

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in children and adults. This study examined the relationship between hepatic nuclear receptor (NR) expression and histologic features of NAFLD. Drugs targeting a variety of NRs for nonalcoholic steatohepatitis (NASH) are in clinical trials. Liver messenger RNA was isolated from 40 children (10‐19 years) undergoing end‐of‐treatment biopsy in the Treatment of NAFLD in Children (TONIC) trial. High‐throughput quantitative polymerase chain reaction assayed NR messenger RNA. Cluster analysis was used to group 36 NRs, and NR levels were related to histologic measures of specific NAFLD features. Cluster analysis determined five groupings of NRs. Significant (P < 0.05) differential expressions of specific NRs associated with histologic measures include farnesoid X receptor alpha and retinoic acid receptor (RARβ and RARβ) for steatosis; estrogen receptor alpha (ERα) and peroxisome proliferator‐activated receptor gamma 3 (PPARγ3) for hepatocellular ballooning; ER and PPARγ2 for lobular inflammation; PPARα/δ/γ1/γ2, ERα, constitutive androstane receptor, chicken ovalbumin upstream promoter transcription factor 1, RARα, RARβ1, retinoid X receptor, pregnane X receptor, thyroid hormone receptors α and β, and nuclear receptor related‐1 for fibrosis; and ERα and RARβ/β1/α for diagnosis of NASH. Conclusion: Differential expression of specific NRs correlates with histologic severity of specific NAFLD features. These NRs are pleiotropic transactivators regulating basal metabolic functions and inflammatory responses. Derangement of activity of these receptors in NAFLD provides a rationale for exploiting their ability with receptor‐specific ligands to ameliorate NASH and its consequences.
Abbreviations
- CAR
constitutive androstane receptor
- cDNA
complementary DNA
- COUP‐TF
chicken ovalbumin upstream promoter transcription factor
- ER
estrogen receptor
- FXR
farnesoid X receptor
- GCNF
germ cell nuclear factor
- LXR
liver X receptor
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH CRN
nonalcoholic steatohepatitis Clinical Research Network
- NASH
nonalcoholic steatohepatitis
- NR
nuclear receptor
- NURR1
nuclear receptor related 1
- OCA
obeticholic acid
- PCR
polymerase chain reaction
- PPAR
peroxisome proliferator‐activated receptor
- PXR
pregnane X receptor
- qPCR
quantitative polymerase chain reaction
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- TONIC
Treatment of Nonalcoholic Fatty Liver Disease in Children
- TR
thyroid hormone receptor
- VDR
vitamin D receptor
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease among preadolescents and adolescents in the United States.( 1 ) NAFLD encompasses a spectrum of histologic features from isolated steatosis (generally nonprogressive) to potentially severe nonalcoholic steatohepatitis (NASH).( 2 ) NASH is characterized at the molecular level by oxidative stress and activation of proinflammatory profibrotic cascades and is defined histologically as steatosis with inflammation and hepatocellular ballooning, often accompanied by fibrosis.( 3, 4 ) NASH can ultimately lead to decompensated cirrhosis or hepatocellular carcinoma.( 2, 5 ) Among adults, the frequency of NASH as an indication for liver transplant increased 800% between 2001 and 2009 and has become the second most common indication overall( 6 ); NASH is on a trajectory to become the most common indication for transplant by 2025.( 7 ) In the United States, the prevalence of NAFLD among teenagers has doubled in the past 20 years and 38% of obese children are reported to have NAFLD.( 8, 9 )
Some patients with NAFLD maintain isolated hepatic steatosis while others develop inflammation, cell injury, and fibrosis (NASH), although the mechanisms underlying this spectrum of outcomes are unclear.( 10, 11 ) The development of NASH is believed to involve insulin resistance, lipotoxicity, and the activation of necro‐inflammatory pathways that lead to mitochondrial dysfunction and the release of factors that trigger apoptosis.( 3, 12 ) Attenuation of these steps is requisite for improving outcomes.
Nuclear receptors (NRs) are ligand‐inducible transcription factors that activate hierarchical transcriptional networks. They coordinate multi‐organ physiologic pathways related to growth, nutrient uptake, metabolic homeostasis, and inflammation.( 13 ) NRs are expressed in a tissue‐dependent, time‐dependent, and developmentally specific manner, and each NR has its own subset of protein targets.( 14 ) NR ligands intimately related to NASH pathophysiology include fatty acids (both endogenous and dietary), bile acids, sex hormones, and vitamins A and D.( 15 )
In the liver, NRs act as sensors of the metabolic milieu, activating transcription of cellular machinery for maintenance of homeostasis. NRs act as pleiotropic transactivators of transcriptional cascades, coordinating hepatic functions, including detoxification, storage and release of glucose, and production and uptake of cholesterol.( 16 ) NRs and their protein targets are intimately involved in pathologic processes underlying the metabolic syndrome and NAFLD, including insulin resistance, hepatic lipid accumulation and inflammation, and increased intestinal permeability.( 15, 17 )
Drugs that modulate NR activity, including thiazolidinediones and other peroxisome proliferator‐activated receptor (PPAR) agonists, are widely used in the treatment of metabolic disease. Several drugs, including obeticholic acid (OCA; a farnesoid X receptor [FXR]α agonist), pioglitazone (a PPARγ agonist), and elafibranor (GFT505, a dual PPARδ/PPARα agonist) have undergone or are undergoing clinical trials in humans for the treatment of adult NAFLD and have demonstrated variable efficacy.
Despite the accumulating evidence for the role of NRs in NASH pathophysiology and treatment, NR expression patterns in children with NAFLD have not been studied. We hypothesized that NRs would be variably expressed in subjects with differing degrees of severity as assessed by histology and that NRs with therapeutic agonists that are currently undergoing testing (i.e., FXRα, PPARγα/δ, thyroid hormone receptor [TR]β) would be most likely to differ with histology. The expression pattern of NRs in NAFLD may be a useful tool in precision medicine to identify and personalize treatment of those at particular risk.
Patients and Methods
Study Population
This ancillary study of the Treatment of NAFLD in Children (TONIC) trial had Institutional Review Board approval at each of the eight clinical centers comprising The National Institute of Diabetes and Digestive and Kidney Diseases‐sponsored NASH Clinical Research Network (NASH CRN) and approval as an ancillary study of the NASH CRN. The NR expression participants were obtained only from the University of California, San Diego site. Written informed consent was obtained from a parent or legal guardian, and written assent was obtained from children 8‐17 years prior to screening and enrollment.
TONIC was a phase IIb, multicenter, double‐blind, randomized, placebo‐controlled trial of either metformin or vitamin E versus placebo in 173 subjects 8‐17 years with biopsy‐proven NAFLD and persistent alanine aminotransferase >60.( 17, 18 ) Exclusion criteria for TONIC have been described and include diabetes, cirrhosis, use of drugs that could cause or treat fatty liver, and bariatric or hepatobiliary surgery. The TONIC trial concluded in March 2010. End‐of‐treatment percutaneous liver biopsies were obtained on enrolled subjects after 96 weeks of oral daily dosing of vitamin E (800 IU/day), metformin (1 g/day), or placebo. Flash‐frozen fragments of the end‐of‐treatment biopsies from the last 40 subjects enrolled at the University of California, San Diego were prepared for profiling for NR expression as described below. The liver tissue used in this study consisted of small fragments of the biopsy taken for assessment of histologic changes, and a second pass was not made to obtain the sample. These 40 subjects had been randomized to receive metformin (n = 12), vitamin E (n = 14), or placebo (n = 14). Ethnic makeup, body mass index, and Tanner staging of the subjects at time of enrollment are summarized in Table 1.
Table 1.
Phenotypic Characterization of 40 Children With and Without NASH With Nuclear Receptor Expression Profiles
| Not NASH (n = 21) | Borderline/Definite NASH (n = 19) | P Value† | |||
|---|---|---|---|---|---|
| n | Mean (SD) / % | n | Mean (SD) / % | ||
| Demographics | |||||
| Female sex | 4 | 19% | 3 | 16% | 1.00 |
| Hispanic ethnicity | 16 | 76% | 18 | 95% | 0.19 |
| Age in years | |||||
| Mean (SD) | 15.0 (2.4) | 14.7 (2.7) | 0.72 | ||
| Minimum, maximum | 11, 19 | 10, 19 | |||
| Tanner stage | 1.00 | ||||
| 1‐3 | 13 | 62% | 11 | 65% | |
| 4‐5 | 8 | 38% | 6 | 35% | |
| Anthropometric | |||||
| Body mass index in kg/m2 | 32.8 (6.4) | 33.3 (5.3) | 0.80 | ||
| Histology* | |||||
| Steatosis score | 0.003 | ||||
| 0 = <5% | 7 | 33% | 0 | 0% | |
| 1 = 5%‐33% | 9 | 43% | 6 | 32% | |
| 2 = 34%‐66% | 2 | 10% | 10 | 53% | |
| 3 = >66% | 3 | 14% | 3 | 16% | |
| Lobular inflammation score | <0.001 | ||||
| 0 = 0 | 2 | 10% | 0 | 0% | |
| 1 = <2 under 20× magnification | 19 | 90% | 6 | 32% | |
| 2 = 2‐4 under 20× magnification | 0 | ‐ | 9 | 47% | |
| 3 = >4 under 20× magnification | 0 | ‐ | 4 | 21% | |
| Ballooning score | <0.001 | ||||
| 0 = none | 18 | 86% | 5 | 26% | |
| 1 = few | 3 | 14% | 6 | 32% | |
| 2 = many | 0 | ‐ | 8 | 42% | |
| Fibrosis stage | <0.001 | ||||
| 0 = none | 10 | 48% | 2 | 11% | |
| 1a = mild, zone 3 perisinusoidal | 2 | 10% | 2 | 11% | |
| 1b = moderate, zone 3 perisinusoidal | 0 | 0% | 1 | 5% | |
| 1c = portal/periportal only | 8 | 38% | 2 | 11% | |
| 2 = zone 3 and periportal | 1 | 5% | 8 | 42% | |
| 3 = bridging | 0 | 0% | 4 | 21% | |
| TONIC treatment | 0.05 | ||||
| Metformin | 5 | 24% | 7 | 37% | |
| Vitamin E | 11 | 53% | 3 | 16% | |
| Placebo | 5 | 24% | 9 | 47% | |
Biopsies at 96 weeks of treatment were selected for NR expression levels.
Based on Fisher’s exact test for categorical variables and t test for continuous variables.
Histologic Evaluation
Biopsy specimens were evaluated, scored, and graded for histologic features of NAFLD by the Pathology Committee of NASH CRN in a centralized consensus review format; they were blinded to all clinical information and used validated criteria by Kleiner et al.( 18 ) Specimens were scored for steatosis (grade 0 [macrovesicular fat in <5% of hepatocytes], grade 1 [5%‐33%], grade 2 [34%‐66%], and grade 3 [>66%]), lobular inflammation (0‐3), ballooning (0‐2), and fibrosis (stage 0, stage 1a [mild zone 3], stage 1b [moderate zone 3 perisinusoidal], stage 1c [portal/periportal fibrosis only], stage 2 [zone 3 and periportal fibrosis], stage 3 [bridging fibrosis], and stage 4 [cirrhosis]).
Additionally, pattern‐based diagnoses were given for each biopsy. The NAFLD pattern of isolated steatosis (“not NASH”) versus “borderline zone 1 pattern” (a pattern more common in pediatric NAFLD), “borderline zone 3 pattern,” or “definite NASH” were determined according to specific criteria published by the committee.( 9 )
NR Profiling
Total liver messenger RNA (mRNA) was isolated using the RNAqueous‐Micro kit (Life Technologies, Grand Island, NY). Complementary DNA (cDNA) amplification was performed using the WT‐Ovation RNA Amplification System (NuGen, San Carlos, CA). The cDNA product yield and purity were assessed using an Agilent 2100 Bioanalyzer. The Total Human Reference RNA was prepared for cDNA synthesis. Xpress Reference Human Universal RNA (SuperArray/Qiagen, Hilden, Germany) was spiked with 1 μL of each NASH CRN sample cDNA and used to generate a quantitative polymerase chain reaction (qPCR) standard curve.
Relative NR expression levels were determined by high‐throughput qPCR. Primers and probes were designed using ABI PrimerExpress software. Sequences of primers and probes used in this study have been reported( 19 ) and were designed to have similar amplification efficiencies. All probes for TaqMan real‐time PCR were 5′ labeled with 6‐carboxyfluorescein and 3′ labeled with 6‐carboxytetramethylrhodamine. PCR reactions were assembled using a Janus automated workstation (PerkinElmer, Shelton, CT) containing 1×TaqMan Universal PCR Master Mix, 300 nM primers, 250 nM probe, and cDNA equivalent to 1 ng total RNA in a 10‐mL volume. PCR was performed in an ABI PRISM 7700 detection system (Perkin Elmer) at 50C for 2 minutes and 95C for 10 minutes, followed by 40 two‐step cycles of 95C for 15 seconds and 60C for 1 minute. Relative mRNA levels were calculated using the comparative delta‐Ct method and normalized against separately measured 50S ribosomal protein L15 mRNA levels in the same total RNA samples.( 20 ) Expression of the 36 NRs known at the time of analysis and that had primer sets validated for amplification efficiency criteria was compared among categories of steatosis, lobular inflammation, ballooning, and NASH diagnosis. The normalized expression level reported for each NR represents the number of qPCR cycles necessary to generate detectable fluorescence of the receptor in the sample. These units are nonlinear representations of the amount of base mRNA substrate.
Statistical Analysis
Demographic, anthropometric, and histologic features were compared between children with borderline or definite NASH versus those without NASH (Table 1). P values were derived from t tests for continuous variables or Fisher’s exact test for categorical variables.
The outcomes of interest for the current investigation were the expression profile of 36 hepatic NRs in pediatric patients with NAFLD. Exploratory analyses included boxplots of the NRs sorted by the median level of normalized mRNA level (Fig. 1).
Figure 1.
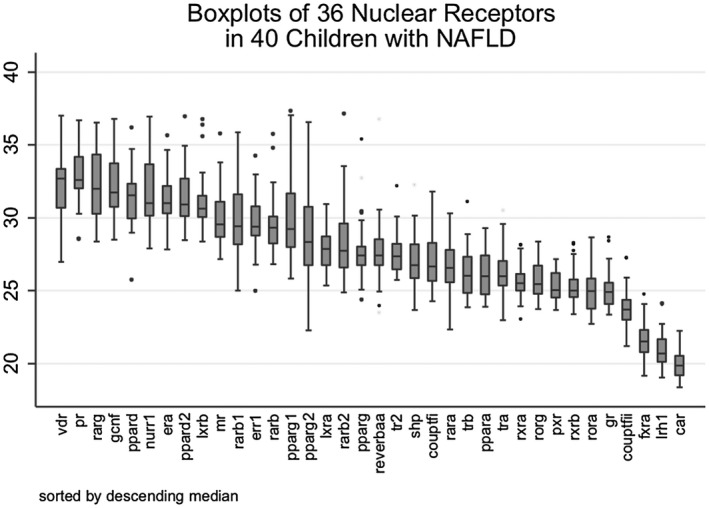
Boxplots of 36 NRs sorted by descending median in 40 children with NAFLD. Boxplots of all 36 NRs illustrate the breadth of expression of each receptor. VDR demonstrates the lowest mean expression (higher number of PCR cycles needed to detect), while CAR demonstrates the highest. Most receptors had at least one outlier. Abbreviations: err; estrogen‐related receptor; gr, glucocorticoid receptor; lrh, liver receptor homolog 1; mr, mineralocorticoid receptor; pr, progesterone receptor; ror, RAR‐related orphan receptor alpha; reverb, nuclear receptor subfamily 1 group D member 1; shp, small heterodimer partner.
Cluster analysis was used to assess the expression profile of NRs. Groupings of the NRs were determined using hierarchical clustering with average linkage based on their normalized expression levels.( 21 ) Twelve NRs had observations with missing data (eight missing vitamin D receptor (VDR), seven missing progesterone receptor, eight missing retinoic acid receptor (RAR)γ, four missing germ cell nuclear factor, one missing PPARδ, three missing nuclear receptor related 1 (NURR1), two missing PPARδ2, one missing liver X receptor (LXR)β, one missing mineralocorticoid receptor, four missing estrogen‐related receptor, one missing PPARγ1, one missing PPARγ2). Missing data were imputed with the median and used for both cluster and regression analyses.
Results of the NR and patient cluster analyses were displayed separately as linear dendrograms (Supporting Figs. S1 and S2) and simultaneously as a heatmap (Fig. 2). The Duda‐Hart criterion for stopping rules was used to determine the number of clusters.
Figure 2.
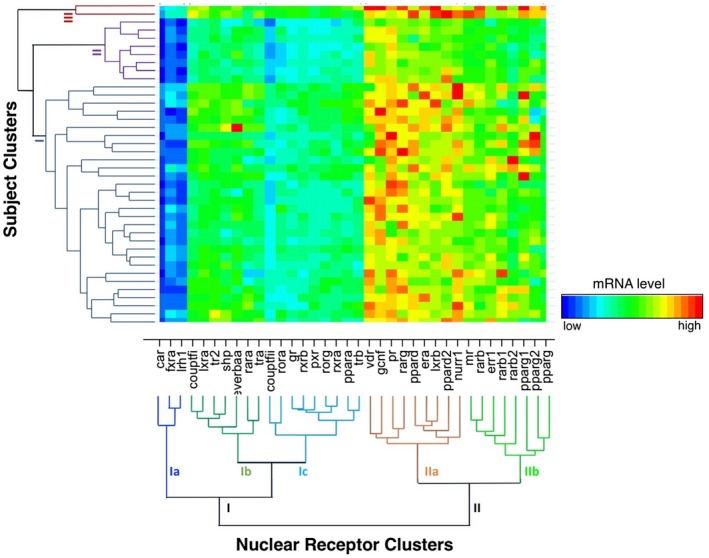
Hierarchical clustering of NR expression levels relative to subject clusters. The normalized mRNA expression levels are displayed as a heatmap, organized by the results of the NR cluster analysis (main x axis) and the subject cluster analysis (main y axis). NRs cluster into two superfamilies and five individual clusters (Ia: CAR, FXRα, LRH1; Ib: COUP‐TF1, LXRα, TR2, REV‐ERBa‐α, RARα, TRα; Ic: COUP‐TFII, RORα, GR, RXRβ, PXR, ROR, RXRα, PPARα, TR; IIa: VDR, GCNF, PR, RARγ, PPARδ, ERα, LXRβ, PPARδ2, NURR1; IIb: MR, RARβ, ERR1, RARβ1, RARβ2, PPARγ1, PPARγ2, PPARγ). Subjects cluster into three clusters: cluster I, 30 subjects; cluster II, 8 subjects; cluster III, 2 subjects. For both dendrograms, the y axis represents the L2 (Euclidean) dissimilarity measure. Abbreviations: ERR, estrogen‐related receptor; GR, glucocorticoid receptor; LRH1, liver receptor homolog 1; MR, mineralocorticoid receptor; PR, progesterone receptor; REV‐ERBa‐α, nuclear receptor subfamily 1 group D member 1; RORα, RAR‐related orphan receptor alpha; shp, small heterodimer partner.
Each of the 36 NRs was compared across the following histologic features: steatosis (grade ≤33% versus grade >33%), steatohepatitis (none versus borderline/definite), fibrosis stage (none/mild versus moderate/advanced), lobular inflammation (<2 versus 2+ foci), and ballooning (none versus few/many). To downweight the effect of outliers, medians were used as summary statistics and robust linear regression was used to derive P values. Logistic regression was used to globally test the set of NRs within each cluster as predictors of each histologic feature dichotomized into higher versus lower severity (Table 2).
Table 2.
Comparison of NRs by Histologic Characteristics Sorted by Dissimilarity Measure From Cluster Analysis
| Steatosis | Ballooning | Lobular Inflammation | Fibrosis | Type of NAFLD | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <33% (n = 22) | 34%+ (n = 18) | P * | None (n = 23) | Any (n = 17) | P * | None/Mild (n = 27) | Mod/Severe (n = 13) | P * | None (n = 12) | Any (n = 28) | P * | Not NASH (n = 21) | NASH (n = 19) | P * | |
| Median† | Median† | Median† | Median† | Median† | |||||||||||
| Cluster IA | |||||||||||||||
| CAR | 19.9 | 19.9 | 0.91 | 19.9 | 19.8 | 1.00 | 19.9 | 19.8 | 0.26 | 20.5 | 19.7 | 0.02 | 19.9 | 19.8 | 0.46 |
| FXRα | 21.8 | 20.9 | 0.03 | 21.5 | 21.6 | 1.00 | 21.7 | 21.3 | 0.76 | 21.2 | 21.6 | 0.35 | 21.7 | 21.3 | 0.70 |
| LRH1 | 21.1 | 20.4 | 0.14 | 20.7 | 20.9 | 0.61 | 20.7 | 20.6 | 0.95 | 20.5 | 20.8 | 0.92 | 20.7 | 20.7 | 0.94 |
| Global test | 0.06 | 0.99 | 0.43 | 0.02 | 0.63 | ||||||||||
| Cluster IB | |||||||||||||||
| COUP‐TFI | 26.6 | 26.9 | 0.85 | 27.2 | 26.6 | 0.35 | 27.0 | 26.6 | 0.24 | 28.6 | 26.3 | 0.003 | 27.2 | 26.6 | 0.07 |
| LXRα | 27.9 | 27.6 | 0.43 | 27.9 | 27.9 | 0.32 | 27.9 | 27.9 | 0.69 | 28.1 | 27.4 | 0.21 | 27.9 | 27.9 | 0.33 |
| TR2 | 27.6 | 27.0 | 0.37 | 27.5 | 27.1 | 0.55 | 27.5 | 27.2 | 0.96 | 27.9 | 27.2 | 0.23 | 27.5 | 27.1 | 0.48 |
| SHP | 26.6 | 27.0 | 0.61 | 26.6 | 27.0 | 0.88 | 26.7 | 27.0 | 0.93 | 27.1 | 26.6 | 0.42 | 26.7 | 26.8 | 0.60 |
| REV‐ERBAα | 28.0 | 27.3 | 0.85 | 28.3 | 27.2 | 0.08 | 27.3 | 27.5 | 0.74 | 28.3 | 27.3 | 0.17 | 28.2 | 27.4 | 0.34 |
| RARα | 26.6 | 26.5 | 0.47 | 27.0 | 26.4 | 0.89 | 26.6 | 26.4 | 0.62 | 27.8 | 26.1 | 0.005 | 27.0 | 26.4 | 0.68 |
| TRα | 26.0 | 26.0 | 0.89 | 26.1 | 25.8 | 0.94 | 26.0 | 25.9 | 0.82 | 26.7 | 25.7 | 0.05 | 26.0 | 26.0 | 0.58 |
| Global test | 0.40 | 0.55 | 0.56 | 0.006 | 0.04 | ||||||||||
| Cluster IC | |||||||||||||||
| COUP‐TFII | 23.8 | 23.7 | 0.37 | 23.9 | 23.6 | 0.43 | 23.9 | 23.6 | 0.26 | 24.2 | 23.5 | 0.06 | 23.9 | 23.6 | 0.12 |
| RORα | 25.3 | 24.6 | 0.12 | 25.0 | 25.0 | 0.83 | 25.0 | 25.2 | 0.77 | 24.8 | 25.1 | 0.59 | 25.0 | 25.2 | 0.95 |
| GR | 25.1 | 24.7 | 0.21 | 24.9 | 24.8 | 0.34 | 24.9 | 24.6 | 0.20 | 24.9 | 24.9 | 0.71 | 24.9 | 24.8 | 0.26 |
| RXRβ | 25.0 | 25.0 | 0.91 | 24.9 | 25.2 | 0.55 | 25.0 | 24.8 | 0.76 | 25.5 | 24.7 | 0.02 | 25.0 | 24.9 | 0.91 |
| PXR | 25.2 | 25.0 | 0.92 | 25.7 | 25.0 | 0.63 | 25.7 | 24.9 | 0.33 | 26.2 | 24.9 | 0.005 | 25.7 | 25.0 | 0.41 |
| RORγ | 25.4 | 25.5 | 0.90 | 25.6 | 25.4 | 0.59 | 25.5 | 25.0 | 0.82 | 26.1 | 25.2 | 0.06 | 25.6 | 25.4 | 0.62 |
| RXRα | 25.7 | 25.3 | 0.28 | 25.7 | 25.4 | 0.30 | 25.5 | 26.0 | 0.60 | 25.7 | 25.5 | 0.72 | 25.7 | 25.3 | 0.32 |
| PPARα | 26.1 | 25.4 | 0.93 | 26.0 | 26.0 | 0.94 | 26.1 | 25.5 | 0.45 | 27.2 | 25.4 | 0.007 | 26.1 | 25.5 | 0.26 |
| TRβ | 26.1 | 25.3 | 0.85 | 26.1 | 25.4 | 0.53 | 26.1 | 25.3 | 0.33 | 27.0 | 25.7 | 0.05 | 26.1 | 25.3 | 0.34 |
| Global test | 0.14 | 0.50 | 0.03 | 0.09 | 0.21 | ||||||||||
| Cluster IIA | |||||||||||||||
| VDR | 32.0 | 32.1 | 0.65 | 33.0 | 32.0 | 0.39 | 32.9 | 32.0 | 0.97 | 31.9 | 32.9 | 0.22 | 33.0 | 32.1 | 0.58 |
| GCNF | 31.4 | 31.8 | 0.65 | 31.8 | 31.7 | 0.86 | 33.1 | 31.3 | 0.17 | 33.9 | 31.4 | 0.02 | 32.8 | 31.5 | 0.41 |
| PR | 32.4 | 32.6 | 0.84 | 32.6 | 32.6 | 0.81 | 32.6 | 32.5 | 0.80 | 33.4 | 32.2 | 0.16 | 32.6 | 32.5 | 0.71 |
| RARγ | 32.3 | 31.3 | 0.54 | 32.3 | 31.3 | 0.20 | 32.3 | 31.3 | 0.15 | 32.4 | 31.4 | 0.20 | 32.9 | 30.9 | 0.02 |
| PPARδ | 31.6 | 31.4 | 0.85 | 31.6 | 31.0 | 0.20 | 31.7 | 30.8 | 0.08 | 32.6 | 31.2 | 0.006 | 31.7 | 30.8 | 0.06 |
| ERα | 31.6 | 30.9 | 0.28 | 31.5 | 30.9 | 0.05 | 31.5 | 30.7 | 0.02 | 32.2 | 30.9 | 0.009 | 31.6 | 30.9 | 0.02 |
| LXRβ | 30.5 | 30.8 | 0.76 | 30.8 | 30.5 | 0.51 | 30.8 | 30.5 | 0.54 | 31.1 | 30.5 | 0.05 | 30.9 | 30.5 | 0.55 |
| PPARδ2 | 31.2 | 30.8 | 0.70 | 30.7 | 31.0 | 0.78 | 30.9 | 31.7 | 0.78 | 30.9 | 31.0 | 0.94 | 30.9 | 31.2 | 0.75 |
| NURR1 | 31.1 | 30.7 | 0.40 | 31.6 | 30.6 | 0.36 | 31.6 | 30.6 | 0.27 | 33.0 | 30.9 | 0.04 | 31.8 | 30.6 | 0.16 |
| Global test | NC§ | 0.42 | 0.18 | 0.21 | 0.01 | ||||||||||
| Cluster IIB | |||||||||||||||
| MR | 29.7 | 29.2 | 0.24 | 29.4 | 29.6 | 0.65 | 29.6 | 29.6 | 0.53 | 30.1 | 29.4 | 0.33 | 29.6 | 29.6 | 0.49 |
| RARβ | 29.6 | 28.4 | 0.05 | 29.5 | 29.0 | 0.21 | 29.4 | 29.0 | 0.64 | 29.2 | 29.4 | 0.73 | 29.5 | 28.3 | 0.05 |
| ERR1 | 29.7 | 29.2 | 0.50 | 29.1 | 29.7 | 0.48 | 29.4 | 29.5 | 0.90 | 29.2 | 29.6 | 0.86 | 29.1 | 29.7 | 0.91 |
| RARβ1 | 30.0 | 28.4 | 0.02 | 30.0 | 29.1 | 0.26 | 30.0 | 28.7 | 0.10 | 31.6 | 29.1 | 0.01 | 30.1 | 28.7 | 0.03 |
| RARβ2 | 27.8 | 27.0 | 0.20 | 27.9 | 27.6 | 0.61 | 27.9 | 27.1 | 0.52 | 28.7 | 27.3 | 0.09 | 27.9 | 27.1 | 0.41 |
| PPARγ1 | 29.6 | 29.1 | 0.48 | 29.0 | 29.7 | 0.78 | 29.6 | 29.0 | 0.33 | 31.8 | 28.9 | 0.001 | 29.7 | 29.0 | 0.20 |
| PPARγ2 | 29.2 | 26.9 | 0.07 | 28.3 | 29.3 | 0.44 | 29.3 | 26.7 | 0.003 | 30.8 | 28.2 | 0.04 | 29.1 | 27.9 | 0.10 |
| PPARγ3 | 27.3 | 27.6 | 0.93 | 27.1 | 27.8 | 0.003 | 27.6 | 26.8 | 0.07 | 27.5 | 27.4 | 0.75 | 27.4 | 27.5 | 0.48 |
| Global test | 0.03 | 0.06 | 0.09 | 0.11 | 0.08 | ||||||||||
Abbreviations: ERR, estrogen‐related receptor; GR, glucocorticoid receptor; LRH1, liver receptor homolog 1; MR, mineralocorticoid receptor; PR, progesterone receptor; REV‐ERBa‐α, nuclear receptor subfamily 1 group D member 1; RORα, RAR‐related orphan receptor alpha; SHP, small heterodimer partner.
Based on robust linear regression.
Normalized mRNA levels.
P values from global test regressing presence of more severe histologic feature on set of NRs in each cluster.
Not calculable, regression did not converge.
Two‐sided P values were nominal and not adjusted for multiple comparisons. Analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC) and Stata version 14 (StataCorp, College Station, TX).
Results
Subject Characteristics and Results of Trial
All 40 subjects received end‐of‐treatment research biopsies on which histologic analyses and NR expression were performed. Subject treatments included placebo, vitamin E, or metformin. No subject was excluded from the analyses or lost to follow‐up. In total, 85% of subjects were Hispanic with a mean body mass index of 33.0 ± 5.8 kg/m2. Although all subjects had biopsy‐proven NAFLD at the time of TONIC enrollment, by the end of treatment, 17.5% (n = 7) had fully resolved (<5% steatosis and no other evidence of NASH). Histologically, 47.5% (n = 19) of subjects exhibited zone 1 or 3 borderline NASH or definite NASH; of that subset, 63% (n = 16) had zone 3 perisinusoidal and periportal fibrosis or had bridging fibrosis (i.e., fibrosis stages 2‐3).
NR Expression Sorted by Median
Boxplots of the 36 NRs illustrate the summary measures (including median and interquartile range) of the distribution of expression of each NR for the 40 patients.
Cluster Analyses and Heat Map
Cluster analysis revealed two superfamilies of NRs that further divided into five individual clusters. Superfamily I NRs consisted of cluster Ia (constitutive androstane receptor [CAR], FXRα, liver receptor homolog 1), cluster Ib (chicken ovalbumin upstream promoter transcription factor 1 [COUP‐TFI], LXRα, TR2, small heterodimer partner, nuclear receptor subfamily 1 group D member 1 [REV‐ERBAα], RARα, TRα), and cluster Ic (COUP‐TFII, RAR‐related orphan receptor alpha, glucocorticoid receptor, retinoid X receptor [RXR]β, pregnane X receptor [PXR], RAR‐related orphan receptor gamma, RXRα, PPARα, TRβ). NR cluster Ia receptors were expressed at the lowest level, on average, across the sample. Superfamily II NRs consisted of IIa (VDR, germ cell nuclear factor [GCNF], progesterone receptor, RARγ, PPARδ, estrogen receptor alpha [ERα], LXRβ, PPARδ2, NURR1) and IIb (mineralocorticoid receptor, RARβ, estrogen‐related receptor 1, RARβ1, RARβ2, PPARγ1, PPARγ2, PPARγ3). NR cluster IIa receptors were expressed at the highest level, on average, across the sample. The degree of the relationship between each NR is designated on the y axis of the dendrogram (shorter branch lengths represent greater similarity between the NRs).
Cluster analysis of the aggregate mRNA expression profiles for each subject revealed three clusters. Cluster I consisted of the majority of the sample (30 subjects). Cluster II contained 8 subjects, and cluster III contained 2 subjects. Compared to clusters I and II, subject cluster III individuals demonstrated higher expression of all NRs, a trend that was especially pronounced among NR superfamily II receptors. Cluster II subjects demonstrated a higher expression of NR cluster Ic receptors but a markedly lower expression of NR cluster superfamily II receptors.
Demographics, histology, and treatment group did not significantly differ among the three patient clusters (I, II, III) derived from the cluster analysis (data not shown).
Comparison of NRs by Histologic Characteristics Sorted by the Dissimilarity Measure from Cluster Analysis
Median expression levels of NRs were compared by steatosis grade (<33% versus ≥33%), hepatocyte ballooning (none versus any), lobular inflammation (none/mild versus moderate/severe), fibrosis (none versus any), and type of NAFLD (not NASH versus NASH). In general, differentially expressed NRs exhibited significantly higher expression (i.e., required fewer qPCR cycles to detect, resulting in a numerically smaller normalized expression level) in pathologic states. The relative expression levels with comparative P values are listed in Table 2.
Three NRs (FXRα, RARβ, and RARβ1), located in NR cluster Ia and cluster IIb, were expressed at significantly higher levels in the state of increased steatosis. Further, steatosis grade was significantly different on a global test incorporating a regression of all cluster IIb receptors. Steatosis grade did not significantly associate with any other NRs.
ERα, located in cluster IIa, was expressed at a higher level in the state of “any” versus “no” ballooning. PPARγ3, in cluster IIb, however, was expressed at a lower level in the state of any versus no ballooning. PPARγ3 was the only NR expressed at a lower level in a pathologic state.
For lobular inflammation, ERα (cluster IIa) and PPARγ2 (cluster IIb) were both expressed at higher levels in the state of moderate/severe versus none/mild. The global test of cluster Ic NRs was also significantly different by inflammation severity.
Comparing NR expression by fibrosis status, 12/36 (33%) of all NRs assayed were expressed at significantly higher levels in “any fibrosis” versus “no fibrosis.” These included members from each cluster: the PPARs (PPARα, PPARγ1, PPARγ2, and PPARδ), two RAR isoforms (RARβ1 and RARα), two endobiotic/xenobiotic sensors (CAR and PXR), two TRs (TRα and TRβ), and two orphan receptors (COUP‐TF1 and NURR1). The global test of cluster Ib NRs was significantly different by fibrosis status.
Comparing the state of NASH versus not NASH overall, the RARs (RARy [cluster 4], RARβ1, RARγ [both cluster IIb]) demonstrated higher expression in the state of NASH versus not NASH. The global tests of cluster Ib and cluster IIa were also significantly different by NASH status.
Discussion
In this study, we stratified 40 pediatric subjects with NAFLD (at time of enrollment) by histologic NAFLD severity to evaluate the differential expression of 36 NRs in liver biopsy tissue taken at the end of treatment from a clinical trial. Although expression levels of NRs may vary based on sex, age/pubertal status,( 22 ) and race/ethnicity, the not NASH and borderline/definite NASH samples were roughly balanced with respect to these features (Table 1).
The discovery that certain NRs are differentially expressed in NASH (versus NAFLD that is not NASH) and in more severe histologic states provides a mechanistic link between the metabolic derangements of NASH and recognized features of liver histology. These findings support the ongoing development of therapeutic NR ligands and may suggest other unexplored therapeutic targets. To our knowledge, no study has quantified normalized expression of hepatic NRs in pediatric NAFLD/NASH.
Cluster analysis groups genes with similar patterns of expression and, in the case of NRs, may suggest the presence of shared transcriptional drivers.( 13 ) Cluster analysis is an exploratory technique aimed at generating (rather than testing) hypotheses. Research using this tool often reveals clusters of genes of related functionality.( 23 ) Encouragingly, the functions of individual NR clusters reported here share significant overlap with the “nuclear receptor ring of physiology” clusters of Bookout et al.( 13 )
As predicted based on the known pathophysiology of NAFLD, both cluster I and cluster II contained NRs with expression that correlated with more severe histologic features. Overall, receptors in cluster II were expressed at higher levels and with more variability between subjects, suggesting a principal role for these NRs in the pathogenesis of pediatric NAFLD. Both cluster I and cluster II contained NRs that are currently under investigation as drug targets or have already been exploited pharmacologically, including PPARα/δ/γ and FXRα. Other receptors, such as ERα, RARy/α/β/β1, and TRα/β, are not yet established as drug targets in NASH but have long been recognized as key modulators of liver disease. The functions of several NRs known to be protective against the development of NASH are shown in Fig. 3. The remaining NRs that demonstrated differential expression by histology (CAR, PXR, COUP‐TFI, GCNF, NURR1) are not well characterized and potentially represent new therapeutic targets.
Figure 3.
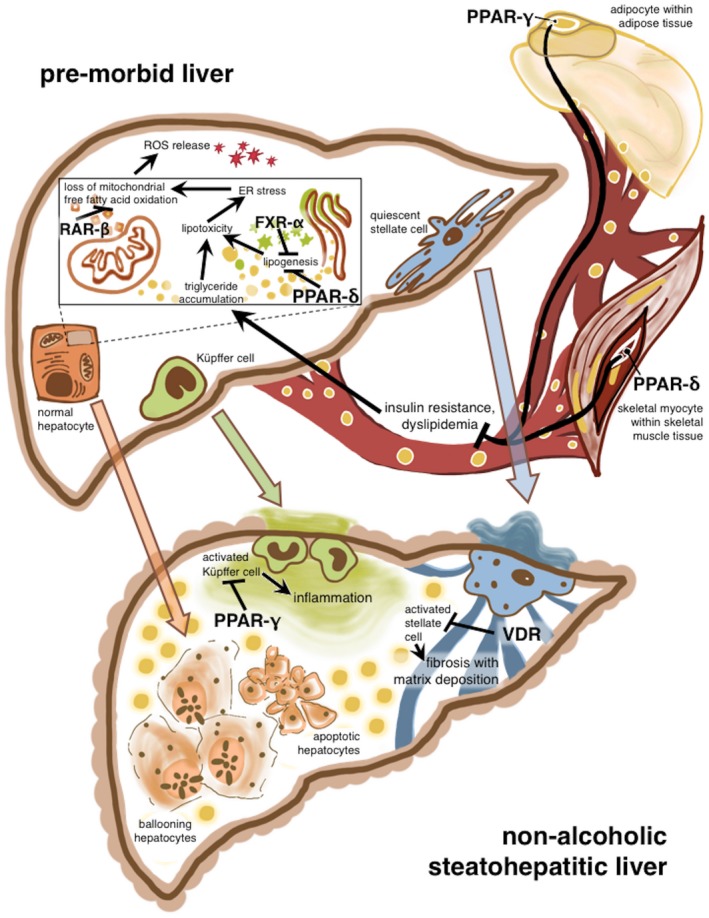
Theoretical model of NR functions in the development of NASH. NR functions (in the presence of their respective ligands) are potentially protective against the development of NASH. Drugs targeting the pictured PPARδ, PPARγ, and FXRα receptors are in trials for the treatment of NASH and the metabolic syndrome. NR–NR heterodimers and interactions are not pictured. (Figure 3 was created by Dr. Nikita Consul, Columbia University College of Physicians and Surgeons, 2017). Abbreviations: ER, endoplasmic reticulum; ROS, reactive oxygen species.
We found that several NRs are expressed at higher levels in more severe histologic states. It is possible that, as in other biologic systems of negative feedback, the presence of an NR ligand (whether endogenous or as a pharmaceutical agent) acts as a transcriptional repressor of the receptor itself to maintain homeostasis. For example, negative feedback is a key aspect of the well‐characterized FXRα/bile acid transcriptional cascade.( 24 ) Negative feedback would account for our finding that differentially expressed NRs have higher expression with more histologically severe disease, i.e., the up‐regulated receptors may be partially compensating for reduced levels of circulating endogenous ligand.
FXRα, in cluster Ia, is a key regulator of the gut–liver–adipose axis and is responsible for initiating systemic responses to the fed state.( 16 ) When activated, FXRα induces hundreds of genes throughout the body that mediate nutrient acquisition and distribution (e.g., fibroblast growth factor 19, PPARα) and inhibit hepatic lipogenesis (e.g., sterol regulatory element‐binding transcription factor 1c).( 25 ) Phase 3 trials of OCA, a synthetic FXRα agonist, are underway following the recent publication of the Farnesoid X Receptor Ligand OCA in NASH Treatment (FLINT) trial, which reported that OCA administration improved lobular inflammation, steatosis, fibrosis, and steatosis but did not affect the diagnosis of NASH.( 26 ) Building on this trial, we report an association between higher levels of steatosis and higher levels of FXRα but no difference in FXRα levels with NASH diagnosis. FXRα directly induces PPARα and PPARγ, linking the cluster I and cluster II NRs.
The PPARs are the only other NRs with agonists undergoing phase 3 trials for treatment of NASH. By targeting multiple PPAR isoforms, the drugs currently on trial for treatment of adult NASH aim to harness the diverse metabolic and anti‐inflammatory effects of this NR class. For example, elafibranor and pioglitazone, each agonists of two PPAR isoforms, are being tested for NASH treatment in adults.( 27, 28 ) In sum, our findings support the current understanding of PPARs as not only linchpins in lipid and glucose metabolism but also anti‐inflammatory and antifibrotic regulators and suggest that pharmacologic targeting of all three isoforms is likely to be effective in pediatric NASH.
Humans express three largely homologous PPAR isoforms (PPARα, PPARδ, and PPARγ), each with multiple tissue‐dependent subisoforms driven by alternative splicing or differential promoters (e.g., PPARγ1, PPARγ2, PPARγ3). The PPARs are lipid sensors that bind specific fatty acids. Adding to the evidence supporting the distinct but related roles of each PPAR isoform, we report that PPARα, PPARδ, and PPARγ each cluster with different groups of genes but that expression of each isoform is elevated in states of increased fibrosis. Our findings contradict Francque et al.’s recent report of decreased PPARα expression with worsened fibrosis among adults with NASH( 29 ), perhaps suggesting a different mechanism for pediatric versus adult NASH or differences between populations studied (primarily Hispanic in this study).
The three subisoforms of PPARγ are distributed into cluster IIb. In addition to its roles in adipocyte development and lipid metabolism, PPARγ is also strongly associated with monocytes and other cells of the immune system where it exerts a potent anti‐inflammatory effect by inhibiting production of tumor necrosis factor alpha.( 30, 31 ) Supporting this role, we found higher expression of PPARγ2 associated with increased lobular inflammation. Overall, our findings of altered PPARγ expression across multiple histologic domains suggest that PPARγ plays an important role in linking adipocyte and monocyte function and adds to the burgeoning evidence connecting NAFLD with systemic inflammation.
Finally, PPARδ is ubiquitous throughout nearly all tissues. In hepatocytes, it inhibits the expression of lipogenic genes (e.g., fatty acid synthase and acetyl coenzyme A decarboxylase). Although it was the last PPAR for which a synthetic ligand was discovered, its increasingly recognized role in preventing insulin resistance, hypertriglycidemia, and immune overactivation has led to excitement about its potential as a treatment for NASH.( 29 ) Although PPARδ has an important metabolic role, its position in cluster II near several immunomodulatory NRs suggests its anti‐inflammatory properties are also salient.
We also report differential expression of the receptors for estrogen, thyroid hormone, and vitamin A, known modulators of NASH. Estrogen is an in vitro antifibrotic agent, as confirmed by several epidemiologic lines of evidence.( 32 ) Given this knowledge, it is not surprising that ER is increased (and possibly therefore estrogen is decreased) in the livers with more ballooning, lobular inflammation, fibrosis, and NASH diagnosis. RAR, the receptor for vitamin A, drives cellular differentiation and regulates mitochondrial and peroxisome oxidation of fatty acids. Animal models support the role of the RAR family of transcription factors in NASH; transgenic mice expressing a dominant negative RARα in hepatocytes are used as an animal model for NASH and hepatocellular carcinoma.( 33 ) Thyroid hormone is a crucial driver of metabolism; hypothyroidism results in insulin resistance and increased circulating low‐density lipoproteins and triglycerides, factors that predispose to the development of NAFLD.( 34 ) Indeed, the prevalence of hypothyroidism among adults with NASH is twice that of healthy controls.( 35 ) Consequently, it was not surprising that both isoforms of TR localized to cluster I (near several metabolism‐related NRs) and that higher expression of both TRα and TRβ was associated with fibrosis. RXRβ, which heterodimerizes with RAR, thyroid hormone, and VDR and is therefore at the center of many NR pathways, was also found to be elevated in states of fibrosis.
In this study, fibrosis was the histologic characteristic that was discriminated by the largest number of NRs. In addition to the PPARs, ERs, RAR/RXRs, and TRs discussed above, CAR, PXR, GCNF, and COUP‐TF1 and NURR1 were also found to be expressed at significantly higher levels in the setting of any versus no fibrosis. Because fibrosis is the only histologic feature that associates with long‐term outcomes of adult NAFLD patients,( 36 ) therapeutic modulation of these receptors may have important clinical implications.
Finally, we found higher NURR1, GCNF, and COUP‐TF1 expression in the setting of fibrosis. The natural ligands of these orphan receptors are unknown, and their role in NAFLD remains similarly undescribed. COUP‐TF1 is at the center of several complex cross‐regulatory circuits of NRs, including FXRα, RXR, and PPARγ, during liver development and possibly beyond.( 37 ) Understanding crosstalk between the NR pathways described above will necessitate a more complete characterization of NURR1 and COUP‐TF1 and may lead to clinically meaningful discoveries.
The intricate regulation of NRs accounts for their exquisite ability to sense and respond to dynamic hormonal and nutritional cues across multiple organ systems in a coordinated fashion. However, their pleiotropic nature also increases the challenge of designing and using NR modulators for therapy. As an illustration of this challenge, recent clinical trials of NR agonists in biopsy‐proven NASH (i.e., FLINT; Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis [PIVENS]; and GOLDEN‐505) demonstrated paradoxical and unexpected decoupling of histologic features (including steatosis, inflammation, and fibrosis) and metabolic effects (including insulin resistance, dyslipidemia, and weight gain).( 38 )
This study is the first to quantify hepatic NR expression in a cohort of children with NAFLD, a population at greatest risk for poor long‐term outcomes. Strengths of this study include the novelty of the population, consensus and blinded review of liver biopsies by NASH CRN pathologists, and the range of histologic severity in the subject biopsies. The use of cluster analysis is also novel. This tool is not only an emerging methodology in systems biology (itself a key framework for understanding the overarching network of the NR superfamily) but could lead to the identification of subtypes of NAFLD that respond differently to therapeutic interventions.
Limitations of this study include the subject sample size and the inherent restrictions of liver biopsy. Liver biopsy represents a sample of mixed cell populations. NR profile variability may thus represent variability between cell types rather than reflecting true pathologic differences.( 39 ) Additionally, this study characterized the NR expression in liver tissue while many NRs are enriched in and exert their primary effects in other tissues. Because this study aimed to characterize the expression profiles of all known hepatic NRs in a discovery cohort, P <0.05 is liberal given the multiple comparisons and should be taken in the context of the multiple NR comparisons evaluated. The preliminary findings of this paper should thus be evaluated in prospective future studies that can also quantify the expression of NR target genes.
Most importantly, the sample size excluded the ability to analyze by treatment group, with potential obfuscation of effects related to treatment effects of vitamin E or metformin. Because vitamin E has been reported to activate PXR( 40 ) and metformin has been reported to affect the expression of several other NRs, including CAR, small heterodimer partner, and TR4,( 41 ) the use of end‐of‐treatment biopsies represents a major limitation of this study; thus, these preliminary results should be interpreted with caution. Finally, the study reports on the histology and NR profiles of posttreatment biopsies, although it is possible that treatment itself altered NR profiles compared to untreated individuals with the same histology.
The association of differential NR expression patterns with pediatric NAFLD of varying histologic severity is a novel finding. NRs are the transcriptional key at the center of the gut–liver–adipose axis. Their integrated actions coordinate metabolism, immune function, and cellular activation and differentiation, processes that become dysregulated in NAFLD and NASH. NRs found to be variably expressed in this study correspond to previously recognized therapeutic targets of drugs for NAFLD and the metabolic syndrome that are being investigated in preclinical development through phase 3 trials. Ongoing efforts to identify the ligands for remaining orphan receptors and the development of ligands that specifically target hepatic or adipose NRs show promise in the treatment of NASH.
Potential conflict of interest
Dr. Lavine consults for Alexion, Allergan, Amarin, Merck, and Takeda. Dr. Brunt consults for Arix, NGM, and Cymabay. The other authors have nothing to report.
Supporting information
Acknowledgment
Members of the NASH CRN are listed in the Supporting Material.
SEE EDITORIAL ON PAGE https://doi.org/10.1002/hep4.1260
References
- 1. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388‐1393. [DOI] [PubMed] [Google Scholar]
- 2. Mencin AA, Lavine JE. Nonalcoholic fatty liver disease in children. Curr Opin Clin Nutr Metab Care 2011;14:151‐157. [DOI] [PubMed] [Google Scholar]
- 3. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2016;150:1769‐1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non‐alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012;55:2005‐2023. [DOI] [PubMed] [Google Scholar]
- 5. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non‐alcoholic fatty liver disease in children: a follow‐up study for up to 20 years. Gut 2009;58:1538‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 7. Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011;141:1249‐1253. [DOI] [PubMed] [Google Scholar]
- 8. Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr 2013;162:496‐500.e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patton HM, Lavine JE, Van Natta ML, Schwimmer JB, Kleiner D, Molleston J. Nonalcoholic steatohepatitis clinical research network. Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008;135:1961‐1971.e1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neuschwander‐Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp‐Arida A, Tonascia J, et al. NASH Clinical Research Network. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 2010;52:913‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 2010;363:1341‐1350. [DOI] [PubMed] [Google Scholar]
- 12. Luyckx FH, Lefebvre PJ, Scheen AJ. Non‐alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. DiabetesMetab 2000;26:98‐106. [PubMed] [Google Scholar]
- 13. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006;126:789‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. GenomeBiol 2011;12:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evans Ronald M, Mangelsdorf DJ. Nuclear receptors, RXR, and the big bang. Cell 2014;157:255‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta 2016;1859:1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. Jama 2011;305:1659‐1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 19. Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell 2006;126:801‐810. [DOI] [PubMed] [Google Scholar]
- 20. Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L. Normalization of qRT‐PCR data: the necessity of adopting a systematic, experimental conditions‐specific, validation of references. J Exp Bot 2009;60:487‐493. [DOI] [PubMed] [Google Scholar]
- 21. Caliński T, Harabasz J. A dendrite method for cluster analysis. Comm Stat 1974;3:1‐27. [Google Scholar]
- 22. Whitfield GK, Remus LS, Jurutka PW, Zitzer H, Oza AK, Dang HT, et al. Functionally relevant polymorphisms in the human nuclear vitamin D receptor gene. Mol Cell Endocrinol 2001;177:145‐159. [DOI] [PubMed] [Google Scholar]
- 23. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci U S A 1998;95:14863‐14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang L, Lee Y‐K, Bundman D, Han Y, Thevananther S, Kim C‐S, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2002;2:721‐731. [DOI] [PubMed] [Google Scholar]
- 25. Cariou B, Staels B. FXR: a promising target for the metabolic syndrome? Trends Pharmacol Sci 2007;28:236‐243. [DOI] [PubMed] [Google Scholar]
- 26. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐α and ‐δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147‐1159.e1145. [DOI] [PubMed] [Google Scholar]
- 28. Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest 2006;116:590‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non‐alcoholic steatohepatitis. J Hepatol, 2015;63:64‐173. [DOI] [PubMed] [Google Scholar]
- 30. Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, et al. Peroxisome proliferator‐activated receptor alpha protects against alcohol‐induced liver damage. Hepatology 2004;40:972‐980. [DOI] [PubMed] [Google Scholar]
- 31. Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator‐activated receptors (PPARs): tissue distribution of PPAR‐alpha, ‐beta, and ‐gamma in the adult rat. Endocrinology 1996;137:354‐366. [DOI] [PubMed] [Google Scholar]
- 32. Yang JD, Abdelmalek MF, Pang H, Guy CD, Smith AD, Diehl AM, et al. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 2014;59:1406‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shiota G. Loss of function of retinoic acid in liver leads to steatohepatitis and liver tumor: a NASH animal model. Hepatol Res 2005;33:155‐160. [DOI] [PubMed] [Google Scholar]
- 34. Pramfalk C, Pedrelli M, Parini P. Role of thyroid receptor beta in lipid metabolism. Biochim Biophys Acta 2011;1812:929‐937. [DOI] [PubMed] [Google Scholar]
- 35. Huang Y‐Y, Gusdon AM, Qu S. Cross‐talk between the thyroid and liver: a new target for nonalcoholic fatty liver disease treatment. World J Gastroenterol 2013;19:8238‐8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, associates with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev 2006;20:2293‐2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol 2015;62(Suppl.):S65‐S75. [DOI] [PubMed] [Google Scholar]
- 39. Gascon‐Barré M, Demers C, Mirshahi A, Néron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology 2003;37:1034‐1042. [DOI] [PubMed] [Google Scholar]
- 40. Traber MG. Vitamin E, nuclear receptors and xenobiotic metabolism. Arch Biochem Biophys 2004;423:6‐11. [DOI] [PubMed] [Google Scholar]
- 41. Chai SC, Cherian MT, Wang YM, Chen T. Small‐molecule modulators of PXR and CAR. Biochim Biophys Acta 2016;1859:1141‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials