

Abstract
Natural killer cells (NK cells) play an essential role in the immunological mechanism underlying chronic hepatitis C (CHC). Impairment of NK cell function facilitates persistent infection with hepatitis C virus (HCV) and hepatocellular carcinogenesis. However, the mechanism by which NK cell activity is suppressed in CHC is not completely understood. In this study, we focused on carcinoembryonic antigen–related cell‐adhesion molecule 1 (CEACAM1). CEACAM1 is thought to suppress NK cell function. We examined the effect of CEACAM1 on NK cell function in CHC. We investigated the function of CEACAM1 in vitro using Huh7.5.1 cells and the HCV‐Japanese fulminant hepatitis (JFH)‐1 strain. We analyzed serum CEACAM1 level, NK cell function, and CEACAM1 messenger RNA (mRNA) level in human liver samples. Levels of CEACAM1 on the cell surface, CEACAM1 mRNA levels, and soluble CEACAM1 levels in supernatants were significantly higher in Huh7.5.1 cells infected with JFH‐1 (Huh7.5.1/JFH‐1 cells) than in Huh7.5.1 cells. Significantly higher NK cell cytotoxicity was observed toward K562 cells after coculture with CEACAM1 knockout Huh7.5.1/JFH‐1 cells than after coculture with Huh7.5.1/JFH‐1 cells. CEACAM1 expression was induced by the HCV E2 glycoprotein in HCV infection. Significantly higher serum CEACAM1 levels were detected in patients with CHC compared with healthy subjects and patients who achieved sustained virological responses. The expression of CD107a on NK cells from patients with CHC was negatively correlated with serum CEACAM1 levels. Significantly higher levels of CEACAM1 mRNA were detected in HCV‐infected livers compared with uninfected livers. Conclusion: CEACAM1 expression was induced in hepatocytes following HCV infection and decreased NK cell cytotoxicity. These results demonstrate a possible role for CEACAM1 in the pathogenesis of CHC and hepatocellular carcinoma progression.
Abbreviations
- 7‐AAD
7‐aminoactinomycin D
- APC
allophycocyanin
- BILN
HCV nonstructural protein 3/4A protease inhibitor BILN2061
- CEACAM1
carcinoembryonic antigen–related cell‐adhesion molecule 1
- CFSE
carboxy fluorescein succinimidyl ester
- CHB
chronic hepatitis B
- CHC
chronic hepatitis C
- DAA
direct‐acting antiviral agents
- DMEM
Dulbecco’s modified Eagle’s medium
- FCS
fetal calf serum
- ELISA
enzyme‐linked immunosorbent assay
- FGR
full genomic replicon
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HLA
human leukocyte antigen
- ICAM
intercellular adhesion molecule
- IFN
interferon
- JFH
Japanese fulminant hepatitis
- KO
knockout
- mAb
monoclonal antibody
- mRNA
messenger RNA
- MIC
major histocompatibility complex class I chain‐related gene
- NK cells
natural killer cells
- NS5A
nonstructural protein 5A
- PBMC
peripheral blood mononuclear cell
- PE
phycoerythrin
- qRT‐PCR
reverse‐transcription real‐time PCR
- SGR
subgenomic replicon
- PVR
poliovirus receptor
- SVR
sustained virological response
- ULBP
UL16 binding protein
Hepatitis C virus (HCV) infection carries a risk of progression to liver cirrhosis or hepatocellular carcinoma (HCC), which is a poor prognostic factor for patients with chronic hepatitis C (CHC).1 Recently, direct‐acting antiviral agents (DAAs) have become standard treatments for HCV, and DAA therapy produces a sustained virological response (SVR) in 90% to 95% of patients.2 Although DAA therapy has improved the SVR rate, investigation of immunoregulatory mechanisms in patients with CHC is required to develop new therapies that can achieve complete eradication of HCV. The role of the immunological surveillance system in the development of HCC requires attention and further investigation to elucidate new strategies for HCC treatment.
Natural killer cells (NK cells) play crucial roles in chronic liver diseases.3, 4, 5 The activation of NK cell function inhibits HCV replication, suggesting that impairment of NK cell function leads to persistent infection with HCV.6 According to Guerra et al., knockout of the NKG2D gene inhibits NK cell cytotoxicity and increases tumor burden in a spontaneous tumor model.7 In patients with HCC, both cytotoxicity and interferon‐γ (IFN‐γ) production in intrahepatic and peripheral NK cells are impaired.8 Impaired NK cell function in patients with CHC is believed to lead to persistent HCV infection and HCC development. Therefore, elucidating the mechanism by which NK cell activity is suppressed in response to HCV infection is essential for improving prognosis in patients with CHC.
NK cells are controlled by a balance of activation and inhibition.3, 9 Altered NK receptor expression on NK cells is associated with pathogenesis of CHC4, 6, 10, 11, 12 However, ligands for NK receptors have not yet been fully investigated in patients with CHC. Carcinoembryonic antigen–related cell‐adhesion molecule 1 (CEACAM1) inhibits NK cell function.13 CEACAM1 is regarded as both a receptor and ligand for NK cells. Generally, CEACAM1 is expressed on lymphocytes, myelocytes, dendritic cells, epithelial cells, and endothelial cells. CEACAM1 is associated with inflammation, angiogenesis, and immune response to cancer and infections.14 CEACAM1 expression in the liver is associated with disease progression of HCC.15 However, the role of CEACAM1 in CHC is not completely understood.
In this study, we examined the role of CEACAM1 in NK cell function in patients with CHC. CEACAM1 expression was induced by the HCV E2 glycoprotein in HCV infection and subsequently reduced NK cell cytotoxicity, which may be associated with persistent infection and the development of HCC.
Materials and Methods
Cells and Viruses
Huh7.5.1 cells, HEK293T cells, and Huh7.5.1 cells infected with JFH‐1 (Huh7.5.1/JFH‐1 cells) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Japan) containing 10% heat‐inactivated fetal calf serum (FCS), 100 U/mL penicillin, and 100 μg/ml streptomycin. Huh7 cells harboring the HCV subgenomic replicon of the JFH‐1 (SGR) or the full genomic replicon of the JFH‐1 (FGR) were cultured in DMEM supplemented with 10% heat‐inactivated FCS and 1 mg/mL G418 (Nacalai Tesque).16 K562 cells and human NK cells were cultured in RPMI‐1640 medium (DMEM; Sigma, St. Louis, MO) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% heat‐inactivated FCS. Further details are described in the Supporting Information.
Knockout of CEACAM1 in Huh7.5.1 Cells
We generated CEACAM1 knockout (KO) Huh7.5.1 cells using the CRISPR/Cas9 system according to previously described methods.17 We confirmed CEACAM1 deficiency in monoclonal cells by flow cytometry and enzyme‐linked immunosorbent assay (ELISA).
Separation of Human Peripheral Blood Mononuclear and NK Cells
Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll‐Hypaque density centrifugation. NK cells were isolated from PBMCs using an NK isolation kit (Miltenyi Biotech, CA). We defined NK cells as CD3‐CD56+ cells and confirmed that purity was greater than 90% by flow cytometry.
Flow Cytometry
Huh7.5.1 cells and Huh7.5.1/JFH‐1 cells were incubated for 20 minutes with the following antibodies: BAT3 (EPR9223), intercellular adhesion molecule 1 (ICAM1)‐phycoerythrin (PE) (LB‐2), ICAM2‐PE (CBR‐1C2/2), nectin‐2‐PE (R2.525), major histocompatibility complex class I chain‐related gene (MIC)A/B‐PE (6D4), human leukocyte antigen (HLA)‐A, B, C‐PE (G46‐2.6), poliovirus receptor (PVR)‐PE (SKII.4), HLA‐E‐PE (3D12), HLA‐G‐PE (87G), UL16 binding protein 1 (ULBP1)‐allophycocyanin (APC) (170818), ULBP2/5/6‐PE (165903), ULBP3‐APC (166510), and CEACAM1‐PE (283340). Only BAT3 required a secondary antibody (Alexa Fluor 488 [Cell Signaling Technology, Danvers, MA]). We analyzed the expression of these proteins using a FACS Canto II flow cytometer (BD Biosciences, Franklin Lakes, NJ) and DIVA software (BD Biosciences).
ELISA
Serum CEACAM1 levels and CEACAM1 in supernatants were examined using a CEACAM1 ELISA kit (R&D Systems, CA).
Reverse‐Transcription Real‐Time PCR and RNA Silencing
See Supporting Information for details.
Evaluation of NK Cytotoxicity
Carboxy fluorescein succinimidyl ester (CFSE)‐labeled K562 cells (1.5 × 105/well) were added to naïve NK cells (1.5 × 105/well) at an effector cell to target cell (E/T) ratio of 1:1 and incubated at 37°C for 6 hours. After incubation, 7‐aminoactinomycin D (7‐AAD) was added to the cells, and cytotoxicity was measured by flow cytometry. NK cytotoxicity toward CFSE‐labeled K562 cells was calculated using a previously described method.18
CD107a Degranulation Assay and Evaluation of Intracellular IFN‐γ Expression
K562 cells and human NK cells were cocultured at an E/T ratio of 1:1. We added an antihuman CD107a monoclonal antibody (mAb) and incubated the coculture for 1 hour. Subsequently, we added GolgiPlug (1:1000, BD Biosciences) to the coculture and incubated the cells for an additional 3 hours, followed by staining with CD3 and CD56 mAbs. Next, we permeabilized the cells with Cytofix/Cytoperm and Perm/Wash Buffer (BD Bioscience) and stained the cells with an IFN‐γ mAb. CD107a expression on NK cells and intracellular IFN‐γ expression were evaluated by flow cytometry.
Transwell Assay
Huh7.5.1/JFH‐1 cells (5.0 × 104/well) were cultured in the bottom of a 96‐well Transwell cell culture system (pore size 3.0 μm; Corning, Japan), and NK cells (1.5 × 105/well) were cultured on the membranes of the Transwell cell culture inserts. After 24 hours of coculture in Transwells, CFSE‐labeled K562 cells (1.5 × 105/well) were added to the membranes of Transwell cell culture inserts in each well, and the plate was incubated for 6 hours. Subsequently, the cytotoxicity toward CFSE‐labeled K562 cells was calculated.
Reagents
ICAM2‐PE, nectin‐2‐PE, MICA/B‐PE HLA‐A, B, C‐PE CD3‐BV510, CD56‐BV421, and CD107a‐APC antibodies were purchased from BD Biosciences. CEACAM1‐PE, ULBP1‐APC, ULBP2/5/6‐PE, and ULBP3‐APC antibodies were purchased from R&D Systems. The 7‐AAD Viability Staining Solution, PVR‐PE, HLA‐E‐PE, and HLA‐G‐PE were purchased from BioLegend (San Diego, CA). The BAT3 antibody was purchased from Abcam (Cambridge, UK). The ICAM1‐PE antibody was purchased from Becton Dickinson Immunocytometry Systems (San Jose, CA). We purchased the NK Cell Isolation Kit from Miltenyi Biotec (California), the Vybrant CFDA SE Cell Tracer Kit from Life Technologies (Ontario, Canada), and the HCV nonstructural protein 3/4A protease inhibitor BILN2061 (BILN) from Acme Bioscience (Belmont, CA).19
Overexpression of the HCV Core Protein and HCV E1 and E2 Glycoproteins in Huh7.5.1 Cells Using a Lentiviral Vector System
We designed complementary DNAs encoding the HCV core protein, E1 glycoprotein, and E2 glycoprotein. We generated the overexpression models using a previously described method.20
Immunofluorescence Staining of Huh7.5.1/JFH‐1 Cells
Immunostaining for nonstructural protein 5A (NS5A) in Huh7.5.1/JFH‐1 cells cultured in 48‐well plates was performed as previously described.19, 21 Stained cells were analyzed using a fluorescence microscope (BZ‐9000; Keyence, Japan).
Human Subjects
This study enrolled 24 healthy subjects, 27 patients with CHC, 46 patients who were treated with PEGylated IFN‐α plus ribavirin and achieved an SVR, 9 patients with chronic hepatitis B (CHB), 19 patients with NASH, 8 patients who achieved an SVR with sofosbuvir/ledipasvir, and 23 patients with HCC. For analysis of NK cell function, 23 patients with CHC were enrolled to analyze NK cell function. Eleven healthy subjects and 46 patients with CHC were enrolled to analyze messenger RNA (mRNA) levels in human liver tissues. All of the healthy subjects were negative for HCV and hepatitis B virus and had no history of immunological diseases, liver diseases, or malignant diseases. This human study was approved by the Ethics Committee of Osaka University Graduate School of Medicine. Written informed consent was received from all of the participants.
Statistical Analysis
For comparison of two groups, Wilcoxon signed‐rank tests, Mann‐Whitney tests, paired t‐test, and linear regression analyses were performed with the JMP 13.0 software package (SAS Institute, North Carolina). For multiple comparison, one‐way analysis of variance was performed followed by the Tukey honestly significant difference test with the JMP 13.0 software package. P values less than 0.05 were considered significant.
Results
CEACAM1 Expression in JFH‐1 Infected Huh7.5.1 Cells
First, we examined the expression levels of molecules regulating NK cell function on Huh7.5.1 cells and JFH‐1‐infected Huh7.5.1 cells (Huh7.5.1/JFH‐1 cells). The analyzed ligands that activate or inhibit receptors on NK cells were as follows: BAT3, ICAM‐1, ICAM‐2, nectin‐2, PVR, MICA/B, ULBP1, ULBP2/5/6, ULBP3, HLA‐A, B, C, ‐E, HLA‐G, and CEACAM1. Among these ligands, CEACAM1 expression was clearly increased in cells infected with JFH‐1 (Fig. 1A,B), which encouraged us to focus on CEACAM1 expression. We measured the concentration of CEACAM1 in the supernatant and the level of CEACAM1 mRNA in Huh7.5.1/JFH‐1 cells. Significantly higher levels of CEACAM1 were detected in the supernatant of Huh7.5.1/JFH‐1 cells than in the supernatant of Huh7.5.1 cells, and significantly higher levels of CEACAM1 mRNA were observed in Huh7.5.1/JFH‐1 cells than in Huh7.5.1 cells (Fig. 1B). Both CEACAM1 expression on the cells and CEACAM1 levels in the supernatant depended on the time of JFH‐1 infection, and CEACAM1 levels in the supernatant correlated with the surface CEACAM1 level (Fig. 1C,D). We treated Huh7.5.1/JFH‐1 cells with BILN to determine the influence of HCV on CEACAM1 expression. NS5A expression was decreased in Huh7.5.1/JFH‐1 cells treated with BILN compared with untreated cells (Fig. 1E). Significantly lower levels of CEACAM1 were expressed on Huh7.5.1/JFH‐1 cells treated with BILN than on untreated Huh7.5.1/JFH‐1 cells. Consistent with this finding, significantly lower levels of CEACAM1 mRNA were observed in Huh7.5.1/JFH‐1 cells treated with BILN than in untreated Huh7.5.1/JFH‐1 cells (Fig. 1F). These data suggest that HCV regulates CEACAM1 expression.
Figure 1.
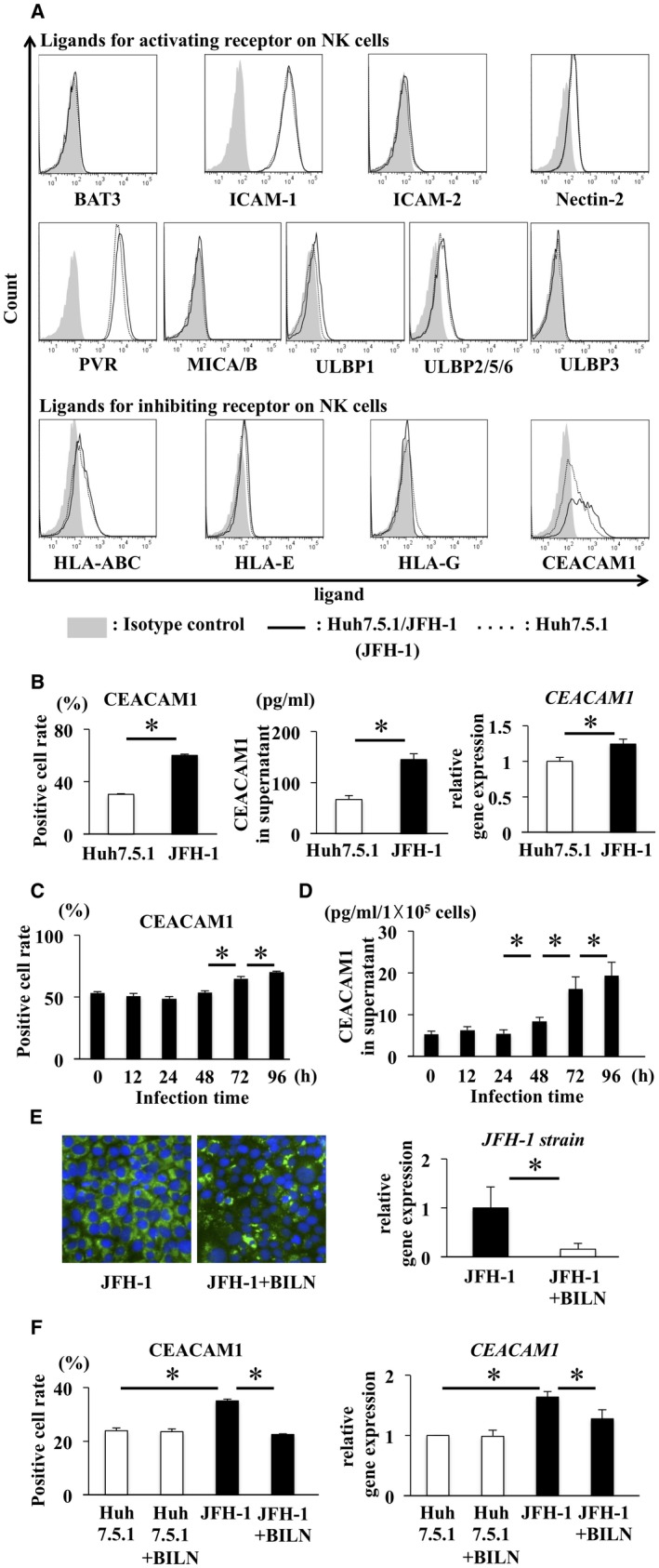
CEACAM1 expression increased on Huh7.5.1 cells infected with JFH‐1 and decreased after treatment. (A) Expression of ligands for NK cells, such as BAT3, ICAM‐1, ICAM‐2, nectin‐2, PVR, MICA/B, ULBP1, ULBP2/5/6, ULBP3, HLA‐A, B, C, HLA‐E, HLA‐G and CEACAM1, was analyzed on Huh7.5.1 cells and Huh7.5.1 cells infected with JFH‐1 (Huh7.5.1/JFH‐1 cells). The dotted line represents Huh7.5.1 cells, the solid line represents Huh7.5.1/JFH‐1 cells, and the shaded area represents the isotype control. (B) CEACAM1 expression on Huh7.5.1 cells and Huh7.5.1/JFH‐1 cells was analyzed by flow cytometry (left panel, means ± SD, n = 4). CEACAM1 levels in the supernatants of Huh7.5.1 cells and Huh7.5.1/JFH‐1 cells were analyzed by ELISA (middle panel, means ± SD, n = 4). In addition, levels of the CEACAM1 mRNA in Huh7.5.1 cells and Huh7.5.1/JFH‐1 cells were analyzed by qRT‐PCR (right panel, means ± SD, n = 4). (C,D) CEACAM1 levels at the cell surface and in supernatants of Huh7.5.1/JFH‐1 cells were measured by flow cytometry and ELISA, respectively, at the indicated times after infection (means ± SD, n = 4). (E) Immunofluorescence staining for NS5A (green) and 4’,6‐diamidino‐2‐phenylindole (DAPI; blue) was performed in Huh7.5.1/JFH‐1 (left panel) cells and Huh7.5.1/JFH‐1 cells treated with BILN (right panel). Levels of JFH‐1 mRNA in Huh7.5.1/JFH‐1 cells before and after treatment were evaluated by qRT‐PCR (means ± SD, n = 4). (F) Analysis of Huh7.5.1 cells, Huh7.5.1 cells + BILN, Huh7.5.1/JFH‐1 cells, and Huh7.5.1/JFH‐1 cells + BILN. CEACAM1 expression on these cells was analyzed by flow cytometry (means ± SD, n = 4). Levels of CEACAM1 mRNA in these cells were evaluated by qRT‐PCR (means ± SD, n = 4). * P < 0.05.
Naïve NK Cells Displayed Increased Cytotoxicity Toward K562 Cells After Coculture With CEACAM1 Knockout Huh7.5.1/JFH‐1 Cells Compared With Huh7.5.1/JFH‐1 Cells
We generated CEACAM1 KO Huh7.5.1 cells and CEACAM1 KO Huh7.5.1/JFH‐1 cells using the CRISPR/Cas9 system to determine whether CEACAM1 expression on Huh7.5.1 cells controls NK cell activity. According to the results of the flow cytometry analysis, neither CEACAM1 KO Huh7.5.1 cells nor CEACAM1 KO Huh7.5.1/JFH‐1 cells expressed CEACAM1 on their surfaces (Fig. 2A). The CEACAM1 protein was not detected in the supernatants from CEACAM1 KO Huh7.5.1 cells or CEACAM1 KO Huh7.5.1/JFH‐1 cells (Fig. 2B). Immunohistochemistry staining for NS5A revealed an equivalent efficiency of JFH‐1 infection between Huh7.5.1/JFH‐1 cells and CEACAM1 KO Huh7.5.1/JFH‐1 cells (Fig. 2C). With the exception of CEACAM1, the expression of ligands for receptors that activate and inhibit NK cells did not differ between CEACAM1 KO Huh7.5.1 cells and Huh7.5.1 cells. Similar results were observed in CEACAM1 KO Huh7.5.1/JFH‐1 cells and Huh7.5.1/JFH‐1 cells (Supporting Fig. S1).
Figure 2.
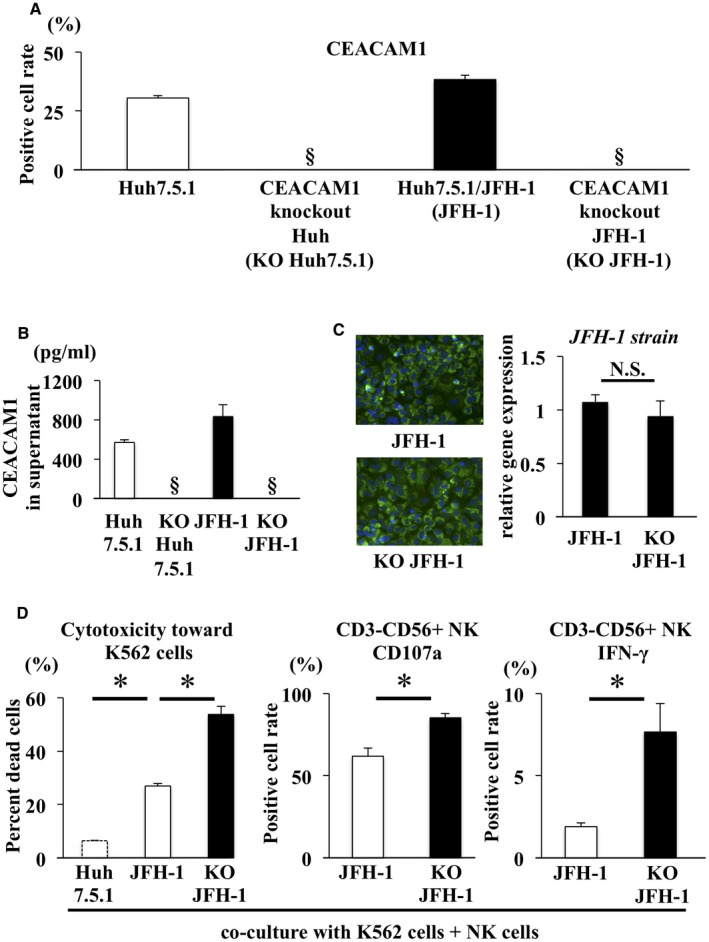
Naïve NK cell cytotoxicity toward K562 cells was increased by coculture with CEACAM1 KO Huh7.5.1/JFH‐1 cells compared with Huh7.5.1/JFH‐1 cells. (A,B) CEACAM1 KO Huh7.5.1 cells were generated using the CRISPR/Cas9 system. Flow cytometry and ELISA were used to confirm that the CEACAM1 KO Huh7.5.1 cells and CEACAM1 KO Huh7.5.1/JFH‐1 cells did not express CEACAM1 at the cell surface and in supernatants, respectively. (C) Immunofluorescence staining for NS5A (green) and DAPI (blue) was performed in Huh7.5.1/JFH‐1 cells (left panel) and CEACAM1 KO Huh7.5.1/JFH‐1 cells (right panel). The efficiency of infection in Huh7.5.1/JFH‐1 cells was similar to CEACAM1 KO Huh7.5.1/JFH‐1 cells, as determined by qRT‐PCR (means ± SD, n = 4). (D) CFSE‐labeled K562 cells were added after 24 hours of NK cell coculture with Huh7.5.1 cells, Huh7.5.1/JFH‐1 cells, or CEACAM1 KO Huh7.5.1/JFH‐1 cells, and cocultures were incubated at 37°C. After 6 hours of incubation, 7‐AAD was added, and cytotoxicity was measured by flow cytometry (means ± SD, n = 4). NK cells were incubated with K562 cells and subjected to flow cytometry, to evaluate the expression of CD107a (means ± SD, n = 4) and the intracellular expression of IFN‐γ (means ± SD, n = 4). Representative data obtained from the peripheral NK cells of healthy subjects are shown. * P < 0.05. §Not detected. Abbreviation: N.S., not significant.
Naïve NK cells were isolated from healthy subjects and cocultured with Huh7.5.1 cells, Huh7.5.1/JFH‐1 cells, or CEACAM1 KO Huh7.5.1/JFH‐1 cells. NK cells displayed significantly greater cytotoxicity toward K562 cells, an NK‐sensitive cell line, after coculture with CEACAM1 KO Huh7.5.1/JFH‐1 cells than coculture with Huh7.5.1/JFH‐1 cells (Fig. 2D). Significantly higher levels of the cytotoxic marker CD107a and IFN‐γ were expressed in NK cells cocultured with CEACAM1 KO Huh7.5.1/JFH‐1 cells than in NK cells cocultured with Huh7.5.1/JFH‐1 cells (Fig. 2D). Thus, CEACAM1 expressed on HCV‐infected Huh7.5.1 cells controls the cytotoxicity and IFN‐γ production of NK cells.
Cell–Cell Contact Was Required to Suppress NK Cell Function
We applied the Transwell culture system to analyze the mechanism by which NK cell function is suppressed. In the Transwell experiments, NK cell cytotoxicity was similar following coculture with Huh7.5.1/JFH‐1 compared with coculture with CEACAM1 KO Huh7.5.1/JFH‐1 cells. In the absence of the Transwell, NK cells cocultured with Huh7.5.1/JFH‐1 cells displayed significantly less cytotoxicity than NK cells cocultured with CEACAM1 KO Huh7.5.1/JFH‐1 cells (Fig. 3A). CD107a expression on NK cells and intracellular IFN‐γ production in NK cells showed similar results to the cytotoxicity assay (Fig. 3B,C). Based on these results, cell–cell contact is required to suppress NK cell function.
Figure 3.
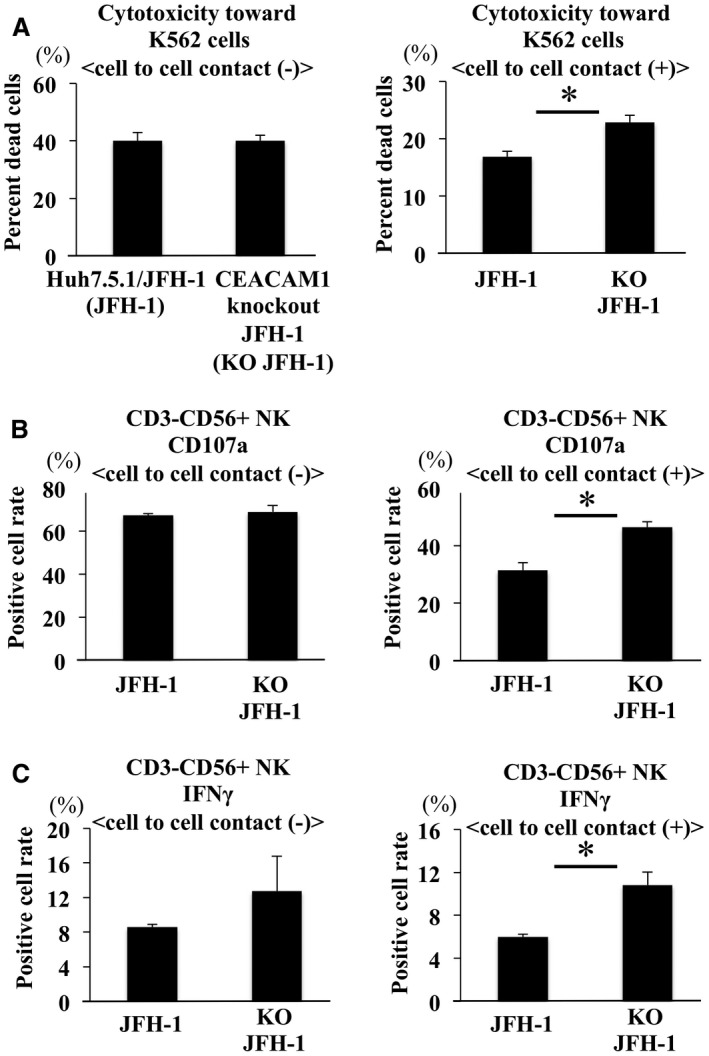
CEACAM1‐mediated suppression of NK cell function required cell–cell contact. (A‐C) Huh7.5.1/JFH‐1 cells were cultured in the bottom of the 96‐well Transwell cell culture system, and NK cells were cultured on the membrane of the Transwell cell culture inserts or in the bottom of the plate. After 24 hours of coculture in a Transwell, CFSE‐labeled K562 cells were added to NK cells and incubated for 6 hours. We subsequently measured the degree of cytotoxicity toward K562 cells (A), CD107a expression on NK cells (B), and IFN‐γ expression in NK cells (C) using flow cytometry (means ± SD, n = 4). Representative data obtained from the peripheral NK cells of healthy subjects are shown. * P < 0.05.
HCV E2 Glycoprotein Increased CEACAM1 Expression on Huh7.5.1/JFH‐1 Cells
CEACAM1 expression on SGR cells was equivalent to that of SGR cells treated with DAAs (Fig. 4A), but significantly higher levels of CEACAM1 were expressed on FGR cells than on FGR cells treated with DAAs (Fig. 4B). FGR cells expressed the HCV core protein, E1 glycoprotein and E2 glycoprotein, but SGR cells did not. We transiently overexpressed the HCV core protein, E1 glycoprotein, and E2 glycoprotein in Huh7.5.1 cells using lentiviral vectors, and CEACAM1 expression on these cells was measured by flow cytometry. Higher levels of the CEACAM1 protein were detected in cells overexpressing the HCV E2 glycoprotein but not with overexpression of the HCV core protein or HCV E1 glycoprotein (Fig. 4C). Therefore, the HCV E2 glycoprotein may increase CEACAM1 expression.
Figure 4.

The HCV E2 glycoprotein may upregulate CEACAM1 expression on Huh7.5.1/JFH‐1 cells. (A) CEACAM1 expression on untreated and treated SGR cells was analyzed by flow cytometry (means ± SD, n = 4). (B) CEACAM1 expression on untreated and treated FGR cells was measured by flow cytometry (means ± SD, n = 4). (C) We overexpressed the HCV core protein, E1 glycoprotein, and E2 glycoprotein in Huh7.5.1 cells using a lentiviral vector system. CEACAM1 expression on these cells was evaluated by flow cytometry (means ± SD, n = 4). The shaded area represents the isotype control, the solid line represents mock‐infected cells, and the dotted line represents overexpressing cells. * P < 0.05.
Serum CEACAM1 Levels and NK Cell Functions in Patients With CHC
We analyzed serum CEACAM1 levels in 24 healthy subjects, 27 patients with CHC, 46 patients who achieved an SVR treated with IFN, 9 patients with CHB, and 19 patients with NASH using an ELISA (Table 1). Significantly higher serum CEACAM1 levels were detected in patients with CHC than in healthy subjects, patients with CHB, and patients with NASH. Significantly lower serum CEACAM1 levels were detected in patients who achieved an SVR than in patients with CHC (Fig. 5A). In the 27 patients with CHC, serum CEACAM1 levels correlated significantly with serum HCV viral loads (Fig. 5B). We also examined serum CEACAM1 levels of patients who achieved an SVR with sofosbuvir/ledipasvir using sequential stored serum samples. Serum CEACAM1 levels at 24 weeks after the end of treatment were same as those at the end of treatment, and both were significantly lower than those at pretreatment (Supporting Fig. S2). Thus, elevated serum CEACAM1 levels may be associated with HCV infection.
Table 1.
Clinical Characteristics of Patient‐Examined Serum CEACAM1 Levels
| HS | CHC | SVR | CHB | NASH | |
|---|---|---|---|---|---|
| Number of patients | 23 | 27 | 46 | 9 | 19 |
| Sex, male/female | 13/10 | 13/14 | 26/20 | 5/4 | 5/14 |
| Age, years, median (range) | 69 (41‐78) | 65 (29‐81) | 63.5 (25‐74) | 51 (31‐78) | 65 (30‐80) |
| AST, IU/L, median (range) | 21 (12‐33) | 67 (20‐365)* | 22 (12‐188) | 33 (20‐108) | 54 (23‐137)* |
| ALT, IU/L, median (range) | 16 (9‐41) | 58 (10‐313)* | 20 (6‐214) | 32 (14‐199) | 72 (18‐181)* |
| HCV‐RNA, log IU/mL, median (range) | Not tested | 6.4 (4.6‐7.4) | Undetected | Not tested | Not tested |
* P < 0.05 versus HS and SVR.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; HS, healthy subject.
Figure 5.

Serum CEACAM1 levels and CD107a expression on NK cells in patients with CHC. (A) Serum CEACAM1 levels were measured in healthy subjects, patients with CHC, patients who achieved an SVR, patients with CHB, and patients with NASH using an ELISA. (B) In patients with CHC, serum CEACAM1 levels correlated significantly with serum HCV‐RNA loads. (C) We examined serum CEACAM1 levels using an ELISA and CD107a expression on NK cells cocultured with K562 cells by flow cytometry. In 23 patients with CHC, serum CEACAM1 levels were inversely correlated with CD 107a expression on NK cells. (D) We measured CD107a expression on naïve NK cells in 15 of 23 patients with CHC before and after treatment by flow cytometry. (E) Serum CEACAM1 levels in all 15 patients shown in (D) were analyzed before and after treatment using an ELISA. (F) Using qRT‐PCR, we examined levels of CEACAM1 mRNA in 11 liver tissues obtained from uninfected patients through a hepatectomy and from 46 liver tissues obtained from patients with CHC through a liver biopsy.
We next analyzed NK cell functions in 23 patients with CHC (Table 2). NK cell functions were evaluated by determining the expression of CD107a, a marker of NK cytotoxicity, on NK cells. Serum CEACAM1 levels were negatively correlated with CD107a expression on NK cells in patients with CHC (R = 0.488, P < 0.05) (Fig. 5C). Based on these data, elevated CEACAM1 levels are associated with decreased NK cell function in patients with CHC. Matched NK cells and serum samples were collected from 15 of the 23 patients with CHC before and after treatment. The expression of CD107a on NK cells in these 15 samples was analyzed at 48 weeks after treatment (Table 2). CD107a expression on NK cells increased in 14 of 15 patients after treatment compared with the pretreatment level (Fig. 5D). Serum CEACAM1 concentrations of all 15 patients decreased after treatment (Fig. 5E).
Table 2.
Clinical Characteristics of Patient‐Examined NK Cell function
| Pretreatment | Posttreatment | |
|---|---|---|
| Number of patients | 23 | 15 |
| Sex, male/female | 7/16 | 3/12 |
| Age, years, median (range) | 68 (48‐90) | 69 (49‐75) |
| AST, IU/L, median (range) | 34 (20‐116) | 20 (18‐31) |
| ALT, IU/L, median (range) | 32 (14‐106) | 14 (7‐25) |
| HCV‐RNA, log IU/mL, median (range) | 6.3 (3.8‐7.1) | Undetected |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Higher Levels of the CEACAM1 mRNA Were Detected in Liver Tissues From Patients With CHC Than in Tissues From Healthy Subjects
We also examined liver expression of CEACAM1 in patients with CHC. Levels of CEACAM1 mRNA were analyzed in 11 healthy subjects and 46 patients with CHC using reverse‐transcription real‐time PCR (qRT‐PCR) (Table 3). In healthy subjects, we extracted total RNA from liver tissues (no tumor) obtained during a hepatectomy, and we extracted total RNA from liver biopsy samples obtained from patients with CHC. Significantly higher levels of the CEACAM1 mRNA were found in patients with CHC than in healthy subjects (Fig. 5F).
Table 3.
Clinical Characteristics of Patient‐Examined CEACAM1 mRNA Levels in Liver Tissues
| HS | CHC | P Value | |
|---|---|---|---|
| Number of patients | 11 | 46 | — |
| Sex, male/female | 8/3 | 18/28 | NS |
| Age, years, median (range) | 60 (38‐80) | 65 (29‐81) | NS |
| AST, IU/L, median (range) | 17 (13‐25) | 48.5 (20‐365) | < 0.05 |
| ALT, IU/L, median (range) | 15 (11‐22) | 48 (10‐313) | < 0.05 |
| HCV‐RNA, log IU/mL, median (range) | Not tested | 6.5 (4.1‐7.4) | — |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase HS, healthy subject; NS, not significant.
Discussion
Few studies have identified ligands inhibiting NK cell function in patients with CHC. 22, 23, 24 HLA‐E, a ligand that suppresses NK cell function, weakens NK cell activity in patients with CHC.11, 25 In this study, CEACAM1 was expressed at higher levels on Huh7.5.1 cells infected with HCV in vitro. Thus, we focused on the role of CEACAM1 in controlling NK cell function and found that HCV‐induced CEACAM1 expression was associated with the suppression of NK cell function. CEACAM1 weakens the activity of NK cells in melanoma cells.13 In this study, cells infected with HCV‐JFH‐1 exhibited increased levels of the CEACAM1 mRNA, CEACAM1 protein on the cell surface, and soluble CEACAM1 in the supernatant. The increased level of CEACAM1 mRNA in infected cells was reversed in cells treated with the NS3/4A protease inhibitor. Thus, HCV infection controls the increase in CEACAM1 expression. We do not exclude the possibility that other inhibitory ligands have roles in suppressing NK cell activity in patients with CHC. However, in our study, both in vitro and clinical data revealed that CEACAM1 is important in suppressing NK cell activity in patients with CHC.
We focused on the function of CEACAM1 in HCV infection. We constructed CEACAM1 KO Huh7.5.1 cells. CEACAM1 was not expressed on the surface of CEACAM1 KO Huh7.5.1 cells, and CEACAM1 was not detected in the supernatant of the cells. Changes in levels of other ligands on the cells were not detected, and the same infection efficacy was observed between the original cells and the CEACAM1 KO Huh7.5.1 cells. NK cells cocultured with CEACAM1 KO Huh7.5.1/JFH‐1 cells exhibited increased cytotoxicity toward K562 cells, higher expression of CD107a on the NK cells, and greater production of IFN‐γ in NK cells than Huh7.5.1/JFH‐1 cells. Similar results were observed in the experiment in which CEACAM1 was knocked down in uninfected Huh7.5.1 cells (Supporting Fig. S3). We examined the expression of NK receptors, such as CEACAM1, NKG2D and NKG2A, on NK cells cocultured with CEACAM1 KO Huh7.5.1/JFH‐1 cells. The expression of these molecules did not change (Supporting Fig. S4). According to Markel et al., CEACAM1 is an inhibitory ligand that binds to another CEACAM1 molecule on NK cells.13 Because a blocking antibody for CEACAM1 is not commercially available, we cannot conclude that CEACAM1 expressed on NK cells inhibited NK cell function based on our results. However, we speculate that CEACAM1 expressed on HCV‐infected cells might interact with CEACAM1 expressed on NK cells. We examined whether soluble CEACAM1 or cell–cell contact had a greater influence on NK cells cocultured with CEACAM1 KO Huh7.5.1/JFH‐1 cells. Based on the results of the Transwell assay, cell–cell contacts were required to inhibit NK cell function. Thus, membrane‐bound CEACAM1 on target cells contributes more to the inhibition of NK cell function than does soluble CEACAM1.
We examined the mechanism by which HCV infection induced CEACAM1 expression. CEACAM1 expression on FGR cells decreased following treatment with DAAs. In contrast, CEACAM1 expression on SGR cells did not change following treatment with DAAs. FGR cells expressed the HCV core protein and E1 and E2 glycoproteins, but SGR cells did not. We overexpressed each HCV protein in Huh7.5.1 cells. Only cells overexpressing the HCV E2 glycoprotein exhibited increased CEACAM1 expression. Thus, the HCV E2 glycoprotein might also be involved in inducing CEACAM1 expression on HCV‐infected cells. As shown in the study by Taylor et al., the HCV E2 glycoprotein inhibits the IFN‐inducible protein kinase PKR,26, 27 and Crotta et al. reported the inhibition of NK cell activity by the binding of HCV E2 glycoprotein to CD81 on NK cells.27 In addition to these mechanisms, CEACAM1 expression induced by the HCV E2 glycoprotein might be related to the inhibition of NK cell function. CEACAM1 is upregulated by IFN‐γ, TNF‐α),28 and IFN‐α.29 However, E2 overexpression increased CEACAM1 (Fig. 4C), but not IFN‐α or TNF‐α (data not shown), in Huh7.5.1 cells. IFN‐γ mRNA was not detected in E2 glycoprotein‐overexpressing Huh7.5.1 cells. These data suggest that IFN‐γ, IFN‐α, or TNF‐α are not involved in the underlining mechanisms by which E2 glycoproteins induce CEACAM1. Further study will be needed to clarify the mechanisms of E2‐induced CECAM1 expression.
Based on the clinical data, significantly higher CEACAM1 levels were detected in the sera of patients with CHC than in healthy subjects, patients who achieved an SVR, patients with CHB, and patients with NASH. In patients with CHC, serum CEACAM1 levels correlated with the HCV RNA load. In addition, significantly higher levels of CEACAM1 mRNA were detected in liver tissues from HCV‐infected patients than in liver tissues from uninfected patients. We also investigated the association between serum CEACAM1 levels and NK cell function. Serum CEACAM1 levels were inversely correlated with CD107a expression on NK cells from patients with CHC. Based on these results, the elevated CEACAM1 levels observed in patients with CHC might be associated with impaired cytotoxicity of NK cells. A previous report used immunohistochemical analysis to reveal that liver CEACAM1 expression correlated with the progression of HCC.15 We examined serum CEACAM1 levels in patients with HCC (Supporting Tables S1 and S2). The median serum level (100.8 ng/mL) in HCC patients was significantly higher than that in CHC patients (Fig. 5A). Therefore, CEACAM1 may be involved in the development of HCC in patients with CHC.
In this study, we focused on CEACAM1 expression in CHC and revealed that CEACAM1 expression during HCV infection decreased NK cell cytotoxicity, which may be related to the development of HCC. Inhibition of CEACAM1 expression could become a new strategy for HCC treatment.
Supporting information
View this article online at wileyonlinelibrary.com.
Supported by a grant‐in‐aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and a grant‐in‐aid for research on hepatitis from the Japan Agency for Medical Research and Development under Grant Number JP18fk0210021.
Potential conflicts of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol 2006;41:17‐27. [DOI] [PubMed] [Google Scholar]
- 2. Mishra P, Murray J, Birnkrant D. Direct‐acting antiviral drug approvals for treatment of chronic hepatitis C virus infection: scientific and regulatory approaches to clinical trial designs. Hepatology 2015;62:1298‐1303. [DOI] [PubMed] [Google Scholar]
- 3. Tatsumi T, Takehara T. Impact of natural killer cells on chronic hepatitis C and hepatocellular carcinoma. Hepatol Res 2016;46:416‐422. [DOI] [PubMed] [Google Scholar]
- 4. Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009;119:1745‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rehermann B. Natural killer cells in viral hepatitis. Cell Mol Gastroenterol Hepatol 2015;1:578‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krämer B, Körner C, Kebschull M, Glässner A, Eisenhardt M, Nischalke HD, et al. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology 2012;56:1201‐1213. [DOI] [PubMed] [Google Scholar]
- 7. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D‐deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008;28:571‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008;129:428‐437. [DOI] [PubMed] [Google Scholar]
- 9. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 2006;55:869‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrison RJ, Ettorre A, Little AM, Khakoo SI. Association of NKG2A with treatment for chronic hepatitis C virus infection. Clin Exp Immunol 2010;161:306‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pembroke T, Christian A, Jones E, Hills RK, Wang EC, Gallimore AM, et al. The paradox of NKp46+ natural killer cells: drivers of severe hepatitis C virus‐induced pathology but in‐vivo resistance to interferon α treatment. Gut 2014;63:515‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, et al. CD66a interactions between human melanoma and NK cells: a novel class I MHC‐independent inhibitory mechanism of cytotoxicity. J Immunol 2002;168:2803‐2810. [DOI] [PubMed] [Google Scholar]
- 14. Gray‐Owen SD, Blumberg RS. CEACAM1: contact‐dependent control of immunity. Nat Rev Immunol 2006;6:433‐446. [DOI] [PubMed] [Google Scholar]
- 15. Kiriyama S, Yokoyama S, Ueno M, Hayami S, Ieda J, Yamamoto N, et al. CEACAM1 long cytoplasmic domain isoform is associated with invasion and recurrence of hepatocellular carcinoma. Ann Surg Oncol 2014;21(Suppl 4):S505‐S514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999;285:110‐113. [DOI] [PubMed] [Google Scholar]
- 17. Fukuhara T, Tamura T, Ono C, Shiokawa M, Mori H, Uemura K, et al. Host‐derived apolipoproteins play comparable roles with viral secretory proteins Erns and NS1 in the infectious particle formation of Flaviviridae. PLoS Pathog 2017;13:e1006475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishio A, Tatsumi T, Nawa T, Suda T, Yoshioka T, Onishi Y, et al. CD14(+) monocyte‐derived galectin‐9 induces natural killer cell cytotoxicity in chronic hepatitis C. Hepatology 2017;65:18‐31. [DOI] [PubMed] [Google Scholar]
- 19. Wen X, Abe T, Kukihara H, Taguwa S, Mori Y, Tani H, et al. Elimination of hepatitis C virus from hepatocytes by a selective activation of therapeutic molecules. PLoS One 2011;6:e15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukuhara T, Yamamoto S, Ono C, Nakamura S, Motooka D, Mori H, et al. Quasispecies of hepatitis C virus participate in cell‐specific infectivity. Sci Rep 2017;7:45228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fukuhara T, Kambara H, Shiokawa M, Ono C, Katoh H, Morita E, et al. Expression of microRNA miR‐122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol 2012;86:7918‐7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Golden‐Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high‐risk individuals and inhibits replication in vitro. Hepatology 2010;52:1581‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sene D, Levasseur F, Abel M, Lambert M, Camous X, Hernandez C, et al. Hepatitis C virus (HCV) evades NKG2D‐dependent NK cell responses through NS5A‐mediated imbalance of inflammatory cytokines. Plos Pathogens 2010;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoon JC, Lim JB, Park JH, Lee JM. Cell‐to‐cell contact with hepatitis C virus‐infected cells reduces functional capacity of natural killer cells. J Virol 2011;85:12557‐12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jinushi M, Takehara T, Tatsumi T, Kanto T, Miyagi T, Suzuki T, et al. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell‐induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol 2004;173:6072‐6081. [DOI] [PubMed] [Google Scholar]
- 26. Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon‐inducible protein kinase PKR by HCV E2 protein. Science 1999;285:107‐110. [DOI] [PubMed] [Google Scholar]
- 27. Crotta S, Stilla A, Wack A, D'Andrea A, Nuti S, D'Oro U, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exper Med 2002;195:35‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi H, Okai Y, Paxton RJ, Hefta LJ, Shively JE. Differential regulation of carcinoembryonic antigen and biliary glycoprotein by gamma‐interferon. Cancer Res 1993;53:1612‐1619. [PubMed] [Google Scholar]
- 29. Klaile E, Klassert TE, Scheffrahn I, Müller MM, Heinrich A, Heyl KA, et al. Carcinoembryonic antigen (CEA)‐related cell adhesion molecules are co‐expressed in the human lung and their expression can be modulated in bronchial epithelial cells by non‐typable Haemophilus influenzae, Moraxella catarrhalis, TLR3, and type I and II interferons. Respir Res 2013;14:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials