

Mitochondrial myopathies are associated with mutations of both mitochondrial and nuclear DNA. Deoxyguanosine kinase (DGUOK) is a nuclear gene responsible for maintaining the mitochondrial deoxynucleotide pool required for replication and maintenance of mitochondrial DNA (mtDNA). Homozygous or compound heterozygous DGUOK mutations are associated with decreased activity of the mtDNA-encoded respiratory chain complexes, mtDNA depletions1–4 and deletions.5 We describe a case of juvenile-onset mitochondrial myopathy associated with DGUOK-related multiple mtDNA deletions.
Case report
A 38-year-old woman presented with painless slowly progressive symmetrical proximal muscle weakness since she was 8 years old. On initial presentation, she reported difficulty climbing, holding her arms upright, running, and competing with her classmates. Her weakness was largely attributed to her being overweight. For 1 year before presentation, she had also developed diplopia in far vision. However, no primary eye disease was found. She denied shortness of breath, exercise-induced muscle cramps, hearing impairment, palpitations, seizures, and speech difficulty. Her medical history was significant for obsessive-compulsive disorder and Raynaud phenomena. Her family history included her mother and a brother with big calf muscles who were positive for proximal muscle weakness.
Her neurologic examination was notable for limited eye abduction bilaterally and mild right lid ptosis. Other cranial nerves were intact. She had mild calf hypertrophy and scapular winging. She had mild proximal muscle weakness graded on the Oxford scale as 4/5 in deltoids, 4/5 in supraspinatus muscles, 3/5 in hip flexors, and 5/5 elsewhere. Ankle reflexes were diminished with no gait abnormality. Cardiac and ear examinations were normal.
Her creatine kinase was elevated to 503 U/L. MRI of the pelvis and thighs without contrast was performed, which showed symmetric fatty infiltration of pelvic and thigh muscles with reduced muscle bulk. EMG did not reveal a myogenic or neurogenic pattern. Biopsy of the left biceps was consistent with a mitochondrial myopathy, with many ragged red and blue fibers, moderate variation in fiber morphology, and many cytochrome oxidase–negative and succinate dehydrogenase–positive fibers (figure, A).
Figure. COX:succinate dehydrogenase (SHD) stain and deoxyguanosine kinase (DGUOK) primary sequence.
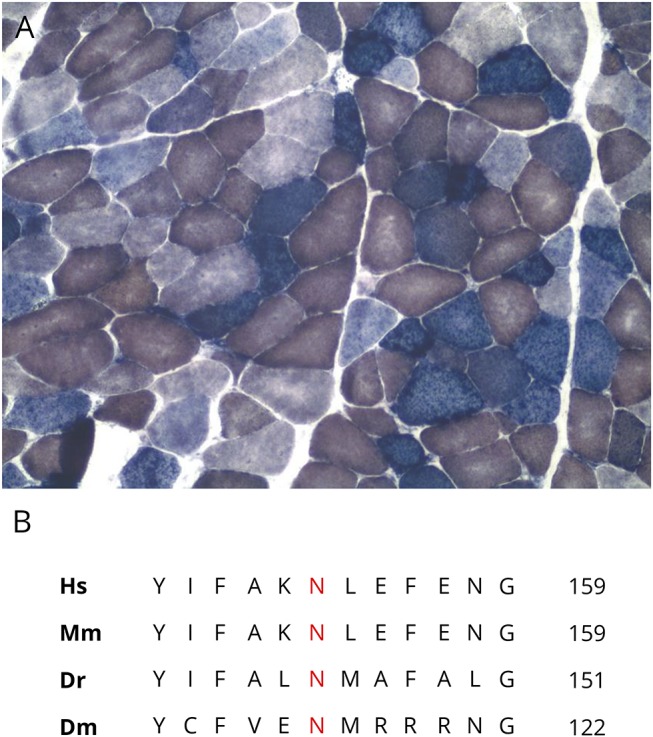
(A) Muscle biopsy showing many cytochrome oxidase–negative/SHD-positive (blue stained) fibers (×100 magnification). (B) Primary sequence of DGUOK showing the position of asparagine (N) in different species including humans (Hs), mouse (Mm), zebrafish (Dr), and Drosophila (Dm).
Next-generation sequencing and deletion/duplication analysis of 319 nuclear genes using the blood sample revealed 2 mutations in the DGUOK gene: c.195G>A and c.462T>A in exon 2 and 4, respectively. Parental data were not available to confirm cis or trans configuration. Mitochondrial genome sequencing and deletion analysis was performed using the muscle sample, which identified 3 large deletions of the mitochondrial genome: 10.7, 12.6, and 9.6 kb. The sum total of heteroplasmy of these deletions was estimated to be less than 15 percent as it was confirmed by sequencing and not by array comparative genomic hybridization.
The patient was diagnosed with DGUOK-related autosomal recessive multiple mitochondrial deletion syndrome, producing proximal muscle weakness and external ophthalmoparesis. She was counseled about exercise, dietary, and supportive measures.
Discussion
Mitochondrial myopathy is a disease of skeletal muscles, with or without CNS involvement, caused by defective mitochondrial metabolism. It is produced by defects in nuclear or mtDNA. Several nuclear genes are responsible for replication and maintenance of mtDNA including POLG, POLG2, C10ORF2, TYMP, TK2, RR2MB, and DGUOK.6 Defects in these genes affect mtDNA content (number of copies) or cause mtDNA deletions.
Loss-of-function mutations in DGUOK are associated with autosomal recessive inheritance of 3 main phenotypes: mtDNA depletion syndrome-3; noncirrhotic portal hypertension; and autosomal recessive progressive external ophthalmoplegia (PEO) with mtDNA deletions. Mutations in DGUOK have largely been described in mtDNA depletion syndromes, producing a multisystem illness or isolated liver disease.1,2,4 Myopathy has rarely been observed with early onset of symptoms.3 Moreover, compared with cases with decreased mtDNA content, only a few cases with mtDNA deletions producing myopathy have been reported. Six such cases were reported in 2012, in which age at onset of limb or extraocular weakness ranged from 20 to 69 years, and the majority (5) were older than 40 years.5 In contrast, our case presented with proximal muscle weakness at age 8 years and remained free of liver disease. Our participant harbors 2 mutations in the DGUOK gene, which have both been known to occur only in trans to other pathogenic variants, suggesting a trans configuration in our patient. The c.195G>A mutation is predicted to produce a p.Trp65Ter nonsense pathogenic variant. It has a frequency of 8.12 × 10−6 in a control population of normal individuals sequenced by next-generation sequencing (NGS).7 It has been reported with neonatal hepatocerebral disease in trans to another truncating mutation.4. The second variant, c.462T>A, is predicted to result in the Asn154Lys substitution, with a significantly higher frequency of 1.34 × 10−4 in normal controls.7 It has been reported in cases of adult-onset PEO in trans with other pathogenic variants.5 The asparagine at position 154 (figure, B) is highly conserved in evolution from Drosophila melanogaster to humans, suggesting an important role in maintaining protein structure or function.
Our case expands the phenotypic spectrum of DGUOK mutations and highlights the importance of NGS in children and adults to timely diagnose mitochondrial myopathy. The markedly slow progression of symptoms observed in our case may have contributed to the delay in diagnosis. Longitudinal studies are needed to further investigate the course and predict the outcomes in patients harboring DGUOK mutations.
Author contributions
F.N.U. Komal: drafting the manuscript. P.M. Moretti: critical revision of the manuscript for important intellectual content and preparation of the figure. A.I. Shaibani: acquisition of data and study supervision.
Study funding
This was not an industry-supported study. All authors have reported no financial conflicts of interest. There was no investigational or off-label use.
Disclosure
This was not an industry-supported study. All authors have reported no financial conflicts of interest. There was no investigational or off-label use. F.N.U. Komal reports no disclosures. P.M. Moretti has received research support from the NIH, the New Jersey Commission on Brain Injury Research, and the Department of Veterans Affairs. A.I. Shaibani receives publishing royalties from Oxford University Press. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013;10:186–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freisinger P, Fütterer N, Lankes E, et al. . Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol 2006;63:1129–1134. [DOI] [PubMed] [Google Scholar]
- 3.Buchaklian AH, Helbling D, Ware SM, Dimmock DP. Recessive deoxyguanosine kinase deficiency causes juvenile onset mitochondrial myopathy. Mol Genet Metab 2012;107:92–94. [DOI] [PubMed] [Google Scholar]
- 4.Dimmock D, Zhang Q, Dionisi-Vici C, et al. . Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat 2008;29:330–331. [DOI] [PubMed] [Google Scholar]
- 5.Ronchi D, Garone C, Bordoni A, et al. . Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 2012;135:3404–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol 2012;47:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek M, Karczewski KJ, Minikel EV, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285. [DOI] [PMC free article] [PubMed] [Google Scholar]