

Abstract
Medically refractory, severe, cholestasis‐induced pruritus in Alagille syndrome may be improved by surgical interruption of the enterohepatic circulation. This multicenter trial (NCT02057692) tested the hypothesis that the intestinal bile acid transport inhibitor maralixibat would similarly reduce pruritus in Alagille syndrome. Thirty‐seven children with Alagille syndrome were randomly assigned to double‐blinded administration of placebo, 70, 140, or 280 µg/kg/day of maralixibat for 13 weeks. Pruritus was assessed by caregiver (itch‐reported outcome instrument [ItchRO]) and clinician report (range, 0‐4 [severe]). Liver chemistries and serum bile acids were measured. The primary outcome was the change from baseline to week 13 in ItchRO relative to placebo. In the a priori first analysis of the primary efficacy endpoint, the mean adjusted difference between participants receiving 140 or 280 µg/kg/day and placebo was –0.47 (95% confidence interval [CI], –1.14, 0.20; P = 0.16). Statistically significant decreases were observed with doses of 70 and 140 µg/kg/day (mean adjusted difference, –0.89; 95% CI, –1.70, –0.08; P = 0.032; and mean adjusted difference, –0.91; 95% CI, –1.62, –0.19; P = 0.014) but not 280 µg/kg/day (mean adjusted difference, –0.04; 95% CI, –0.94, 0.86; P = 0.44) or all doses combined (mean adjusted difference, –0.61; 95% CI, –1.24, 0.20; P = 0.055). A 1‐point reduction in pruritus was more common in maralixibat‐treated versus placebo‐treated participants (caregiver ItchRO, 65% versus 25%; P = 0.06; clinician score, 76% versus 25%; P = 0.01). There were no significant changes in liver chemistries or bile acids relative to placebo. Adverse and serious adverse events were similar between maralixibat and placebo. Conclusion: Although the prespecified primary analyses of ItchRO were not all statistically significant, the data suggest that maralixibat is safe and may reduce pruritus in Alagille syndrome.
Abbreviations
- AE
adverse event
- ALGS
Alagille syndrome
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- ASBT
apical sodium‐dependent bile acid transporter
- ASBTi
ASBT inhibitor
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- CI
confidence interval
- CSS
clinical scratch score
- DB
direct bilirubin
- DILI
drug‐induced liver injury
- GGT
gammaglutamyl transpeptidase
- IE
ileal exclusion
- IMAGO
Safety and efficacy study of LUM001 in the treatment of cholestatic liver disease in patients with Alagille syndrome
- ItchRO
itch‐reported outcome
- ITT
intention‐to‐treat population
- LDL‐C
low‐density lipoprotein cholesterol
- mITT
modified intention‐to‐treat population
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- Obs
observation
- PBC
primary biliary cholangitis
- PEBD
partial external bile diversion
- SBA
serum bile acid
- TB
total bilirubin
Alagille syndrome (ALGS) is a rare autosomal dominant disorder, classically manifested by cholestatic liver disease and variable involvement of the heart, eyes, face, skeleton, kidneys, and vasculature.( 1 ) The liver disease of ALGS can present in infancy with marked cholestasis and fat malabsorption and with the subsequent development of intense pruritus that may be debilitating, causing cutaneous mutilation and disruption of sleep and school activities. In cholestatic liver disease, pruritus appears to be associated with elevated total serum bile acids (SBAs), although the specifics of the relationship are not well understood.
The management of pruritus in ALGS is challenging, and a variety of therapies are often used. These include antihistamines, rifampin, ursodeoxycholic acid, cholestyramine, naltrexone, and sertraline. Clinical experience suggests that these drugs have variable efficacy in reducing pruritus; however, no prospective clinical trial has quantified the effect of any of these therapies, either alone or in combination. Partial external bile diversion (PEBD) or ileal exclusion (IE), both of which interrupt the enterohepatic circulation, have had moderate success in reducing pruritus in patients with ALGS; however, the procedures require surgery, and biliary diversion presents the long‐term burden of caring for a stoma.( 2, 3 ) Thus, refractory pruritus is, in some circumstances, an indication for liver transplantation in ALGS. Hence, there is an unmet need for the development and testing of improved medical therapies for ALGS‐associated pruritus.
Maralixibat (SHP625, LUM001; Shire) is a potent inhibitor of the ileal bile acid transporter/apical sodium‐dependent bile acid transporter (ASBT) (SLC10A2), which was initially developed as a cholesterol‐lowering agent. This transporter mediates the uptake of conjugated bile acids across the brush border membrane of the ileal enterocyte from where they are ultimately transported to the liver in the enterohepatic circulation. ASBT expression is under negative feedback regulation by luminal bile acids; thus, in the setting of cholestasis and reduced intraluminal bile acid concentrations, ASBT is maladaptively up‐regulated.( 4, 5 ) Therefore, inhibiting the ileal reabsorption of bile acids may represent a useful strategy for reducing SBAs in cholestatic disease and potentially reducing pruritus.( 6 ) Because PEBD and IE have been shown to reduce SBAs and improve pruritus and xanthomas in ALGS, pharmacologic blockade of intestinal re‐uptake of bile acids with an ASBT inhibitor (ASBTi) may be a viable alternative to surgical intervention for pruritus in ALGS.( 3, 7 )
We conducted a randomized, double‐blind, placebo‐controlled, parallel group, multicenter trial of maralixibat in children with ALGS and pruritus. The study was designed to determine the effects of graduated doses of maralixibat for 13 weeks compared with placebo on pruritus, SBAs, liver enzymes, and other biochemical markers associated with cholestatic liver disease. The itch‐reported outcome (ItchRO) instrument, a novel tool to assess pruritus, was administered through an electronic diary to capture twice‐daily pruritus scores, the primary endpoint for this study.( 8 )
Participants and Methods
STUDY POPULATION
This study enrolled children aged 1 year through 18 years who had cholestasis and pruritus caused by ALGS, which was diagnosed based on study criteria (Supporting Table S1) and confirmed by JAGGED1 or NOTCH2 genotyping. Eligibility (i.e., presence of significant pruritus) was determined using twice‐daily caregiver‐based assessment of pruritus by ItchRO observation of child reported by parent/guardian/caregiver (Obs).( 8 ) ItchRO scores range from 0 to 4, with higher scores indicating increasing pruritus severity. The average daily score was derived from the highest score of the morning and evening observations, which reflects the worst pruritus of that day. Eligibility for this study required an average daily ItchRO(Obs) score of ≥2 for 2 consecutive weeks. Patients were excluded if they had chronic diarrhea requiring intervention, surgical interruption of the enterohepatic circulation, prior liver transplant, alanine aminotransferase (ALT) >15 times the upper limit of normal or decompensated cirrhosis (full inclusion and exclusion criteria are listed in Supporting Table S2).
Written informed consent was obtained from caregivers, and assent was obtained when appropriate from the child according to local institutional review board rules. This study was approved by local institutional review boards, complied with the Declaration of Helsinki and Good Clinical Practice Guidelines, and was registered at ClinicalTrials.gov (NCT02057692). The study was developed with and conducted in collaboration with Lumena Pharmaceuticals, now part of the Shire Group of Companies, in the context of a Cooperative Research and Development Agreement with the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Lumena/Shire was not involved in the data analysis or the reporting and interpretation of the results, which were independently performed by the NIDDK‐funded Childhood Liver Disease Research Network. As set out by the Cooperative Research and Development Agreement, Lumena/Shire was permitted to read and comment on the manuscript prior to submission.
STUDY DESIGN
This was a double‐blind, randomized, placebo‐controlled phase 2b trial conducted at 12 NIDDK‐funded Childhood Liver Disease Research Network sites (listed in Supporting Table S3). Originally, participants were randomized to one of three treatment arms in a 2:1 randomization ratio between maralixibat and placebo (n = 8 each in placebo, 70 µg/kg/day, or 140 µg/kg/day), with the primary comparison between the pooled maralixibat groups and placebo. Approximately 1 year after the start of the study (after 9 participants had begun investigational drug administration), an additional higher dose arm (280 µg/kg/day, n = 8) was added to the study based on preliminary results from a similar but smaller study conducted in the United Kingdom (Safety and efficacy study of LUM001 in the treatment of cholestatic liver disease in patients with Alagille syndrome [IMAGO]; design and preliminary results reported [NCT01903460]). To maintain the original design features of a 2:1 randomization ratio between placebo and active drug, 4 additional participants were randomized to placebo (n = 12 total), and the primary comparison became the pooled two highest tolerated active doses versus placebo. A dose was considered “not tolerated” if >50% of participants in that dose cohort did not tolerate the treatment, as evidenced by dose reduction, suspension, or discontinuation due to gastrointestinal tolerability related to maralixibat.
Randomization was performed by the central pharmacy using schedules (original and amended) that were prepared by a clinical research organization. Permuted blocks of size 3 and size 9 were used for the original design and the amended study, respectively. The caregivers, participants, investigators, and the sponsor were unaware of treatment assignment until the last participants completed week 13, at which time the database was locked and the blind broken.
The study drug was administered once daily in the morning at least 30 minutes prior to breakfast. Dosing was escalated over 5 weeks to enhance tolerability of the study drug (Fig. 1). The final dose of the study drug was then maintained for 8 weeks. Study visits occurred at weeks 0, 2, 4, 8, and 13 and phone interviews at weeks 1, 3, and 6. Changes in the use of antipruritic medications during the study were not permitted.
Figure 1.
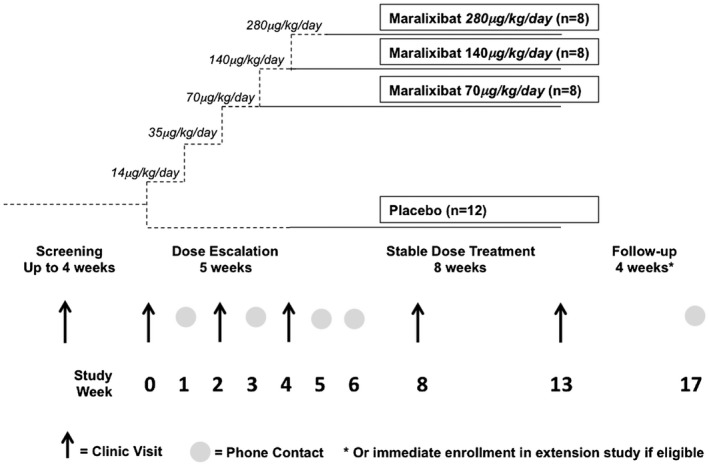
Study schema.
The primary endpoint was the change in pruritus as measured by the ItchRO(Obs). The average daily ItchRO(Obs) score for 7 days pretreatment was compared with the last 7 days of treatment (i.e., change from baseline to week 13 or end of treatment for those who discontinued early [designated week 13]). Secondary endpoints included changes from baseline to week 13 for SBAs, ALT, aspartate aminotransferase, alkaline phosphatase (ALP), gammaglutamyl transpeptidase (GGT), total bilirubin (TB), and direct bilirubin (DB).
Other efficacy endpoints included changes from baseline over time (weeks 2, 4, 8, and 13) for ItchRO(Obs), SBAs, ALT, ALP, GGT, TB, DB, total cholesterol, low‐density lipoprotein cholesterol (LDL‐C), and 7α‐hydroxy‐4‐cholesten‐3‐one (C4; surrogate marker of bile acid biosynthesis). Changes from baseline in the clinical scratch score (CSS; Supporting Table S4), which has been used in pediatric studies( 9 ) and is based on a scale developed by Whitington,( 7 ) were examined. Changes from baseline for ItchRO completed by participants if ≥9 years old or by caregivers with input from participants 5‐8 years old as well as xanthoma severity (Supporting Table S5) were also assessed.( 2 ) SBAs were quantified by stable‐isotope dilution analysis using liquid chromatography‐electrospray ionization‐mass spectrometry (Division of Pathology and Laboratory Medicine, Cincinnati Children's Hospital Medical Center, Cincinnati, OH).( 10 )
Adverse events (AEs), serious AEs, treatment discontinuations due to AEs, and AEs of special interest (e.g., gastrointestinal symptoms, liver injury, fat‐soluble vitamin level abnormalities, and growth retardation) were used to characterize the safety and tolerability of maralixibat.
STATISTICAL METHODS
Sample Size Determination
The planned sample size of 36 evaluable subjects with ALGS was based on practical considerations rather than a desired power for a prespecified difference. With the proposed sample of 28 subjects for the primary efficacy analyses (16 maralixibat from the two highest tolerated doses and 12 placebo), there would be 80% power to detect an effect size of ≥1.12, using a two‐sided type I error of 5% and a two‐sample t test. Analyses were not adjusted for multiple comparisons, and nominal P values are reported.
Analytic Methods
Efficacy analyses were performed using the modified intention‐to‐treat population (mITT), defined as all participants randomized, receiving at least one dose of study drug, and having at least one postbaseline efficacy assessment. Participants were analyzed by assigned treatment. Sensitivity analyses were performed using the per protocol population, defined as the mITT population that did not have a major protocol violation and the pure ITT population (all randomized and dosed participants) if it differed from the mITT (which it did not). For participants who prematurely discontinued from the study, a last observation carried forward approach, which only used values within 7 days of the last dose of study drug, was used to impute missing efficacy values. Safety, subject disposition, and baseline characteristics were analyzed using the safety population, defined as all randomized participants who received at least one dose of study drug. Participants were analyzed by treatment received. Statistical significance was defined as P < 0.05; no adjustments for multiplicity were applied in this phase 2b study.
For efficacy analyses, the first statistical test performed for each primary and secondary outcome measure was the comparison between the two highest tolerated active‐dose groups combined (designated hereafter as maralixibat*) and placebo. In addition, all active doses combined (designated as maralixibata) as well as each individual dose were compared with placebo.
Treatment comparisons of the primary endpoint and of secondary and exploratory efficacy endpoints that were continuous were made using an analysis of covariance model with treatment and baseline measures as covariates. Estimates of least squares mean changes and associated SEMs and 95% confidence intervals (CIs) were reported. Active treatment groups (combined and individual) were tested against the placebo group, with adjusted mean treatment difference, SEM, 95% CI, and pairwise P value reported. In addition, changes from baseline to each visit were summarized and tested. Analyses of discrete outcomes were analyzed using the Fisher’s exact test or the Cochran‐Mantel‐Haenszel test.
Exploratory responder analyses were defined a priori for ItchRO(Obs) (responder if change from baseline to week 13 was ≤–1 or ≤–2), CSS (responder if change from baseline to week 13 was ≤–1), and clinician xanthoma scale (responder if change from baseline to week 13 was ≤–1) and analyzed using the Fisher’s exact test.
Results
PARTICIPANT CHARACTERISTICS
Fifty‐three participants were enrolled and assessed for eligibility. Of these, 37 were randomized to the investigational drug between November 24, 2014, and November 16, 2016 (Fig. 2). Fifteen participants failed the screening ItchRO(Obs) criteria and therefore were ineligible. Twenty‐five participants received maralixibat (8‐70 µg/kg/day, 11‐140 µg/kg/day, 6‐280 µg/kg/day) and 12 received placebo. All but 2 participants completed the 13‐week treatment period; 1 participant on placebo was lost to follow‐up on day 28 and 1 participant who was randomized to 70 µg/kg/day withdrew on day 1 with a rash and elevated liver biochemistries after receiving one dose (14 µg/kg). The mean age of participants was 6.8 years, and the majority (65%) were between 2 and 8 years old. All had a history of antipruritic medication use, prescribed as per clinical practice prior to enrollment (antihistamines, 73%; ursodeoxycholic acid, 84%; rifampin, 68%). Laboratory parameters were characteristic of individuals with cholestasis and ALGS (mean SBAs, 216.3 µM; ALT, 158.7 IU/L; GGT, 494.9 IU/L; TB, 5.3 mg/dL; total cholesterol, 405.7 mg/dL). Baseline characteristics were similar among the four groups (Supporting Table S6).
Figure 2.
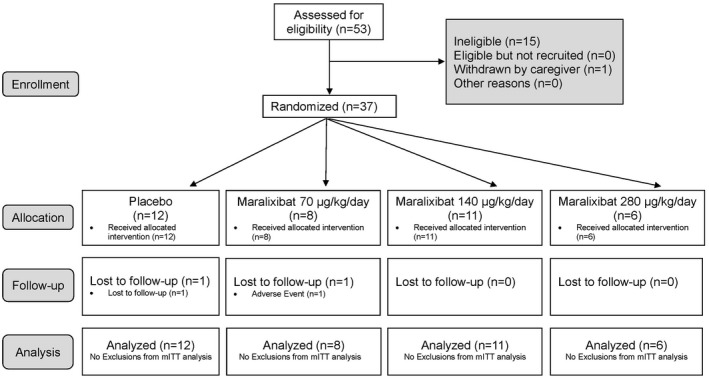
Consolidated standards of reporting trials diagram.
EFFICACY
In this phase 2b study, maralixibat*, maralixibata, and each individual dose of maralixibat were compared relative to placebo for the primary efficacy endpoint of change in pruritus, as measured by ItchRO(Obs) from baseline to week 13 (Table 1). In the first analysis of the primary efficacy endpoint, the mean adjusted difference between maralixibat* and placebo was –0.47 (95% CI, –1.14, 0.20; P = 0.16). Relative to placebo, significant decreases were observed with the individual doses of 70 and 140 µg/kg/day (mean adjusted difference, –0.89; P = 0.032; and mean adjusted difference, –0.91; P = 0.014, respectively). No change was observed in the group receiving 280 µg/kg/day (mean adjusted difference, –0.04; P = 0.44). The change in maralixibata relative to placebo was not statistically significant (mean adjusted difference, 0.61; P = 0.055).
Table 1.
Analysis of Primary Endpoint: Change From Baseline to Week 13 in ItchRO(Obs)
| Change From Baseline | Treatment Compared to Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
|
Outcome Measure Treatment Group Maralixibat |
n | Baseline Mean (SEM) | LS Means Change (SEM) | 95% CI | P value | Difference in LS Means (SEM) | 95% CI | P value |
| ItchRO(Obs) average daily score | ||||||||
| 70 μg/kg/day | 8 | 3.2 (0.23) | –1.5 (0.30) | (–2.1, –0.9) | <.001 | –0.89 (0.40) | (–1.70, –0.08) | 0.032 |
| 140 μg/kg/day | 11 | 2.7 (0.16) | –1.5 (0.26) | (–2.0, –1.0) | <.001 | –0.91 (0.35) | (–1.62, –0.19) | 0.014 |
| 280 μg/kg/day | 6 | 3.3 (0.24) | –0.6 (0.36) | (–1.3, 0.1) | 0.093 | –0.04 (0.44) | (–0.94, 0.86) | 0.930 |
| Maralixibat* | 17 | 2.9 (0.15) | –1.1 (0.21) | (–1.5, –0.6) | <.001 | –0.47 (0.33) | (–1.14, 0.20) | 0.159 |
| Maralixibata | 25 | 3.0 (0.13) | –1.2 (0.18) | (–1.6, –0.8) | <.001 | –0.61 (0.31) | (–1.24, 0.01) | 0.055 |
| Placebo | 12 | 2.8 (0.15) | –0.6 (0.25) | (–1.1, –0.1) | 0.024 | |||
Abbreviation: LS, least squares.
Compared with baseline, the placebo group had a significant decrease in ItchRO(Obs) at week 13 (mean adjusted difference, –0.58; P = 0.024). Individual responses over time to maralixibat or placebo are illustrated in Fig. 3. The mean reduction from baseline in ItchRO(Obs) was similar among groups in the first 2‐4 weeks of the study during dose escalation as all participants received the same doses of study drug. There was accentuation of the response after 4 weeks in the groups receiving 70 and 140 µg/kg/day when participants were receiving their maximal dose of maralixibat (Fig. 4).
Figure 3.
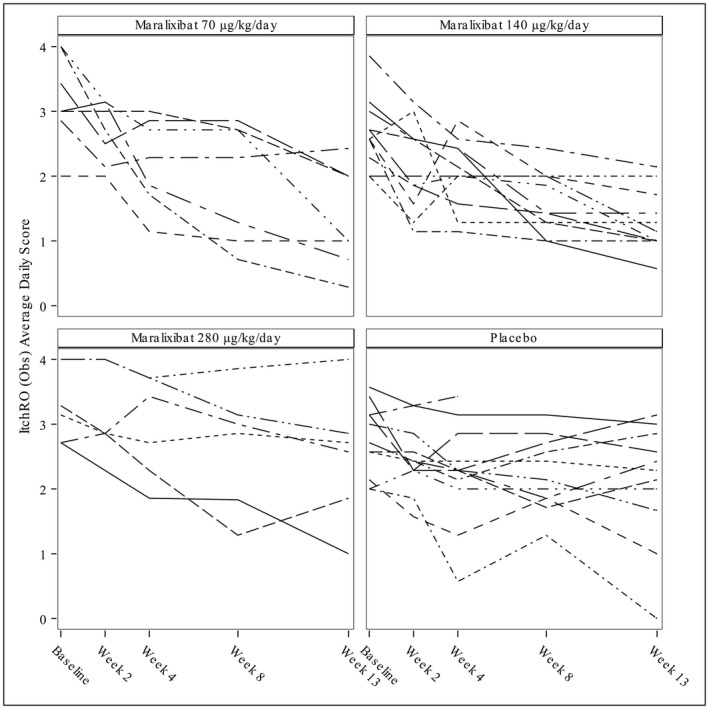
Spaghetti plots of changes in ItchRO(Obs) over time. Each line represents an individual participant. The participants are grouped by the target dose of the study drug.
Figure 4.
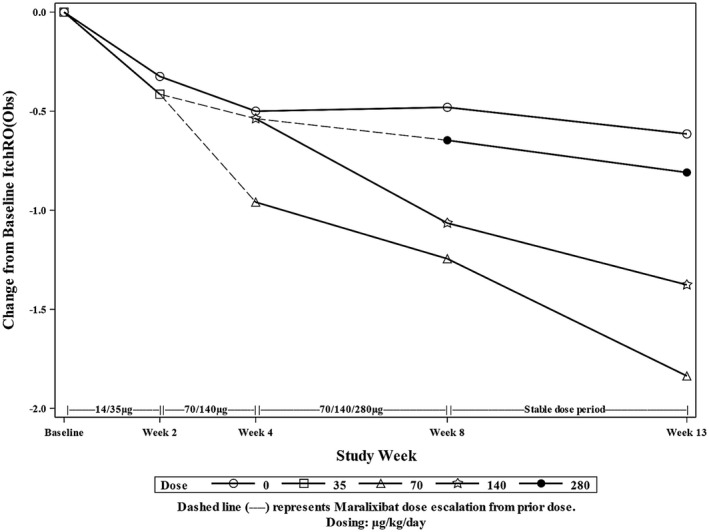
Pooled analysis of change from baseline of ItchRO(Obs). Mean change from baseline in ItchRO(Obs) was pooled among participants receiving the same dose of maralixibat. Dashed lines represent the transition of some participants to a new dose of maralixibat.
Changes from baseline to week 13 in SBAs and liver biochemistries were assessed as secondary endpoints (Table 2; Supporting Table S7). Individual responses over time for SBAs and C4 are shown in Supporting Fig. S1. TB and DB tended to decrease in participants receiving maralixibat, with significant decreases from baseline to week 13 observed for maralixibat* and maralixibata (Table 2; Supporting Table S7); however, when these changes were compared with the placebo group, which showed no statistically significant change in TB or DB, the changes in the maralixbat* and maralixibata groups were no longer statistically significant. For ALT, there were statistically insignificant increases during maralixibat treatment (Table 2). Pooled data over time for TB and ALT for maralixibat‐treated and placebo‐treated participants are presented in Fig. 5. No changes in GGT, ALP, or total cholesterol were observed (Supporting Table S7). Individual responses over time for TB and ALT are shown in Supporting Fig. S1. LDL‐C but not total cholesterol was significantly reduced relative to placebo with maralixibat* treatment (Supporting Table S7). SBAs were variable and were not significantly reduced during maralixibat treatment (Table 2; Fig. 5). C4 tended to increase with therapy, although the changes were not significant and the levels were highly variable (Table 2; Fig. 5). One participant receiving 70 µg/kg/day of maralixibat had extraordinarily high baseline SBAs (1,014 µmol/L) and C4 (161.9 ng/mL). Pooled data for those participants receiving maralixibat revealed a reduction in SBAs in the first 4 weeks of the study, with a potentially compensatory increase in C4 by week 8 (Fig. 5).
Table 2.
Analysis of Secondary Efficacy Endpoints: Change From Baseline to Week 13
| Change From Baseline | Treatment Compared to Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Outcome Measure Treatment Group | n | Baseline Mean (SEM) | LS Means Change (SEM) 1 | 95% CI | P value | Difference in LS Means (SEM) | 95% CI | P value |
| Serum bile acid (μmol/L) | ||||||||
| 70 μg/kg/day | 7 | 392 (126.8) | –117 (46.2) | (–212, –23) | 0.016 | –107 (57.2) | (–224, 10) | 0.071 |
| 140 μg/kg/day | 11 | 151 (37.3) | –40 (34.9) | (–111, 31) | 0.256 | –30 (47.5) | (–127, 67) | 0.534 |
| 280 μg/kg/day | 6 | 188 (44.0) | –27 (46.3) | (–122, 67) | 0.558 | –17 (56.5) | (–132, 98) | 0.766 |
| Maralixibat* | 17 | 164 (28.2) | –34 (29.2) | (–93, 26) | 0.255 | –23 (43.6) | (–112, 65) | 0.594 |
| Maralixibata | 24 | 231 (45.6) | –62 (23.9) | (–111, –13) | 0.015 | –51 (40.6) | (–134, 32) | 0.216 |
| Placebo | 12 | 205 (46.9) | –10 (32.7) | (–77, 56) | 0.751 | |||
| Total bilirubin (mg/dL) | ||||||||
| 70 μg/kg/day | 7 | 7.96 (3.39) | –0.29 (0.38) | (–1.06, 0.48) | 0.447 | –0.39 (0.47) | (–1.35, 0.56) | 0.407 |
| 140 μg/kg/day | 11 | 3.36 (1.06) | –0.35 (0.30) | (–0.97, 0.26) | 0.251 | –0.46 (0.42) | (–1.31, 0.40) | 0.284 |
| 280 μg/kg/day | 6 | 4.22 (2.10) | –0.80 (0.40) | (–1.62, 0.02) | 0.054 | –0.91 (0.49) | (–1.92, 0.10) | 0.076 |
| Maralixibat* | 17 | 3.66 (0.97) | –0.58 (0.25) | (–1.09, –0.06) | 0.029 | –0.68 (0.38) | (–1.47, 0.10) | 0.086 |
| Maralixibata | 24 | 4.92 (1.23) | –0.48 (0.21) | (–0.91, –0.06) | 0.027 | –0.59 (0.35) | (–1.31, 0.13) | 0.107 |
| Placebo | 12 | 6.41 (1.95) | 0.10 (0.28) | (–0.48, 0.68) | 0.719 | |||
| Alanine aminotransferase (U/L) | ||||||||
| 70 μg/kg/day | 7 | 155 (33.7) | 15 (17.9) | (–22, 51) | 0.422 | 27 (22.8) | (–20, 73) | 0.253 |
| 140 μg/kg/day | 11 | 117 (17.4) | 13 (14.9) | (–17, 44) | 0.383 | 25 (21.0) | (–18, 68) | 0.241 |
| 280 μg/kg/day | 6 | 191 (42.4) | 30 (19.6) | (–10, 70) | 0.142 | 41 (23.7) | (–7, 90) | 0.090 |
| Maralixibat* | 17 | 143 (20.0) | 21 (12.1) | (–3, 46) | 0.086 | 33 (18.6) | (–5, 71) | 0.082 |
| Maralixibata | 24 | 146 (16.9) | 19 (10.0) | (–1, 40) | 0.066 | 31 (17.3) | (–4, 66) | 0.082 |
| Placebo | 12 | 188 (26.9) | –12 (14.0) | (–40, 17) | 0.400 | |||
Abbreviation: LS, least squares.
Figure 5.
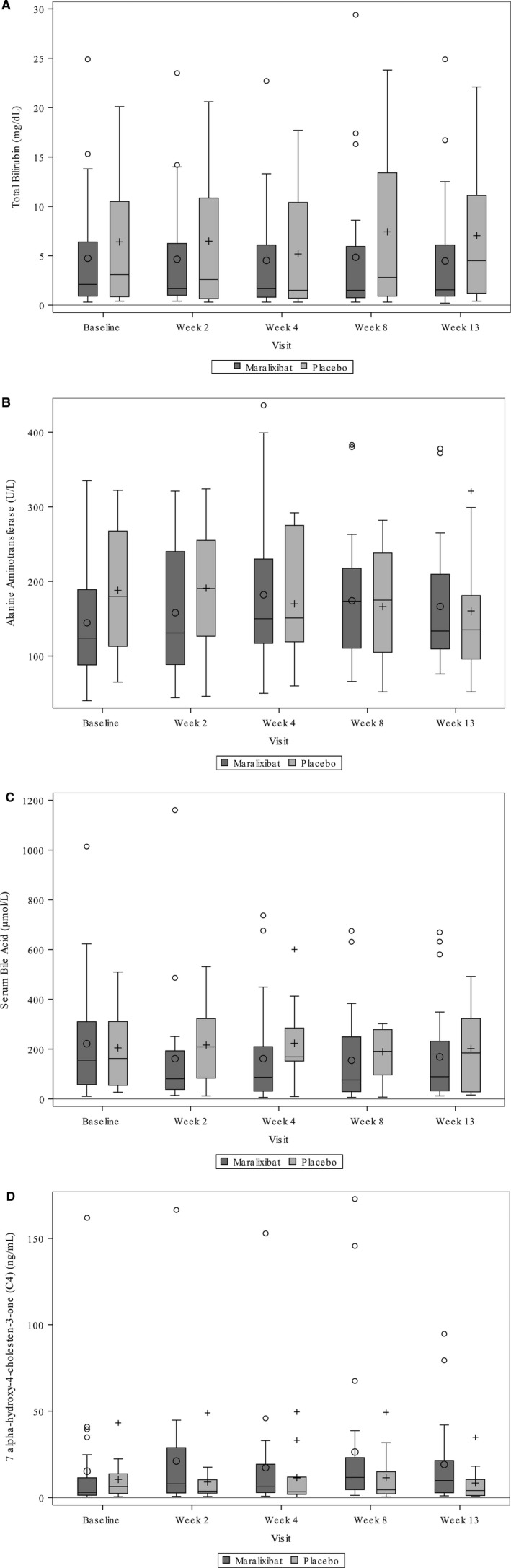
Box and whisker plots of changes in measured laboratory parameters over time. (A) Total bilirubin; (B) alanine aminotransferase; (C) serum bile acids; (D) C4. Maralixibata was compared to placebo. Data are represented as follows: mean, o or + inside the box; median, line inside the box; box, interquartile range (25‐75 percentiles); whiskers, values within 1.50 times interquartile range; outliers, individual data points beyond the whiskers.
Additional responder analyses were conducted to further assess the impact of maralixibat on pruritus. Only 5 participants (4 on maralixibat and 1 on placebo) had a decrease of at least 2 units in ItchRO(Obs), and none of the maralixibat groups yielded a significant difference relative to placebo (Supporting Table S8). With a less stringent threshold of –1, overall response rates were higher and significantly greater in maralixibata (68% versus 25%, P = 0.03; Supporting Table S8). ItchRO completed by participants if ≥9 years old or by caregivers with input from participants 5‐8 years old could only be assessed in 23 of the study participants (Supporting Table S9). The improvement in the placebo group was nearly the same as maralixibat* and maralixibata (mean adjusted difference, –1.189; SEM, 0.3734; P = 0.843; and mean adjusted difference, –1.281; SEM, 0.2831; P = 0.685, respectively). Maralixibat had a significant impact on changes from baseline to week 13 in CSS. Improvement was significantly greater in maraxibat*, maralixibata, 140, and 280 µg groups relative to placebo (Supporting Table S10). Using a predefined responder analysis at a cutoff of ≤–1 and a post‐hoc cutoff of ≤–3, significant changes from baseline to week 13 were observed for maralixibat* versus placebo (76% versus 25%, P = 0.01; and 35% versus 0%, P = 0.028, respectively; Supporting Table S10). Individual changes over time for CSS are shown in Fig. 6. Changes in xanthomas with maralixibat* treatment were not significant (31% versus 9%, P = 0.350; Supporting Table S11).
Figure 6.
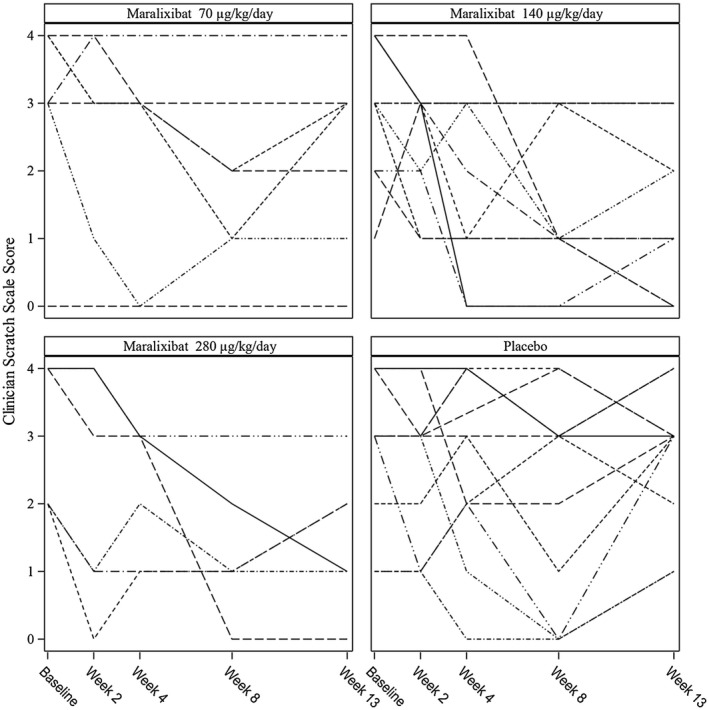
Spaghetti plots of changes in the clinician scratch scale over time. Each line represents an individual participant. The participants are grouped by the target dose of the study drug.
SAFETY
No deaths occurred during the study. One serious AE of vomiting occurred that led to hospitalization in a participant receiving 70 µg/kg/day of maralixibat; this was not felt to be related to the study drug. Maralixibat was stopped in the first week of the study for 1 participant who developed a rash and elevated ALT; the investigator considered this unlikely to be related to the study drug. Treatment‐emergent AEs were common and comparable in participants receiving maralixibat and placebo (Supporting Table S12). Given the proposed mechanism of action of maralixibat, gastrointestinal AEs, including diarrhea and abdominal pain, were of special interest and found to occur at similar rates in maralixibata‐treated and placebo‐treated participants (overall gastrointestinal, 52% versus 58%; diarrhea, 32% versus 50%; abdominal pain, 16% versus 17%, respectively). There was no clinically significant difference in change in weight from baseline to week 13 in maralixibat‐treated versus placebo‐treated participants (maralixibata, 0.73 ± 0.12 kg; placebo, 0.69 ± 0.17 kg; mean ± SEM, P = 0.842).
Criteria for identification of drug‐induced liver injury (DILI) or therapy‐related hepatotoxicity in the setting of chronic cholestasis are not well defined either in the literature or at a regulatory level. For the purposes of this study, specific changes from baseline were developed a priori as potential safety signals requiring enhanced monitoring and stopping rules were established (Supporting Table S13). There were no participants in either the maralixibat‐treated or placebo‐treated groups who met these stopping criteria (data not shown). The criteria were also not met if baseline was defined as the average of screening and baseline laboratory values (data not shown).
Fat‐soluble vitamin levels were also examined as a potential safety signal given the profound cholestasis in these participants, the high dosing requirements of fat‐soluble vitamins as part of their routine care, and the potential for changes in intestinal luminal bile acid concentrations. Changes were characterized relative to baseline and as shifts from sufficient to insufficient or from insufficient to sufficient, as defined (Supporting Table S14).( 11 ) In general, the number of participants who changed their sufficiency status during the course of the study was low. In the maralixibat group, vitamin D levels changed in equal percentages to insufficient and sufficient during the study (13% for each). Vitamin A levels became insufficient in 1 participant treated with maralixibat, while vitamin E levels became sufficient in 2 participants treated with maralixibat. Excess levels of vitamin A (n = 4) and vitamin D (n = 1) were observed at week 13 of maralixibat therapy. In 3 participants treated with maralixibat, the international normalized ratio increased as a potential marker of change in vitamin K sufficiency. None of these changes were observed in the placebo group.
Discussion
Data from this randomized placebo‐controlled trial in children with ALGS suggest that maralixibat is generally safe, well‐tolerated, and may reduce pruritus. The first predefined analysis of the primary endpoint did not meet the predefined statistical level for efficacy, while other analyses of the primary endpoint did. The reason for the heterogeneous responses cannot be determined from these investigations, and further study to assess safety and efficacy of maralixibat as a treatment for cholestasis‐associated pruritus in children with cholestasis is warranted.
The expected therapeutic benefit of maralixibat is based on the response of patients with ALGS to surgical interruption of the enterohepatic circulation, primarily through PEBD or IE.( 2, 3, 7 ) This response may not be as dramatic as has been observed in some individuals with progressive familial intrahepatic cholestasis. In general, responses tend to be favorable, with improvement in pruritus, xanthomas, and hypercholesterolemia. There are less clear or minimal effects on SBAs, bilirubin, and ALT.( 3 ) In most cases, surgery is performed for severe pruritus equivalent to CSS grade 4. Not all patients have complete resolution of their pruritus, but most have clinically significant improvement, equivalent to a reduction in CSS of 2 or more. ItchRO was not available to assess response in any of these published experiences. The favorable response to surgery has been generally documented over the first 12 to 24 months after surgery. None of the published studies of surgical intervention have examined results at 3 months after surgery, so direct comparison to this study is not possible. The relatively short time course of the current study may not have fully captured the potential efficacy of maralixibat. Ongoing long‐term follow‐up studies (NCT02047318, NCT02117713) may shed light on this matter, although these studies are not placebo controlled, with the exception of a 4‐week, randomized, placebo‐controlled withdrawal of maralixibat (NCT02160782).
The primary endpoint for this study was pruritus as assessed by ItchRO(Obs). This endpoint was chosen in recognition of the profound impact of pruritus on children with ALGS and on their families. Pruritus is notoriously difficult to objectively assess in clinical settings, and this challenge is amplified in research trials. Instruments for assessing pruritus can broadly be categorized into patient‐reported outcome tools and those that are independent of patient reporting, such as the CSS. An intermediate level of reporting is by parental/guardian observation of children, as was reported in this study. No single instrument, including actigraphy, has been fully validated to capture the multidimensional features of pruritus and its change over time, especially in children. To address this for this clinical trial, a novel tool, ItchRO, was developed using a rigorous tool development methodology to assess pruritus in children with cholestasis, specifically those with ALGS.( 8 ) Detailed analyses of ItchRO and the effect of maralixibat on quality of life, which is complex and beyond the scope of this report, will be the subject of a future report from this study.( 12 )
The placebo effect in this study was evident and highlights the critical need for a double‐blind placebo‐controlled study design in clinical trials addressing pruritus. It is well recognized that somatic symptoms, such as pain and fatigue, can be improved by placebo due to positive expectations, but the effect of placebo on itch in cholestasis has not been extensively studied. In the placebo‐treated arm of a study assessing the effect of another ASBTi (GSK2330672) on pruritus in patients with primary biliary cholangitis (PBC), there was a 23% improvement in itch on a 10‐point numerical rating scale.( 13 ) A similar reduction of 0.6 in ItchRO was observed in the unpublished results of IMAGO (NCT01903460).
Pharmacologic inhibition of ASBT is an evolving potential approach to the treatment of constipation, cholestasis, diabetes, and fatty liver disease.( 14 ) The findings of the current study should be considered in the context of recent related investigations of cholestatic liver disease. The only peer‐reviewed published report of an ASBTi is in adults with PBC.( 13 ) This was a 2‐week, double‐blind, randomized, placebo‐controlled, crossover phase 2a trial of 22 patients receiving GSK2330672 (NCT01899703). The primary endpoints of the study were safety and tolerability, which were deemed acceptable. Pruritus, quantified using three separate scores, was reduced by 30% to 57% with 2 weeks of therapy. Decreased SBAs and compensatory increases in C4 supported the expected biological effect on intestinal bile acid transport. The biochemical characteristics of cholestasis at baseline in the adult participants of the PBC study were less severe than in the children with ALGS in the current study (e.g., mean TB was 12.2 µM for PBC and 90.6 µM for ALGS; SBAs were 48.6 µM for PBC and 216.3 µM for ALGS). Additional studies of an ASBTi in cholestasis have been preliminarily reported as abstracts or as registered trials. An open‐label dose‐ranging phase II study of A4250 (Albireo Pharma) in 19 children with a variety of cholestatic conditions demonstrated improvement in pruritus in 14 children, as assessed by a visual itch score recorded by caretakers.( 15 ) The small sample size (14 participants with maralixibat and 6 participants with placebo) in IMAGO, which was conducted in the United Kingdom, may have limited the power of the study to identify a potential effect (NCT01903460).
SBA levels are an attractive choice as an endpoint for the use of an ASBTi in pediatric cholestasis. Levels represent a complex dynamic interplay of intestinal absorption and hepatic extraction, synthesis, and excretion. The experiences in this study of ALGS raise important concerns about this possible endpoint. First, there is significant variability in SBAs in children with ALGS, and this necessitates a fairly large sample size to demonstrate a potential therapeutic effect. Ursodeoxycholic acid, which was used by 84% of the participants, is not actively transported by ASBT; as such, its common use in ALGS may contribute significantly to SBAs and thus complicate the use of SBAs to define the ASBTi effect. Fecal bile acid determination, which would be a direct assay of ASBTi activity, is cumbersome and was not employed in this study. Changes in C4 are used as an alternative surrogate marker of ASBTi activity because diminished ileal absorption is predicted to lead to reduced fibroblast growth factor 19‐mediated ileal signaling to the liver and derepression of bile acid biosynthesis reflected by elevated C4. In this study, C4 levels were highly variable and may in part explain the lack of a statistically significant increase with maralixibat. The magnitude of changes in C4 suggests that a maximal ASBTi effect was not induced by the doses of drug used in this study. Alternatively, luminal bile acids in children with severe cholestasis with ALGS may be so low that an effect on C4 is difficult to demonstrate. LDL‐C levels were reduced, consistent with enhanced conversion of cholesterol to bile acids, as has been observed in surgical interruption of the enterohepatic circulation.( 16 ) Total cholesterol levels, which are reflective of lipoprotein X accumulation, would not be expected to be impacted as quickly by an ASBTi.
No significant safety issues were identified in this study of maralixibat. One of the potentially attractive features of an ASBTi is that it can act at the luminal ileal brush border membrane without significant systemic absorption, thereby reducing potential risk of toxicity. The major potential predicted side effect of an ASBTi relates to sequelae of bile acid malabsorption with related diarrhea and abdominal pain. Approximately half of the children who received maralixibat had gastrointestinal symptoms, none of which were severe. The critical importance of a blinded placebo control was reconfirmed by the finding of similar gastrointestinal problems in the placebo‐treated group. One of the complexities of clinical trials in cholestatic liver disease is the lack of understanding of approaches to monitoring for adverse effects on the liver itself.( 17 ) DILI is a major concern in the development of new drugs but is poorly characterized in chronic liver disease, especially cholestatic disease. Cholestatic features can be the most worrisome for serious drug‐related injury. Given the exceptionally limited systemic absorption of maralixibat, DILI was of limited concern. Despite this, prospective methods for monitoring potential drug toxicity needed to be established. The parameters chosen in this study (Supporting Table S13) did not reveal hepatotoxicity related to this treatment approach. These parameters might be considered for future studies in ALGS. The placebo treatment group along with the screening and enrollment laboratory studies provide additional novel insight into the natural variability of key liver parameters in ALGS that may guide future clinical studies of novel pharmacologic agents in pediatric cholestasis.
A major limitation of this study is the unexpected lack of response in the children receiving 280 µg/kg/day of maralixibat. It is unlikely that this is the result of an excessive dose of drug, especially in light of the relatively limited increase in C4. ALGS is a rare disorder, and the number of participants in the study was selected more on practical rather than power considerations. The randomization process in this small phase 2b study allocated only 6 participants to 280 µg/kg/day dosing, and this limited sample size may have skewed the findings. Both the placebo and 280‐µg/kg/day groups were characterized by a preponderance of children younger than 5 years of age (Supporting Table S6). ItchRO is a new tool that has had limited performance experience, and it is unclear if it performs equally at all ages. The positive CSS response in the 280‐µg/kg/day group suggests that this could be an issue.
Despite the inconsistent findings of the analyses of the primary endpoint of this study, the data in total suggest that maralixibat is generally safe and well tolerated and may reduce pruritus in ALGS. There is a clear unmet and significant need in the management of pruritus in ALGS and other cholestatic liver diseases in children. Continued investigation of maralixibat is warranted. These studies should continue to consider ongoing potential placebo effects and seek methods to determine dose responses on ileal ASBT activity and their relationship to biochemical and symptomatic effects.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgment
Heather Van Doren, M.F.A., senior medical editor with Arbor Research Collaborative for Health, provided editorial assistance on this manuscript. Peter F. Whitington, M.D., who retired prior to submission of this manuscript, contributed to the development, execution, and interpretation of this study.
Funding
Supported by U01 grants from the National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases: (U01DK103149 to B.L.S. & D.H.L.); (U01DK103135, U01DK062456‐13, U01DK062456‐15 to B.K.); (U01DK062456 to C.S. & J.C.M.); (U01DK062436 to L.M.B.); (U01DK062497 to K.D.S. & A.M.); (U01DK084536 to J.P.M.); (U01DK062466 to R.H.S.); (U01DK084575 to K.F.M.); (U01DK062481 to K.M.L.); (U01DK062500 to P.R.); (U01DK062470 to S.J.K.); (U01DK103140 to S.L.G.); (U01DK084538 to D.T.); (U01DK062453 to C.L.M. & R.J.S.); also supported (in part or in full) by the National Center for Advancing Translational Sciences of the NIH under the following Clinical and Translational Science Award grants: Cincinnati Children’s Hospital Medical Center (DK 62497, UL1TR0000777 to K.D.S. & A.M.), Children’s Healthcare of Atlanta (DK 62470, UL1TR002378 to S.J.K.), Children’s Hospital of Philadelphia (DK 62481, UL1TR001878 to K.M.L.), University of Michigan (DK 62456 to C.S. & J.C.M.), Riley Hospital for Children (DK 84536, UL1TR001108 to J.P.M.), Seattle Children’s Hospital (DK 84575, UL1TR000423 to K.F.M.), University of California San Francisco Children’s Hospital (DK 62500, UL1TR001872 to P.R.), Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center (DK 62503, UL1TR000005 to R.H.S.), Children’s Hospital Colorado (DK 62453, UL1TR001082 to C.L.M. & R.J.S.), Children’s Hospital Los Angeles (DK 62452, UL1TR000130 to D.T.), Ann and Robert H. Lurie Children’s Hospital of Chicago (DK 62436, UL1TR001422 to L.M.B.), Texas Children’s Hospital (DK103149 to B.L.S. & D.H.L.), Hospital for Sick Children (DK103135, DK062456‐13, DK062456‐15 to B.M.K.), and University of Utah (DK103140, UL1TR001067 to S.L.G.). See Supporting Table S3 for full listing of sites, principal investigators, and grant numbers.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Clinical trials registration number NCT02057692.
Potential conflict of interest: Dr. Kamath consults for Retrophin. Dr. Murray received grants from Gilead and Shire and owns stock in Merck. Dr. Miethke consults and received grants from Shire. Dr. Molleston consults for Lilly and received grants from AbbVie, Gilead, and Shire. Dr. Spino consults for Albireo. Dr. Leung consults for Merck and received grants from Bristol‐Myers Squibb, Gilead, AbbVie, and Roche. Dr. Rosenthal consults and is on the speakers’ bureau for Retrophin; he consults for Gilead, AbbVie, Intercept, Alexion, Albireo, and Audentes and received grants from Gilead, AbbVie, Bristol‐Myers Squibb, and Roche. Dr. Karpen consults for Intercept, Albireo, and Retrophin. Dr. Setchell consults for Retrophin and received grants from Retrophin and Shire; he owns stock in Asklepion. Dr. Sokol consults for Shire, Retrophin, Albireo, and Alexion. The other authors have nothing to report.
References
- 1. Emerick K, Rand E, Goldmuntz E, Krantz I, Spinner N, Piccoli D. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29:822‐829. [DOI] [PubMed] [Google Scholar]
- 2. Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology 2002;35:1501‐1506. [DOI] [PubMed] [Google Scholar]
- 3. Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, et al. Childhood Liver Disease Research Network (ChiLDReN). Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology 2017;65:1645‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neimark E, Chen F, Li X, Shneider BL. Bile acid‐induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 2004;40:149‐156. [DOI] [PubMed] [Google Scholar]
- 5. Hofmann AF. Inappropriate ileal conservation of bile acids in cholestatic liver disease: homeostasis gone awry. Gut 2003;52:1239‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neimark E, Shneider B. Novel surgical and pharmacological approaches to chronic cholestasis in children: partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. J Pediatr Gastroenterol Nutr 2003;36:296‐297. [DOI] [PubMed] [Google Scholar]
- 7. Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology 1988;95:130‐136. [DOI] [PubMed] [Google Scholar]
- 8. Kamath BM, Abetz‐Webb L, Kennedy C, Hepburn B, Gauthier M, Johnson N, et al. Development of a novel tool to assess the impact of itching in pediatric cholestasis. Patient 2018;11:69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Karrer FM. Use of rifampin for severe pruritus in children with chronic cholestasis. J Pediatr Gastroenterol Nutr 1999;29:442‐447. [DOI] [PubMed] [Google Scholar]
- 10. Zhang W, Jha P, Wolfe B, Gioiello A, Pellicciari R, Wang J, et al. Tandem mass spectrometric determination of atypical 3beta‐hydroxy‐delta5‐bile acids in patients with 3beta‐hydroxy‐delta5‐C27‐steroid oxidoreductase deficiency: application to diagnosis and monitoring of bile acid therapeutic response. Clin Chem 2015;61:955‐963. [DOI] [PubMed] [Google Scholar]
- 11. Shneider BL, Magee JC, Bezerra JA, Haber B, Karpen SJ, Raghunathan T, et al. Childhood Liver Disease Research Education Network (ChiLDREN). Efficacy of fat‐soluble vitamin supplementation in infants with biliary atresia. Pediatrics 2012;130:e607‐e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamath BM, Shneider BL, Spino C, Magee JC, Whitingon PF, Setchell KD, et al. Unraveling the relationship between itching, scratch scales, and biomarkers in children with Alagille syndrome. Hepatology 2017;66:653A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegade VS, Kendrick SF, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double‐blind, randomised, placebo‐controlled, crossover, phase 2a study. Lancet 2017;389:1114‐1123. [DOI] [PubMed] [Google Scholar]
- 14. Slijepcevic D, van de Graaf SF. Bile acid uptake transporters as targets for therapy. Dig Dis 2017;35:251‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baumann U, Lacaille F, Sturm E, Gonzales E, Arnell H, Jorgensen MH, et al. The ileal bile acid transport inhibitor A4250 decreases pruritus and serum bile acids in cholestatic liver diseases‐an ongoing multiple dose, open‐label, multicentre study. J Hepatol 2017;66(Suppl):S91. [Google Scholar]
- 16. Buchwald H, Varco RL, Matts JP, Long JM, Fitch LL, Campbell GS, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the program on the surgical control of the hyperlipidemias (POSCH). New Engl J Med 1990;323:946‐955. [DOI] [PubMed] [Google Scholar]
- 17. Chalasani N, Regev A. Drug‐induced liver injury in patients with preexisting chronic liver disease in drug development: how to identify and manage? Gastroenterology 2016;151:1046‐1051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials