research (87).
Kinetic experiments often shed light on mechanism. One could visualize intracellular movements of receptors, transporters, and escorted or abducted proteins as they travel from the ER to axons and dendrites in response to drug actions (16,88). Time-resolved proteomic and transcriptomic studies could test whether antipsychotic and antidepressant drugs produce early effects on ER stress and UPR pathways. These would be conducted both in the soma, for GPCRs and SSRIs, and in dendrites for ketamine.
Cell-free systems can report on drug-receptor interactions. Few contemporary articles continue to report the binding of psychiatric drugs to purified endoplasmic reticulum from brain (19,20), but this seems crucial for understanding how each class of agonist, antagonist, inverse agonist, open-channel blocker, or allosteric modulator produces chaperoning and/or matchmaking at GPCRs, transporters, or ligand-gated channels. In analogous experiments at nAChRs, agonists chaperone and upregulate at concentrations far lower than required to activate but far higher than the equilibrium binding constant (16). For antagonists, as well, the chaperoning effects occur at concentrations far higher than the equilibrium binding constant. Evidently, the chaperoned state(s) require further characterization. Because we suggest that key chaperoning events occur at the stage of ER exit and endoplasmic reticulum-Golgi intermediate compartment (ERGIC), experiments should incorporate reconstituted systems for membrane budding and fusion (10).
If the hypotheses gain further support, psychiatric diseases will belatedly join the list of diseases that are approached therapeutically by manipulating early exocytotic pathways (2,89). Strategies for designing more effective psychiatric drugs could incorporate the experiments described in this section. An appropriate challenge would be to decrease the cognitive deficits of schizophrenia (6), perhaps by enhancing the early exocytotic pathways of neocortical chandelier and basket cell somata. Although these cells have relatively short axons, their complex and numerous axonal terminals might place heavy demands on early exocytotic pathways (90,91).
Comment on Pathophysiology
Three large classes of proteins may participate in pharmacological chaperoning, matchmaking, escorting, abduction, ER stress, unfolded protein responses, and related mechanisms: class A—one third of a typical cell’s protein species enter the ER; class B—many additional cytoplasmic protein species govern proteostasis of ER proteins and transport of vesicles in the early and late endocytotic pathways; and class C—many additional proteins enter the nucleus to govern chromatin structure, initiation, and transcription of the genes and messenger RNA processing of the proteins in class A and B. A sizeable fraction of human genes encode neuronal proteins in class A, B, or C, and noncoding regions may also play a role. Malfunctions in classes A, B, or C could well lead to a deterioration of neural function.
A marked, but neither apoptotic nor necrotic, deficit of important proteins that pass through the ER could account for the observations that reduced gray matter volume occurs in schizophrenia, depression, or bipolar disease, but also that this reduction is more subtle than in neurodegenerative disease. The accompanying functional deterioration might also be subtle enough to avoid ER stress and/or UPR activation, but resilient function (92) could still be partially restored by the pharmacological chaperoning, matchmaking, or abduction effects of existing antipsychotic and antidepressant drugs. Even if each hypothesis of this article is proven, we would hesitate to infer that all psychiatric patients have a neuronal population showing cellular stress, according to this phrase’s contemporary biomedical connotations. Therefore, inside-out therapeutic mechanisms are compatible with the idea that various polygenic, multifactorial, and partially penetrant processes underlie psychiatric diseases. However, this compatibility remains vexingly general.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (MH-086383 and MH-088550). Our laboratory’s work on nicotinic receptors has been supported by the National Institutes of Health (AG-33954, DA-17279, DA-19375), the Michael J. Fox Foundation, the Joint Center for Translational Medicine, the California Tobacco-Related Disease Research Program (17RT-0127, 18FT-0066, 19KT-0032), and Targacept, Inc. In the past, our laboratory’s work on G protein-coupled receptors has been supported by GM-081662, on neurotransmitter transporters by DA-09121, and on intracellular messengers by GM-29836.
HAL, JMM, and RS wrote the article. HAL developed the concepts. JMM contributed ideas on lynx. RS contributed scholarship on the unfolded protein response.
We thank Hadassah Tamir for suggesting that axonal transport underlies some aspects of psychiatric drug effects and for discussion, Johannes Schwarz for pointing out aspects of dyskinesia, and Luke Wiseman for tutorials on the unfolded protein response. We also thank Nathan Dascal, Dennis Dougherty, Hesso Farhan, Robert Farley, RobertFreedman, Ege Kavalali, Odile Kellermann, Jun Li, John Lowe, Stefan McDonough, J. Michael McIntosh, Randy Schekman, Darryle Schoepp, Andrew Steele, Teagan Wall, and the anonymous referees for comments. HAL thanks members of our research group for discussion and for allowing the time to assemble this essay.
HAL has submitted patent applications covering pharmacological chaperones in neurodegenerative and psychiatric disease. JMM is founder and shareholder of Ophidion, Inc. She has applied for US patents 10322359 and 20080221013 on the use of lynx for therapeutic purposes. RS reports no biomedical financial interests or potential conflicts of interest.
Footnotes
Supplementary material cited in this article is available online.
References
- 1.Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE (2010): The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci 31:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Routledge KE, Gupta V, Balch WE (2010): Emergent properties of proteostasis-COPII coupled systems in human health and disease. Mol Membr Biol 27:385–397. [DOI] [PubMed] [Google Scholar]
- 3.Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ (2005): Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron 46:595–607. [DOI] [PubMed] [Google Scholar]
- 4.Kuryatov A, Luo J, Cooper J, Lindstrom J (2005): Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol Pharmacol 68:1839–1851. [DOI] [PubMed] [Google Scholar]
- 5.Lester HA, Xiao C, Srinivasan R, Son C, Miwa J, Pantoja R, et al. (2009): Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 11:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miwa JM, Freedman R, Lester HA (2011): Neural systems governed by nicotinic acetylcholine receptors: Emerging hypotheses. Neuron 70: 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balch WE, Morimoto RI, Dillin A, Kelly JW (2008): Adapting proteostasis for disease intervention. Science 319:916–919. [DOI] [PubMed] [Google Scholar]
- 8.Walter P, Ron D (2011): The unfolded protein response: From stress pathway to homeostatic regulation. Science 334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 9.Wiseman RL, Haynes CM, Ron D (2010): SnapShot: The unfolded protein response. Cell 140:590–590.e2. [DOI] [PubMed] [Google Scholar]
- 10.Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R (2012): COPII and the regulation of protein sorting in mammals. Nat Cell Biol 14:20–28. [DOI] [PubMed] [Google Scholar]
- 11.Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N(2011): Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci U S A 108:7832–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P, Burdette AJ, Porter JC, Ricketts JC, Fox SA, Nery FC, et al. (2010): The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum Mol Genet 19:3502–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hummasti S, Hotamisligil GS (2010): Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 107:579–591. [DOI] [PubMed] [Google Scholar]
- 14.Matus S, Glimcher LH, Hetz C (2011): Protein folding stress in neurodegenerative diseases: A glimpse into the ER. Curr Opin Cell Biol 23:239–252. [DOI] [PubMed] [Google Scholar]
- 15.Nassif M, Matus S, Castillo K, Hetz C (2010): Amyotrophic lateral sclerosis pathogenesis: A journey through the secretory pathway. Antioxid Redox Signal 13:1955–1989. [DOI] [PubMed] [Google Scholar]
- 16.Srinivasan R, Richards CI, Xiao C, Rhee D, Pantoja R, Dougherty DA, et al. (2012): Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the ER stress response. Mol Pharmacol 81:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mancias JD, Goldberg J (2008): Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J 27:2918–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young WS 3rd, Wamsley JK, Zarbin MA, Kuhar MJ (1980): Opioid receptors undergo axonal flow. Science 210:76–78. [DOI] [PubMed] [Google Scholar]
- 19.Roth BL, Laskowski MB, Coscia CJ (1982): Microsomal opiate receptors differ from synaptic membrane receptors in proteolytic sensitivity. Brain Res 250:101–109. [DOI] [PubMed] [Google Scholar]
- 20.Roth BL, Coscia CJ (1984): Microsomal opiate receptors: Characterization of smooth microsomal and synaptic membrane opiate receptors. J Neurochem 42:1677–1684. [DOI] [PubMed] [Google Scholar]
- 21.Cho DI, Zheng M, Kim KM (2010): Current perspectives on the selective regulation of dopamine D2 and D3 receptors. Arch Pharm Res 33:1521–1538. [DOI] [PubMed] [Google Scholar]
- 22.Sesack SR, Aoki C, Pickel VM (1994): Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci 14:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornea-Hebert V, Watkins KC, Roth BL, Kroeze WK, Gaudreau P, Leclerc N, Descarries L (2002): Similar ultrastructural distribution of the 5-HT2A serotonin receptor and microtubule-associated protein MAP1A in cortical dendrites of adult rat. Neuroscience 113:23–35. [DOI] [PubMed] [Google Scholar]
- 24.Cornea-Hebert V, Riad M, Wu C, Singh SK, Descarries L (1999): Cellular and subcellular distribution of the serotonin 5-HT2A receptor in the central nervous system of adult rat. J Comp Neurol 409:187–209. [DOI] [PubMed] [Google Scholar]
- 25.Doherty MD, Pickel VM (2000): Ultrastructural localization of the serotonin 2A receptor in dopaminergic neurons in the ventral tegmental area. Brain Res 864:176–185. [DOI] [PubMed] [Google Scholar]
- 26.Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R (2011): The endoplasmic reticulum stress sensor, ATF6_, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem 286:7947–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth BL (2011): Irving Page Lecture: 5-HT2A serotonin receptor biology: Interacting proteins, kinases and paradoxical regulation. Neuropharmacology 61:348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav PN, Kroeze WK, Farrell MS, Roth BL (2011): Antagonist functional selectivity: 5-HT2A serotonin receptor antagonists differentially regulate 5-HT2A receptor protein level in vivo. J Pharmacol Exp Ther 339: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malaga-Dieguez L, Yang Q, Bauer J, Pankevych H, Freissmuth M, Nanoff C (2010): Pharmacochaperoning of the A1 adenosine receptor is contingent on the endoplasmic reticulum. Mol Pharmacol 77:940–952. [DOI] [PubMed] [Google Scholar]
- 30.Van Craenenbroeck K, Clark SD, Cox MJ, Oak JN, Liu F, Van Tol HH (2005): Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J Biol Chem 280:19350–19357. [DOI] [PubMed] [Google Scholar]
- 31.Conn PM, Ulloa-Aguirre A(2011): Pharmacological chaperones for misfolded gonadotropin-releasing hormone receptors. Adv Pharmacol 62: 109–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noorwez SM, Ostrov DA, McDowell JH, Krebs MP, Kaushal S (2008): A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Invest Ophthalmol Vis Sci 49:3224–3230. [DOI] [PubMed] [Google Scholar]
- 33.Leskela TT, Lackman JJ, Vierimaa MM, Kobayashi H, Bouvier M, Petaja-Repo UE (2012): Cys-27 variant of human δ-opioid receptor modulates maturation and cell surface delivery of Phe-27 variant via heteromerization. J Biol Chem 287:5008–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaipatikul V, Erickson-Herbrandson LJ, Loh HH, Law PY (2003): Rescuing the traffic-deficient mutants of rat μ-opioid receptors with hydrophobic ligands. Mol Pharmacol 64:32–41. [DOI] [PubMed] [Google Scholar]
- 35.Fan J, Perry SJ, Gao Y, Schwarz SA, Maki RA (2005): A point mutation in the human melanin concentrating hormone receptor 1 reveals an important domain for cellular trafficking. Mol Endocrinol 19:2579–2590. [DOI] [PubMed] [Google Scholar]
- 36.Tao YX (2010): The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocr Rev 31:506–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawtin SR (2006): Pharmacological chaperone activity of SR49059 to functionally recover misfolded mutations of the vasopressin V1a receptor. J Biol Chem 281:14604–14614. [DOI] [PubMed] [Google Scholar]
- 38.Robert J, Auzan C, Ventura MA, Clauser E (2005): Mechanisms of cell surface rerouting of an endoplasmic reticulum-retained mutant of the vasopressin V1b/V3 receptor by a pharmacological chaperone. J Biol Chem 280:42198–42206. [DOI] [PubMed] [Google Scholar]
- 39.Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, et al. (2004): Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem 279:47254–47263. [DOI] [PubMed] [Google Scholar]
- 40.Kenakin T (2010): G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res 30:313–321. [DOI] [PubMed] [Google Scholar]
- 41.Kenakin T, Miller LJ (2010): Seven transmembrane receptors as shape-shifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ (2012): Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol 52:179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun J, Singh V, Kajino-Sakamoto R, Aballay A (2011): Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332:729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukasiewicz S, Faron-Gorecka A, Kedracka-Krok S, Dziedzicka-Wasylewska M(2011): Effect of clozapine on the dimerization of serotonin 5-HT2A receptor and its genetic variant 5-HT2AH425Y with dopamine D2 receptor. Eur J Pharmacol 659:114–123. [DOI] [PubMed] [Google Scholar]
- 45.Albizu L, Holloway T, Gonzalez-Maeso J, Sealfon SC (2011): Functional crosstalk and heteromerization of serotonin 5-HT2A and dopamine D2 receptors. Neuropharmacology 61:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, et al. (2008): Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, et al. (2011): Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell 147:1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. (2007): Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized phase 2 clinical trial. Nat Med 13:1102–1107. [DOI] [PubMed] [Google Scholar]
- 49.Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, et al. (2011): A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol 31:349–355. [DOI] [PubMed] [Google Scholar]
- 50.Lowe S, Dean R, Ackermann B, Jackson K, Natanegara F, Anderson S, et al. (2012): Effects of a novel mGlu/receptor agonist prodrug, LY2140023 monohydrate, on central monoamine turnover as determined in human and rat cerebrospinal fluid. Psychopharmacology (Berl) 219:959–970. [DOI] [PubMed] [Google Scholar]
- 51.Hosokawa M (2008): Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 13:412–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jong YJ, Kumar V, O’Malley KL (2009): Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem 284:35827–35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, et al. (2012): Negative allosteric modulation of metabolic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression [published online ahead of print April 21]. Neuropharmacology. [DOI] [PubMed] [Google Scholar]
- 54.Roth BL, Sheffler DJ, Kroeze WK (2004): Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 3:353–359. [DOI] [PubMed] [Google Scholar]
- 55.Magalhaes AC, Dunn H, Ferguson SS (2012): Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol 165:1717–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roche KW, Tu JC, Petralia RS, Xiao B, Wenthold RJ, Worley PF (1999): Homer 1b regulates the trafficking of group I metabotropic glutamate receptors. J Biol Chem 274:25953–25957. [DOI] [PubMed] [Google Scholar]
- 57.Kuwasako K, Hay DL, Nagata S, Hikosaka T, Kitamura K, Kato J (2011): The third extracellular loop of the human calcitonin receptor-like receptor is crucial for the activation of adrenomedullin signalling. Br J Pharmacol 166:137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olson R, Dulac C, Bjorkman PJ (2006): MHC homologs in the nervous system–they haven’t lost their groove. Curr Opin Neurobiol 16:351–357. [DOI] [PubMed] [Google Scholar]
- 59.Tao-Cheng JH, Zhou FC (1999): Differential polarization of serotonin transporters in axons versus soma-dendrites: An immunogold electron microscopy study. Neuroscience 94:821–830. [DOI] [PubMed] [Google Scholar]
- 60.Carneiro AM, Blakely RD (2006): Serotonin-, protein kinase C-, and Hic-5-associated redistribution of the platelet serotonin transporter. J Biol Chem 281:24769–24780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inazu M, Takeda H, Ikoshi H, Sugisawa M, Uchida Y, Matsumiya T (2001): Pharmacological characterization and visualization of the glial serotonin transporter. Neurochem Int 39:39–49. [DOI] [PubMed] [Google Scholar]
- 62.Lester HA, Cao Y, Mager S (1996): Listening to neurotransmitter transporters. Neuron 17:807–810. [DOI] [PubMed] [Google Scholar]
- 63.Tavoulari S, Forrest LR, Rudnick G (2009): Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J Neurosci 29:9635–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larsen MB, Sonders MS, Mortensen OV, Larson GA, Zahniser NR, Amara SG (2011): Dopamine transport by the serotonin transporter: A mechanistically distinct mode of substrate translocation. J Neurosci 31: 6605–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korkhov VM, Milan-Lobo L, Zuber B, Farhan H, Schmid JA, Freissmuth M, Sitte HH (2008): Peptide-based interactions with calnexin target misassembled membrane proteins into endoplasmic reticulum-derived multilamellar bodies. J Mol Biol 378:337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tate CG, Whiteley E, Betenbaugh MJ (1999): Molecular chaperones stimulate the functional expression of the cocaine-sensitive serotonin transporter. J Biol Chem 274:17551–17558. [DOI] [PubMed] [Google Scholar]
- 67.El-Kasaby A, Just H, Malle E, Stolt-Bergner PC, Sitte HH, Freissmuth M, Kudlacek O (2010): Mutations in the carboxyl-terminal SEC24 binding motif of the serotonin transporter impair folding of the transporter. J Biol Chem 285:39201–39210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sucic S, El-Kasaby A, Kudlacek O, Sarker S, Sitte HH, Marin P, Freissmuth M (2011): The serotonin transporter is an exclusive client of the coat protein complex II (COPII) component SEC24C. J Biol Chem 286:16482–16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wendeler MW, Paccaud JP, Hauri HP (2007): Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep 8:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonnon C, Wendeler MW, Paccaud JP, Hauri HP(2010): Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J Cell Sci 123:1705–1715. [DOI] [PubMed] [Google Scholar]
- 71.Kim J, Kleizen B, Choy R, Thinakaran G, Sisodia SS, Schekman RW(2007): Biogenesis of γ-secretase early in the secretory pathway. J Cell Biol 179:951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O (2010): miR-16 targets the serotonin transporter: A new facet for adaptive responses to antidepressants. Science 329:1537–1541. [DOI] [PubMed] [Google Scholar]
- 73.Warner-Schmidt JL, Chen EY, Zhang X, Marshall JJ, Morozov A, Svenningsson P, Greengard P (2010): A role for p11 in the antidepressant action of brain-derived neurotrophic factor. Biol Psychiatry 68:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baudry A, Mouillet-Richard S, Launay JM, Kellermann O (2011): New views on antidepressant action. Curr Opin Neurobiol 21:858–865. [DOI] [PubMed] [Google Scholar]
- 75.Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM (2007): Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55:648–661. [DOI] [PubMed] [Google Scholar]
- 76.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. (2011): NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK (2012): Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry 71:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ye B, Zhang Y, Song W, Younger SH, Jan LY, Jan YN (2007): Growing dendrites and axons differ in their reliance on the secretory pathway. Cell 130:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cui-Wang T, Hanus C, Cui T, Helton T, Bourne J, Watson D, et al. (2012): Local zones of endoplasmic reticulum complexity confine cargo in neuronal dendrites. Cell 148:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aridor M, Guzik AK, Bielli A, Fish KN (2004): Endoplasmic reticulum export site formation and function in dendrites. J Neurosci 24:3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. (2006): A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63: 856–864. [DOI] [PubMed] [Google Scholar]
- 82.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. (2010): mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maxwell CR, Ehrlichman RS, Liang Y, Trief D, Kanes SJ, Karp J, Siegel SJ (2006): Ketamine produces lasting disruptions in encoding of sensory stimuli. J Pharmacol Exp Ther 316:315–324. [DOI] [PubMed] [Google Scholar]
- 84.Clements JA, Nimmo WS (1981): Pharmacokinetics and analgesic effect of ketamine in man. Br J Anaesth 53:27–30. [DOI] [PubMed] [Google Scholar]
- 85.MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, et al. (1991): Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J Physiol 432:483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hille B (1977): Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campenot RB, Lund K, Mok SA (2009): Production of compartmented cultures of rat sympathetic neurons. Nat Protoc 4:1869–1887. [DOI] [PubMed] [Google Scholar]
- 88.Srinivasan R, Pantoja R, Moss FJ, Mackey EDW, Son C, Miwa J, Lester HA. (2011): Nicotine upregulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol 137:59–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aridor M(2007): Visiting the ER: The endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv Drug Deliv Rev 59:759–781. [DOI] [PubMed] [Google Scholar]
- 90.Powell SB, Sejnowski TJ, Behrens MM (2012): Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology 62:1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lewis DA, Curley AA, Glausier JR, Volk DW(2012): Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, Gray AN, et al. (2003): Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 53:707–742. [DOI] [PubMed] [Google Scholar]
- 93.Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, Heintz N (1999): Lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron 23:105–114. [DOI] [PubMed] [Google Scholar]
- 94.Autry AE, Monteggia LM (2012): Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64:238–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duman RS, Voleti B (2012): Signaling pathways underlying the pathophysiology and treatment of depression: Novel mechanisms for rapidacting agents. Trends Neurosci 35:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schwappach B (2008): An overview of trafficking and assembly of neurotransmitter receptors and ion channels. Mol Membr Biol 25:270–278. [DOI] [PubMed] [Google Scholar]
- 97.Nosyreva E, Kavalali ET (2010): Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J Neurosci 30:7358–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G, et al. (2011): Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis 2:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horton AC, Ehlers MD (2003): Dual modes of endoplasmic reticulumto-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23:6188–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jeyifous O, Waites CL, Specht CG, Fujisawa S, Schubert M, Lin EI, et al. (2009): SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat Neurosci 12:1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.