

Abstract
Biliary atresia (BA) is a fibroinflammatory disease of the intra- and extrahepatic biliary tree. Without medical treatment, surgical hepatic portoenterosmy (HPE) may restore bile drainage, but progression of the intrahepatic disease results in complications of portal hypertension and advanced cirrhosis in most children. Recognizing that further progress in the field is unlikely without a better understanding of the underlying cause(s) and pathogenesis of the disease, the National Institutes of Diabetes and Digestive and Kidney Diseases sponsored a research workshop focused on innovative and promising approaches and on identifying future areas of research. Investigators discussed recent advances using gestational ultrasound and results of newborn BA screening with serum direct (conjugated) bilirubin that support a pre-natal onset of biliary injury. Experimental and human studies implicate the toxic properties of environmental toxins (e.g. biliatresone) and of viruses (e.g. CMV) to the biliary system. Among host factors, sequence variants in genes related to biliary development and ciliopathies, a notable lack of a cholangiocyte glycocalyx and of submucosal collagen bundles in the neonatal extrahepatic bile ducts, and an innate pro-inflammatory bias of the neonatal immune system contribute to an increased susceptibility to damage and obstruction following an epithelial injury. These advances form the foundation for a future research agenda focused on identifying the environmental and host factor(s) that cause BA, the potential use of population screening, studies of the mechanisms of prominent fibrosis in young infants, determinations of clinical surrogates of disease progression, and the design of clinical trials that target subgroups of patients with initial drainage following HPE.
Keywords: Liver, children, cholangiocyte, cholestasis, bile
The Challenge
Biliary atresia (BA) is a severe neonatal disease caused by an inflammatory and fibrotic obliteration of the extrahepatic biliary tree resulting in cholestasis and progressive hepatic failure. Left untreated, BA invariably leads to death from end-stage liver disease in the first two years of life. Even with surgical treatment, BA remains the most common cause of end-stage liver disease and most frequent reason for liver transplantation in children. Yet, despite its frequency and serious consequences, the cause(s) and pathogenesis of BA remain unknown (1, 2).
BA is a disease of the intra- and extrahepatic biliary tree and causes liver injury largely as a result of extrahepatic obstruction. Described in the late 19th century after histopathologic studies of children dying of liver disease, BA continues to be a major medical challenge in the 21st century, although the nature of the challenge has changed. Two important advances in management of BA occurred in the second half of the 20th century. The first was the development of the Kasai hepatic portoenterostomy (HPE), in which the extrahepatic biliary system is surgically removed and replaced with a loop of intestine (Roux-en-Y anastomosis) that is connected directly to the portal area of the liver, allowing for restoration of bile flow and relief of obstruction. If done within the first 45 days of life, HPE is associated with a 65.5% survival with the native liver at 2 years of age and 40.5% at 15 years (3). The second major advance, also surgical, was liver transplantation. With the combination of HPE and liver transplantation, most children with BA now survive into adulthood. Major clinical challenges remaining are: 1) early diagnosis, so as to increase the percentage of successful HPEs; 2) prevention of the progressive liver injury and fibrosis that occur even after successful HPE; and 3) management of long-term complications of liver transplantation and immune suppression.
Reversing BA at its earliest stages and preventing it altogether, however, are broader key challenges that persist. The advances in BA management during the last 50 years have been dramatic, but further progress seems unlikely without a better understanding of the underlying cause(s) and pathogenesis of the disease.
For these reasons, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) sponsored a research workshop in June 2017, focused on identifying innovative and promising approaches to understand the pathogenesis of BA and translating this knowledge into treatment and prevention (Supplemental Table: Workshop agenda).
Clinical Research
Epidemiologic studies on BA have provided few clues to its etiology. BA occurs in 1 in 8,000 to 18,000 live births and appears to be more frequent in Asians and Africans than Europeans and more common in females than males. These differences, however, are not great. The incidence of BA may vary by region or time of year, but in general there is limited association with exposure to animals or environmental toxins, parental diet, health status, or occupation. Epidemics of BA in humans have not been described and it rarely appears in clusters, temporal or geographic. Strikingly, BA shows little evidence of being genetic: familial cases are rarely found, and most twin studies show discordance of the disease, even between identical twins.
The clinical features, course and outcome of BA have been well described. Children typically appear normal at birth. Cholestasis may be present early, but it is rarely obvious to the clinician because it overlaps with the common physiologic jaundice seen in more than half of newborns during the first few weeks of life. In children with BA, however, jaundice persists and the appearance of signs of cholestasis such as dark urine and pale stools follow. A provisional diagnosis is occasionally made before 4 weeks of life, but more typically not until 6 to 12 weeks and is based upon exclusion of other causes of neonatal cholestasis and typical liver biopsy findings. The diagnosis is confirmed by identification of an atretic biliary tree at the time of HPE.
The success of HPE for BA is most dependent upon the age at which it is performed, with the best results (60-80% restoration of bile flow) seen when done before 30 to 45 days of age. At present, however, at least half of children still require liver transplantation before 2 years of age. In addition, those with a successful HPE who survive infancy with a native liver often have progressive hepatic dysfunction, and ultimately more than 75% of them will require liver transplantation before 20 years of life (1, 2).
Several clinical variants of BA have been described (Table 1). These include isolated, typical BA (80%), BA with cystic dilations of the biliary tree (5-10%), a variant attributed to cytomegalovirus (CMV) infection (5-10%), and several variants with associated congenital malformations (such as laterality defects or congenital heart, urinary or gastrointestinal diseases) which are generally grouped together as biliary atresia-splenic malformation syndrome (BASM: 5-15%). Whether these different variants have different causes is not clear, but their clinical course and response to HPE may differ (4, 5).
Table 1.
Classification of Biliary Atresia (BA) Phenotypes
| BA Phenotype | Incidence and disease features |
|---|---|
| I. Isolated form | 80% of patients, variable jaundice-free period after birth, absence of other major congenital malformations |
| -CMV variant | Variable incidence based on geography and methodological detection, poor response to HPE |
| II. Congenital Malformation form | 10-20 % of patients with at least one major congenital malformation, early onset of jaundice, poorer outcome after HPE |
| a. BASM variant | 1-10% of patients, associated laterality defects (poly/asplenia, heterotaxia, midline liver, interrupted IVC, intestinal malrotation, preduodenal portal vein, congenital heart disease), poor response to HPE |
| b. BA with major malformation variant (non-laterality defect) | 5-10% of patients, with major cardiovascular, gastrointestinal, and genitourinary malformations that are not related to laterality |
| c. Cystic variant | 8% of patients, might co-exist with BASM, better response to HPE than over variants in this group |
An important debate has been whether BA arises during fetal life or only postnatally. In support of the post-natal nature of BA is the lack of reports of BA in stillborn or aborted fetuses and the limited association with prematurity, low birth weight, and poor Apgar scores; reports are conflicting on whether there are differences in the incidence of BA between premature and term newborns (6, 7). In support of a prenatal etiology, instances of biliary atresia with cystic abnormalities have been recognized by second and third trimester ultrasound examinations (8). Furthermore, recent work, validated by a prospective study, reports that direct bilirubin levels were elevated in 34 of 34 BA patients at 24-72 hours, suggesting that the initial insult is prenatal and that direct bilirubin might be a means of screening for and identifying BA shortly after birth (9, 10). Data on histologic evaluation of the extrahepatic bile ducts at the time of Kasai operation presented at the symposium suggest that the biliary abnormalities of BA are similar in patients undergoing surgery early compared to those done later. These combined findings suggest that BA is a disease of the fetus, becoming manifest only after parturition and loss of the protective physiology of the mother and placenta. In keeping with the onset of cholestasis in the early postnatal period, one can envision the broad use of population-based newborn screening using serum direct bilirubin (as reported in a group of hospitals in Houston; references (9, 10) or a stool color card (as reported in Japan, Taiwan, and Canada; references (11–13). Before such use, it will be important to compare the different screening strategies, their cost-effectiveness, and impact on patient outcomes.
Basic Research
Clinical investigation has provided intriguing clues to the nature of BA, but only with basic research will the underlying cause(s) and the pathogenesis of BA become clear. Both environmental and host factors, including the role of viruses, toxins and environmental exposures, developmental pathology, genetic abnormalities, aberrant neonatal immune responses, and abnormal fibrogenesis, have been investigated. Data presented at the symposium suggest that many of these factors warrant further study.
Infectious causes
Reovirus, rotavirus, and cytomegalovirus (CMV) have been proposed as potential etiologies of BA. The Rhesus rotavirus (RRV)-induced BA mouse model has proved helpful in investigating the role of viruses and inflammation in the pathogenesis of bile duct injury in BA. The largest body of literature supporting a viral infection as an initiating event in BA pathogenesis pertains to CMV. CMV DNA was identified in 60% of BA patients at the time of diagnosis in one study (14) and immunological studies of infants with BA reported a liver memory T cell response to CMV in 56% of patients (15). In another study, a subgroup of BA patients with CMV-IgM positivity at diagnosis had higher rates of jaundice, liver inflammation, fibrosis and need for liver transplant (16). It is plausible that the infection is short-lived, leading to an inability to identify the virus in some cases, and that viral infection of cholangiocytes sets the stage for an aberrant autoimmune response targeting cholangiocytes and leading to progressive biliary injury and cirrhosis. Nonetheless, detailed modern molecular virologic analyses have yet to provide validation of perinatal CMV infection, and new approaches to identify low levels of virus, such as tissue-based universal virus detection for viral metagenomics, should be employed in BA.
Environmental Toxins
Spontaneous outbreaks of a BA-like disease in neonatal livestock in Australia recently led to the identification of a biliary toxin called biliatresone (17). Biliatresone, which was isolated from plants eaten by pregnant dams, causes selective extrahepatic biliary damage in larval zebrafish and the offspring of treated pregnant mice. Although humans are almost certainly not exposed to biliatresone, the identification of key structural motifs may lead to toxins with human relevance. Furthermore, the use of biliatresone in model systems has already yielded important insights into the disturbed cellular pathways involved bile duct damage and repair.
Developmental immaturity of the neonatal bile ducts
BA occurs only in newborns, suggesting that the fetus and young neonate, by virtue of developmental stage, are particularly susceptible to injury and dysregulated repair (fibrosis) after an insult to the biliary tree. The human and murine hepatobiliary systems follow the same general pattern of development, although in relative terms have different time courses (18), with bile flow reaching the intestine at embryo days 75 to 85 and mouse embryo day 17.5. Notably, the intra- and extrahepatic biliary systems are developmentally distinct, with the liver and intrahepatic bile ducts (IHBD) arising from a different region of endoderm than the extrahepatic bile ducts (EHBD). IHBD development is SOX4 and SOX9 dependent, while the EHBD, which has not been as well studied, arises from a PDX1+/SOX17+ domain and then segregates into PDX1+ ventral pancreas and SOX17+ EHBD; Sox17 is required for EHBD development in mice, and Sox17 heterozygous mice have a BA-like phenotype (19, 20). Data from Sox4 and Sox9 knockout mice suggest that the junction between the IHBD and EHBD is within the hilum, the site of anastomosis of HPE.
Anatomical immaturities in the neonatal EHBD may also contribute to an age-limited susceptibility to injury. Cholangiocytes elaborate an apical glycocalyx with a “bicarbonate umbrella”; this layer protects cholangiocytes from bile acid toxicity and may be physiologically relevant in many cholangiopathies (21). Preliminary data presented at the symposium showed that BALB/c mice lack a mature cholangiocyte glycocalyx at birth, and that extrahepatic cholangiocyte cell-cell junctions are similarly poorly developed, suggesting the potential for enhanced injury and resulting bile leakage into the submucosa in neonates.
The submucosa of the neonatal EHBD is also immature in the neonatal mouse. The adult EHBD submucosa consists of dense collagen bundles which are surrounded by interstitial fluid. The newborn mouse submucosa, on the other hand, has almost no collagen; collagen bundles are rapidly deposited during the first week of life, presumably by the many cells (not yet identified) with large amounts of rough endoplasmic reticulum present only at that time. This raises the possibility that bile leaks propagate rapidly in the “open” submucosa of the neonate and stimulate a population of cells “primed” for fibrogenesis. These anatomic features suggest EHBDs have an increased susceptibility to injury and increased potential for a fibrotic response. Future work is needed to extend these findings to human bile ducts and confirm their disease relevance.
Genetics
There is increasing evidence that BA patients have a genetic susceptibility to enhanced biliary injury and pathologic repair. Genetic approaches to understand BA have included candidate gene analyses, copy number variation (CNV) association studies, genome-wide association studies (GWAS) and exome sequencing. Some of the genes, such as FOXA2, have been linked to BA only in members of a single family (22). ADD3 was found in some genome-wide single nucleotide polymorphism association studies, and GPC1 was identified through CNV studies; disruption of either of these genes in zebrafish results in biliary defects (23, 24). Preliminary data presented at the symposium on ongoing genetic studies using whole exome sequencing of BASM trios identified PKD1L1 variants, raising the potential that abnormal ciliary function contributes to disease susceptibility.
Other preliminary findings presented at the meeting included a strong variant signal in patients with and without BASM within an intron of EFEMP1, which encodes the elastic fiber protein fibulin-3, and whole exome sequencing of 30 family trios identifying a loss-of-function mutation in STIP1, encoding a stress-induced HSP90 co-chaperone protein, in one child with BA. Zebrafish with a heterozygous loss-of-function mutation in stip1 had markedly increased susceptibility to the toxin biliatresone.
Work with biliatresone also suggests that variations in human glutathione metabolism genes are potential risk factors for biliary injury. Biliatresone is a strong electrophile that binds reduced glutathione (GSH) in vitro and causes a rapid decrease in levels of GSH in both zebrafish and mouse cholangiocytes. Extrahepatic cholangiocytes (in neonates and adults) have significantly lower baseline levels of GSH than liver; a zebrafish biosensor able to detect the ratio between GSH and oxidized glutathione showed that extrahepatic cholangiocytes but not intrahepatic cholangiocytes or hepatocytes become highly oxidized in response to biliatresone (25, 26). Restoration of GSH by treatment with N-acetyl-L-cysteine significantly delayed biliatresone-induced damage in fish and prevented damage in mouse neonatal bile duct explants and cholangiocytes. Zebrafish with loss-of-function mutations in gclm (the regulatory subunit of the rate-limiting enzyme in GSH synthesis) and mrp2 (the canalicular transporter for GSH) were normal except for markedly increased sensitivity to biliatresone, with damage seen in both intra- and extra-hepatic cholangiocytes. Collectively, these data highlight the relevance of GSH levels to biliary damage, and suggest that genetic susceptibility must be combined with an environmental insult to produce biliary injury – strong support for the hypothesis that BA is multifactorial and requires both exposure and susceptibility (Figure).
Figure.
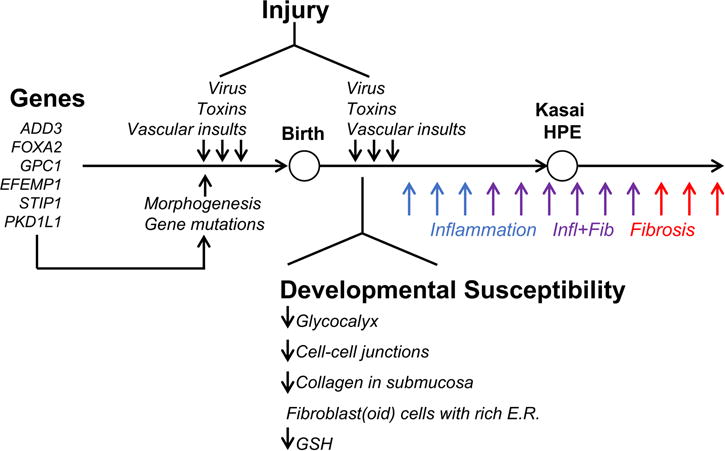
Factors implicated in etiology and pathogenesis of biliary atresia. Susceptibility factors include gene sequence variants and anatomical features that may make extrahepatic bile ducts of the neonate uniquely prone to cellular injury and prominent expression of extracellular matrix. With new findings that infants with biliary atresia have increase in serum conjugated bilirubin in the first days of life, bile duct injury by virus, toxins or vascular insults may also occur prenatally and trigger the inflammatory and fibrosing tissue injury that is typical of the disease.
Immune dysfunction
Whether triggered by a virus, toxin or other factor that injures cholangiocytes, the resultant breach of the epithelium activates the neonatal immune system. Activated immune cells have been identified in BA livers at diagnosis, and have been linked experimentally to autoimmunity, amplification of epithelial injury, and bile duct obstruction (4).
Autoimmune diseases develop from either a primary breakdown in tolerance or secondary to a deficiency in immune regulation. Regulatory T cells (Tregs), characterized by the expression of the transcription factor FOXP3, are essential to control the low levels of autoreactivity that occur normally in humans. In the RRV-induced model of BA and in human BA, defects in the number and function of Tregs have been associated with autoreactive T cells targeting bile duct epithelia (27). One mechanism to explain Treg deficits is hypermethylation of the FOXP3 promoter region, resulting in decreased Treg function (28).
Several lines of evidence support autoimmunity as an important mechanism of disease in BA (see Table 2 for summary of studies related to indirect proof of autoimmunity). While a subject of ongoing debate, no immunosuppressive therapy has been shown to be uniformly effective at decreasing the progression of disease post-HPE. Interestingly, the lack of response to immunosuppressive agents is similar to that seen in other autoimmune biliary diseases (i.e. primary biliary cholangitis [PBC] and primary sclerosing cholangitis [PSC]). Future immunologic and genetic studies should focus on pathways of immune dysregulation, as well as the identification of autoantibodies that may function as biomarkers of disease severity and provide clues to disease pathogenesis.
Table 2.
The relationship between biliary atresia (BA) and autoimmunity
| Criteria for autoimmune disease | Evidence in support of or against autoimmunity in BA |
|---|---|
| Direct Proof: | |
| Transfer of autoantibodies from BA into normal recipient | No direct proof. Anti-enolase autoantibodies in mouse model (57) |
|
| |
| Indirect Proof: | |
| Reproduce BA through adoptive transfer of autoreactive T cells | Adoptive transfer of liver T cells from BA mice into T cell deficient mice results in bile duct targeted inflammatory damage (58, 59) |
|
| |
| Circumstantial Evidence: | |
| 1) Family history of autoimmunity | 44% autoimmunity in 1st degree relatives of BA (5) |
|
| |
| 2) Lymphocytic infiltrate of the target organ, especially if restricted TCR-Vβ usage | Lymphocytic infiltrates of intrahepatic and extrahepatic bile ducts (31, 60, 61); Limited TCR Vβ repertoire, oligoclonal expansion of CD4+ and CD8+ T cells (38) |
|
| |
| 3) Statistical association with HLA genotype or aberrant expression of HLA class II antigens on the affected organ | Japan: increase in HLA-DR2 in BA with linkage disequilibrium, U.S.: No HLA association; Increased expression of HLA class II (HLA DR) on bile duct epithelia (62–64) |
|
| |
| 4) Favorable response to immunosuppression | No response to corticosteroids (37) or IVIg* |
Manuscript under review
Analyses of human tissues have shown that CD4+ and CD8+ lymphocytes are activated in BA, populate the livers at diagnosis and express Th1 cytokines, such as interferon-gamma (IFNγ), interleukin (IL)-2, and tumor necrosis factor-alpha (TNFα) (4, 29). More direct functional insight emerged from the report that CD4+ and CD8+ lymphocytes undergo oligoclonal expansion in the liver and bile duct remnants, and activate upon exposure to CMV antigens (30). In approaches to directly examine how these findings relate to mechanisms of disease, investigators have used the RRV mouse model (4). Findings in this model that have also been validated in liver tissues from infants with BA include the role of cellular factors that recognize viruses (e.g. α2β1 integrins in cholangiocytes), a lack of Treg lymphocytes early in the disease, and the activation of pro-inflammatory signals in liver-resident immune cells (macrophages, dendritic cells, lymphocytes, and NK cells). Experimentally, the depletion of these effector cells individually or the inactivation of key cytokines (e.g. IL-8, IL-15, IFNγ, TNFα) suppresses or prevents epithelial injury of EHBDs, decreases inflammation, and maintains luminal patency (Figure) (4).
Despite a prominent type 1 inflammatory response, studies in humans and mice also show a Th17 commitment and its correlation with disease progression (31). In addition, livers and sera from a subgroup of BA patients have high levels of type 2 cytokines, such as IL-13, IL-33, and IL-4 (4). Taken together, these findings raise the possibility that patients differ molecularly, and that these differences represent biological transitions from a Type 1 to a mixed (Th1-2-17) immune response that drives persistent liver injury and progressive fibrosis.
Fibrosis
Advances in understanding the mechanisms of hepatic fibrosis and the development of anti-fibrotic therapies may benefit a broad array of chronic liver diseases, including BA. However, studies focusing on biliary fibrosis in BA are needed to provide insight into its unusually rapid progression and its onset during early childhood. In EHBDs, the release of the Th2 cytokine IL-13 by innate lymphoid cells induces cholangiocyte proliferation after RRV infection, thus stimulating a proliferative circuit that promotes epithelial repair. In the liver, the dominant result of IL-13 and IL-33 expression is the activation of stellate cell fibrogenesis and the promotion of hepatic fibrosis (4). Interestingly, fibrosis has also been linked to Type 1 immune responses, as demonstrated by the finding that circulating and hepatic lymphocytes have Th1 features, correlate with liver fibrosis, and are able to induce proliferation and activation of stellate cells (32). Resident macrophages also play a role in fibrogenesis with a transition from M1 to M2 profiles. The combination of these cells with the secretion of soluble mediators like IL-8, TGFβ, PDGF, CXCL1 and TLR4 are likely to create an ecosystem conducive to excessive fibrosis in BA (33).
Several non-immune factors have been implicated in triggering and amplifying fibrogenic signals in human cholangiopathies, but few have been systematically studied in BA. Tissue analyses from patients with BA and other cholangiopathies identify “reactive ductular cells,” a population of cholangiocyte-like cells that are thought to participate in tissue repair, and join portal myofibroblasts and stellate cells in the regulation of biliary cirrhosis. Portal fibroblasts and cholangiocytes cross-talk via Wingless(Wnt)-β-catenin, Hedgehog (Hh), Notch, and Hippo/YAP signaling, which constitute important developmental factors and are prime suspects for promotion of fibrosis. In support of this possibility, Hh activity has been associated with epithelial-to-mesenchymal transition, biliary dysmorphogenesis and poor survival in diseased livers (34, 35), with reports that Hh-related signals are important to experimental models of biliary injury in zebrafish and neonatal mice. Although these pathways have not been studied after HPE, the rapid progression to cirrhosis suggests the existence of unique pathogenic processes that are not suppressed after HPE. One example is the increase of serum and liver MMP7 during progression of disease, and its correlation with the fibrosis stage (36).
Innovative Approaches to Treatment
Recent Results
Treatments after HPE with ursodeoxycholic acid, antibiotics, and fat-soluble vitamin preparations are routine, but have not been systematically evaluated and have not substantially improved outcomes. In a double-blind, place-controlled study, corticosteroids begun within 3 days of the HPE (START trial) did not change outcomes and increased the risk of serious adverse effects while receiving corticosteroids compared to placebo (37). The open-label use of corticosteroids in younger BA infants did appear to improve biliary drainage, with yet-to-be proven impact on survival with the native liver (38), raising the possibility that specific subsets of infants may be responsive to treatment. While some clinical centers use corticosteroids after HPE, there is no evidence as of 2017 that its broad use (or the use of other medical therapies) in all BA patients enhances long-term clinical outcomes after HPE.
Current paradigms which pool all BA patients imply that they will be equally responsive to therapeutics. However, approximately 50% of BA patients never achieve effective bile flow after HPE (39), although they are indistinguishable at diagnosis from those who do. Patients with poor bile flow are unlikely to benefit from any medical intervention; better predictors of HPE outcome would enable the design of trials that focus on those patients who clear their jaundice after surgery. Recent reports indicate that those who develop bile flow (defined as a serum total bilirubin level < 2 mg/dL) within 3 months of HPE have a >80% chance to be in the group of BA patients surviving with the natively liver at 2 years of age (40). More importantly, although these are considered “successful” responders to Kasai HPE, at least 75% of them will develop ongoing fibrosis, portal hypertension and cirrhosis, with significant risks of hepatic complications, death and transplant from childhood through young adulthood (41–44). These BA patients are candidates for trials of an array of novel therapeutic agents as they have the potential to respond and stand to benefit significantly.
Potential future therapies
Several new agents are currently being tested in cholestatic and fibrotic liver diseases in adults (45) (Table 3). These include bile acid-based therapeutics such as the FXR agonist obeticholic acid in PBC and the modified bile acid norursodeoxycholic acid, which is currently in Phase 2 trials in adult PSC (46, 47). FXR agonists suppress bile acid synthesis and engage multiple anti-cholestatic adaptive responses in liver, while norursodeoxycholic reduces markers of cholangiocyte damage, suggesting that one, or both, agents might be beneficial in BA. Other agents of potential value are those that interrupt the ileal reabsorption of bile acids, the luminally-restricted ASBT (ileal bile acid transporter) inhibitors. These agents diminish total bile acid flux through the cholestatic liver, reduce hepatic bile acid retention, and alter the composition of bile. In several animal models of biliary tract disease, ASBT inhibition reduced cholestatic and fibrotic liver damage (48, 49). A recently described inhibitor of the hepatocyte basolateral bile acid uptake protein, NTCP, Myrcludex B, may provide a novel approach to reducing bile acid burden in the liver (50). Two agents currently in common clinical use in pediatric liver diseases, bile acid sequestrants (cholestyramine or colesevelam) and ursodeoxycholic acid, have yet to be thoroughly tested in clinical trials in BA (51).
Table 3.
Novel Medical Therapies for Consideration in Biliary Atresia
|
Outside of bile acid-based approaches, directly addressing inflammation or fibrogenesis in this highly fibrotic disease is a potential strategy. Although current anti-inflammatory approaches have yet to bear fruit in BA, it is possible that there are subsets of BA patients who would benefit from selective agents after detailed cellular and molecular immunotyping. There are several anti-fibrotic agents undergoing study in liver diseases, including the CCR2/CCR5 antagonist cenicriviroc, which has improved fibrosis in nonalcoholic steatohepatitis in the recently completed CENTAUR trial (52).
First-step therapies targeting BA patients with good biliary drainage after HPE should be directed towards alleviating cholestasis, cholangiocyte injury, peribiliary fibrosis and portal hypertension. Biomarker development (e.g. liver stiffness measures, serum bile acids or GGT levels) (52) would help evaluate the efficacy of therapeutics, once validated in this population.
Use of Innovative Experimental Models
Among the more exciting aspects of current arcs in liver disease research are those that have begun to discern the molecular underpinnings of hepatocyte and cholangiocyte differentiation. Several groups have used induced pluripotent stem cell technologies or novel in vitro protocols to drive the differentiation of both hepatocytes and cholangiocytes from circulating mononuclear cells, fibroblasts and other cell types. These technologies can uncover specific biological properties of human cholangiocytes derived from patients, something that is problematic, or not possible, in mice. Artificial bile ducts generated from collagen-based scaffolds and biliary organoids have also been developed (53). This interesting technology may be a first step towards patient-specific biliary tract reconstruction. Finally, cutting-edge technologies to craft organoids from hepatocytes, cholangiocytes and various stromal cells may be powerful future strategies to model human disease, develop cell-based therapies, and test therapeutic agents (57) (54–56).
Future Directions in Research
While rare, BA is a heavy burden for the affected child and family, and generates high health care costs to society. Improvements in management, treatment and possibly prevention would have enormous individual and societal benefits. Questions to address in future research on BA include:
Is BA a disease of the fetus that starts in the prenatal period and might be detected before or at birth or shortly thereafter? Identifying means of early, or even pre-natal, detection of BA would be of great benefit. For instance, validating the finding that serum direct (conjugated) bilirubin is increased soon after birth in infants with BA and designing new screening strategies for cholestasis (e.g.: direct bilirubin, stool color card) applicable to the general newborn population, would permit diagnosis at early stages of biliary injury, allowing for early surgical intervention. Early detection might also allow for medical therapies to prevent the progression of injury, such as use of agents that increase GSH or increase bile flow.
Are biliatresone and related environmental toxins relevant to human BA? The use of zebrafish-based screening protocols and the successful culture of primary human cholangiocytes or biliary epithelium/organoids are powerful systems to test candidate toxins in the environment. They may also identify common pathways of biliary injury that may be targeted by new treatment strategies (such as glutathione replacement).
What is the contribution of gene variants to disease susceptibility? Ongoing GWAS and whole exome sequencing (and perhaps future genome scanning) of well characterized large cohorts may identify additional risk alleles that are relevant to specific phenotypes and/or progression of disease. Analysis of gene mutations and epigenetic modifications in the involved tissue, including the residual extrahepatic epithelium from the biliary remnants, could also be informative.
To what extent do the unique anatomical features of the developing extrahepatic bile ducts contribute to the susceptibility to biliary injury and the rapid progression to obliterative fibrosis?
Do inflammatory and immune responses play a role in the progression of biliary fibrosis and liver dysfunction?
Can proteins and other factors residing in the neonatal biliary epithelium be used as biomarkers of disease and disease progression?
The use of high-density immunotyping panels and cell-sequencing of mononuclear cells in peripheral blood, liver and the extrahepatic biliary tree may be particularly effective in identifying cellular cross-talk and molecular targets to block mechanisms of disease.
Can patients who will respond favorably to HPE be identified preoperatively, so that they can be targeted for trials of new therapeutic agents?
What accounts for the progressive hepatic dysfunction that occurs in a high proportion of children with a successful HPE who ultimately require liver transplantation? Is the injury caused by the same factors that cause BA or by secondary events occurring after HPE (such as an autoimmune response or alterations in the microbiome)?
What are the most clinically relevant end-points of disease progression other than liver failure, transplantation or death? Can these end-points be used for discovery and validation of non-invasive methods for monitoring hepatic dysfunction and development of fibrosis and portal hypertension (liver stiffness, serum markers)?
Chief among the drivers of future progress in BA research will be the access to well-phenotyped patients and their tissues, collaboration among basic and clinical investigators, and partnership with industry. Already available are serum, plasma, tissue and DNA samples from a cohort of more than 1,000 subjects with BA enrolled into studies of the Childhood Liver Disease Research Network (https://childrennetwork.org). These subjects are the focus of prospective studies and provide tissues and clinical data for studies of etiology and pathogenesis of disease; the specimens in the NIDDK repository are available to investigators interested in BA research. Short- and long-term studies by the Network and interventional trials will be particularly critical to the development of new strategies to improve the outcome of children with BA.
Supplementary Material
Acknowledgments
The authors would like to thank all investigators who presented at the “Biliary Atresia: A Clinical and Translational Science Research Workshop,” on June 28, 2017 at the National Institutes of Health, Bethesda, MD – Mark Davenport, Nancy Spinner, Benjamin Shneider, Frederic Lemaigre, Michael Pack, Mario Strazzabosco, Claus Petersen, Mikko Pakarinen, Frederick Suchy, Ludovic Valier, and Holger Willenbring.
Funding Support
The research workshop “Biliary Atresia: Clinical and Research Challenges for the 21st Century” was organized and funded by the National Institute of Diabetes and Digestive and Kidney Diseases without outside commercial funding or support. The following grants and Center supported the work: DK064008, 062497 and 083781 (to JAB), DK092111 and the Fred and Suzanne Biesecker Pediatric Liver Center (to RW), DK062453 (to CM), DK062470 (to SK), DK062453 (to RS)
Financial Disclosure
Dr. Bezerra has research funding from Gilead. Dr. Wells: none to report. Dr. Mack: none to report. Dr. Karpen: is a consultant/advisor to Albireo and Intercept. Dr. Sokol has research funding from Shire and is a consultant/advisor for Shire, Alexion, Albireo, and Retrophin.
List of Abbreviations
- BA
biliary atresia
- HPE
hepatic portoenterostomy
- CMV
cytomegalovirus
- BASM
biliary atresia-splenic malformation
- EHBD
extrahepatic bile duct
- IHBD
intrahepatic bile duct
- GWAS
genome wide association study
- GSH
reduced glutathione
- Tregs
regulatory T cells
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
Footnotes
Conflict of Interest
Drs. Hoofnagle and Doo are employees of the Federal Government and have no conflicts of interest to report.
Contributor Information
Jorge A. Bezerra, Email: jorge.bezerra@cchmc.org, Liver Care Center of Cincinnati Children’s Hospital Medical Center and the Department of Pediatrics of the University of Cincinnati College of Medicine, Cincinnati, OH, USA.
Rebecca G. Wells, Email: rgwells@mail.med.upenn.edu, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
Cara L. Mack, Email: cara.Mack@childrenscolorado.org, Pediatric Liver Center, Children’s Hospital Colorado and Department of Pediatrics, University of Colorado School of Medicine, Aurora, CO, USA.
Saul J. Karpen, Email: saul.karpen@emory.edu, Emory University School of Medicine and Children’s Healthcare of Atlanta, GA, USA.
Jay Hoofnagle, Email: hoofnaglej@extra.niddk.nih.gov, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, USA.
Edward Doo, Email: dooe@niddk.nih.gov, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, USA.
Ronald J. Sokol, Email: ronald.Sokol@childrenscolorado.org, Pediatric Liver Center, Children’s Hospital Colorado and Department of Pediatrics, University of Colorado School of Medicine, Aurora, CO, USA.
References
- 1.Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374:1704–1713. doi: 10.1016/S0140-6736(09)60946-6. [DOI] [PubMed] [Google Scholar]
- 2.Sundaram SS, Mack CL, Feldman AG, Sokol RJ. Biliary atresia: Indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl. 2017;23:96–109. doi: 10.1002/lt.24640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serinet MO, Wildhaber BE, Broue P, Lachaux A, Sarles J, Jacquemin E, Gauthier F, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123:1280–1286. doi: 10.1542/peds.2008-1949. [DOI] [PubMed] [Google Scholar]
- 4.Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol. 2015;12:342–352. doi: 10.1038/nrgastro.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz KB, Haber BH, Rosenthal P, Mack CL, Moore J, Bove K, Bezerra JA, et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology. 2013;58:1724–1731. doi: 10.1002/hep.26512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durkin N, Deheragoda M, Davenport M. Prematurity and biliary atresia: a 30-year observational study. Pediatr Surg Int. 2017;33:1355–1361. doi: 10.1007/s00383-017-4193-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Wessel DBE, Boere T, Hulzebos CV, de Kleine RHJ, Verkade HJ, Hulscher JBF, on behalf of the Netherlands Study group of Biliary Atresia R Preterm Infants With Biliary Atresia: A Nationwide Cohort Analysis From The Netherlands. J Pediatr Gastroenterol Nutr. 2017;65:370–374. doi: 10.1097/MPG.0000000000001692. [DOI] [PubMed] [Google Scholar]
- 8.Shen O, Sela HY, Nagar H, Rabinowitz R, Jacobovich E, Chen D, Granot E. Prenatal diagnosis of biliary atresia: A case series. Early Hum Dev. 2017;111:16–19. doi: 10.1016/j.earlhumdev.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics. 2011;128:e1428–1433. doi: 10.1542/peds.2011-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harpavat S, Garcia-Prats JA, Shneider BL. Newborn Bilirubin Screening for Biliary Atresia. N Engl J Med. 2016;375:605–606. doi: 10.1056/NEJMc1601230. [DOI] [PubMed] [Google Scholar]
- 11.Schreiber RA, Masucci L, Kaczorowski J, Collet JP, Lutley P, Espinosa V, Bryan S. Home-based screening for biliary atresia using infant stool colour cards: a large-scale prospective cohort study and cost-effectiveness analysis. J Med Screen. 2014;21:126–132. doi: 10.1177/0969141314542115. [DOI] [PubMed] [Google Scholar]
- 12.Matsui A. Screening for biliary atresia. Pediatr Surg Int. 2017;33:1305–1313. doi: 10.1007/s00383-017-4175-3. [DOI] [PubMed] [Google Scholar]
- 13.Lee M, Chen SC, Yang HY, Huang JH, Yeung CY, Lee HC. Infant Stool Color Card Screening Helps Reduce the Hospitalization Rate and Mortality of Biliary Atresia: A 14-Year Nationwide Cohort Study in Taiwan. Medicine (Baltimore) 2016;95:e3166. doi: 10.1097/MD.0000000000003166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Yu J, Zhang R, Yin Y, Ye J, Tan L, Xia H. The perinatal infection of cytomegalovirus is an important etiology for biliary atresia in China. Clin Pediatr (Phila) 2012;51:109–113. doi: 10.1177/0009922811406264. [DOI] [PubMed] [Google Scholar]
- 15.Brindley SM, Lanham AM, Karrer FM, Tucker RM, Fontenot AP, Mack CL. Cytomegalovirus-specific T-cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells. Hepatology. 2012;55:1130–1138. doi: 10.1002/hep.24807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zani A, Quaglia A, Hadzic N, Zuckerman M, Davenport M. Cytomegalovirus-associated biliary atresia: An aetiological and prognostic subgroup. J Pediatr Surg. 2015;50:1739–1745. doi: 10.1016/j.jpedsurg.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Lorent K, Gong W, Koo KA, Waisbourd-Zinman O, Karjoo S, Zhao X, Sealy I, et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci Transl Med. 2015;7:286ra267. doi: 10.1126/scitranslmed.aaa1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43:245–256. doi: 10.1016/j.biocel.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Gerard C, Tys J, Lemaigre FP. Gene regulatory networks in differentiation and direct reprogramming of hepatic cells. Semin Cell Dev Biol. 2017;66:43–50. doi: 10.1016/j.semcdb.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Uemura M, Ozawa A, Nagata T, Kurasawa K, Tsunekawa N, Nobuhisa I, Taga T, et al. Sox17 haploinsufficiency results in perinatal biliary atresia and hepatitis in C57BL/6 background mice. Development. 2013;140:639–648. doi: 10.1242/dev.086702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP, Beuers U. A biliary HCO3-umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. 2012;55:173–183. doi: 10.1002/hep.24691. [DOI] [PubMed] [Google Scholar]
- 22.Tsai EA, Grochowski CM, Falsey AM, Rajagopalan R, Wendel D, Devoto M, Krantz ID, et al. Heterozygous deletion of FOXA2 segregates with disease in a family with heterotaxy, panhypopituitarism, and biliary atresia. Hum Mutat. 2015;36:631–637. doi: 10.1002/humu.22786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui S, Leyva-Vega M, Tsai EA, EauClaire SF, Glessner JT, Hakonarson H, Devoto M, et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology. 2013;144:1107–1115 e1103. doi: 10.1053/j.gastro.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai EA, Grochowski CM, Loomes KM, Bessho K, Hakonarson H, Bezerra JA, Russo PA, et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum Genet. 2014;133:235–243. doi: 10.1007/s00439-013-1368-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waisbourd-Zinman O, Koh H, Tsai S, Lavrut PM, Dang C, Zhao X, Pack M, et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology. 2016;64:880–893. doi: 10.1002/hep.28599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao X, Lorent K, Wilkins BJ, Marchione DM, Gillespie K, Waisbourd-Zinman O, So J, et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology. 2016;64:894–907. doi: 10.1002/hep.28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mack CL. What Causes Biliary Atresia? Unique Aspects of the Neonatal Immune System Provide Clues to Disease Pathogenesis. Cell Mol Gastroenterol Hepatol. 2015;1:267–274. doi: 10.1016/j.jcmgh.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lages CS, Simmons J, Chougnet CA, Miethke AG. Regulatory T cells control the CD8 adaptive immune response at the time of ductal obstruction in experimental biliary atresia. Hepatology. 2012;56:219–227. doi: 10.1002/hep.25662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233–242. doi: 10.1055/s-2007-985068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mack CL, Falta MT, Sullivan AK, Karrer F, Sokol RJ, Freed BM, Fontenot AP. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133:278–287. doi: 10.1053/j.gastro.2007.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lages CS, Simmons J, Maddox A, Jones K, Karns R, Sheridan R, Shanmukhappa SK, et al. The dendritic cell-T helper 17-macrophage axis controls cholangiocyte injury and disease progression in murine and human biliary atresia. Hepatology. 2017;65:174–188. doi: 10.1002/hep.28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen J, Zhou Y, Wang J, Chen J, Yan W, Wu J, Yan J, et al. Interactions between Th1 cells and Tregs affect regulation of hepatic fibrosis in biliary atresia through the IFN-gamma/STAT1 pathway. Cell Death Differ. 2017;24:997–1006. doi: 10.1038/cdd.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Kerola A, Lampela H, Lohi J, Heikkila P, Mutanen A, Jalanko H, Pakarinen MP. Molecular signature of active fibrogenesis prevails in biliary atresia after successful portoenterostomy. Surgery. 2017;162:548–556. doi: 10.1016/j.surg.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 34.Jung HY, Jing J, Lee KB, Jang JJ. Sonic hedgehog (SHH) and glioblastoma-2 (Gli-2) expressions are associated with poor jaundice-free survival in biliary atresia. J Pediatr Surg. 2015;50:371–376. doi: 10.1016/j.jpedsurg.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Omenetti A, Bass LM, Anders RA, Clemente MG, Francis H, Guy CD, McCall S, et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology. 2011;53:1246–1258. doi: 10.1002/hep.24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerola A, Lampela H, Lohi J, Heikkila P, Mutanen A, Hagstrom J, Tervahartiala T, et al. Increased MMP-7 expression in biliary epithelium and serum underpins native liver fibrosis after successful portoenterostomy in biliary atresia. J Pathol Clin Res. 2016;2:187–198. doi: 10.1002/cjp2.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bezerra JA, Spino C, Magee JC, Shneider BL, Rosenthal P, Wang KS, Erlichman J, et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA. 2014;311:1750–1759. doi: 10.1001/jama.2014.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tyraskis A, Davenport M. Steroids after the Kasai procedure for biliary atresia: the effect of age at Kasai portoenterostomy. Pediatr Surg Int. 2016;32:193–200. doi: 10.1007/s00383-015-3836-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Vries W, de Langen ZJ, Groen H, Scheenstra R, Peeters PM, Hulscher JB, Verkade HJ, et al. Biliary atresia in the Netherlands: outcome of patients diagnosed between 1987 and 2008. J Pediatr. 2012;160:638–644 e632. doi: 10.1016/j.jpeds.2011.09.061. [DOI] [PubMed] [Google Scholar]
- 40.Shneider BL, Magee JC, Karpen SJ, Rand EB, Narkewicz MR, Bass LM, Schwarz K, et al. Total Serum Bilirubin within 3 Months of Hepatoportoenterostomy Predicts Short-Term Outcomes in Biliary Atresia. J Pediatr. 2016;170:211–217. e211–212. doi: 10.1016/j.jpeds.2015.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chardot C, Buet C, Serinet MO, Golmard JL, Lachaux A, Roquelaure B, Gottrand F, et al. Improving outcomes of biliary atresia: French national series 1986-2009. J Hepatol. 2013;58:1209–1217. doi: 10.1016/j.jhep.2013.01.040. [DOI] [PubMed] [Google Scholar]
- 42.Ng VL, Haber BH, Magee JC, Miethke A, Murray KF, Michail S, Karpen SJ, et al. Medical status of 219 children with biliary atresia surviving long-term with their native livers: results from a North American multicenter consortium. J Pediatr. 2014;165:539–546 e532. doi: 10.1016/j.jpeds.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verkade HJ, Bezerra JA, Davenport M, Schreiber RA, Mieli-Vergani G, Hulscher JB, Sokol RJ, et al. Biliary atresia and other cholestatic childhood diseases: Advances and future challenges. J Hepatol. 2016;65:631–642. doi: 10.1016/j.jhep.2016.04.032. [DOI] [PubMed] [Google Scholar]
- 44.Bessho K, Mourya R, Shivakumar P, Walters S, Magee JC, Rao M, Jegga AG, et al. Gene expression signature for biliary atresia and a role for interleukin-8 in pathogenesis of experimental disease. Hepatology. 2014;60:211–223. doi: 10.1002/hep.27045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology. 2017;65:350–362. doi: 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, Schramm C, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67:549–558. doi: 10.1016/j.jhep.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, Drenth JP, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med. 2016;375:631–643. doi: 10.1056/NEJMoa1509840. [DOI] [PubMed] [Google Scholar]
- 48.Baghdasaryan A, Fuchs CD, Osterreicher CH, Lemberger UJ, Halilbasic E, Pahlman I, Graffner H, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64:674–681. doi: 10.1016/j.jhep.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 49.Miethke AG, Zhang W, Simmons J, Taylor AE, Shi T, Shanmukhappa SK, Karns R, et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. 2016;63:512–523. doi: 10.1002/hep.27973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slijepcevic D, Roscam Abbing RLP, Katafuchi T, Blank A, Donkers JM, van Hoppe S, de Waart DR, et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology. 2017;66:1631–1643. doi: 10.1002/hep.29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davenport M. Adjuvant therapy in biliary atresia: hopelessly optimistic or potential for change? Pediatr Surg Int. 2017 doi: 10.1007/s00383-017-4157-5. [DOI] [PubMed] [Google Scholar]
- 52.Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, Francque S, et al. A Randomized, Placebo-Controlled Trial of Cenicriviroc for Treatment of Nonalcoholic Steatohepatitis with Fibrosis. Hepatology. 2017 doi: 10.1002/hep.29477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sampaziotis F, de Brito MC, Geti I, Bertero A, Hannan NR, Vallier L. Directed differentiation of human induced pluripotent stem cells into functional cholangiocyte-like cells. Nat Protoc. 2017;12:814–827. doi: 10.1038/nprot.2017.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asai A, Aihara E, Watson C, Mourya R, Mizuochi T, Shivakumar P, Phelan K, et al. Paracrine signals regulate human liver organoid maturation from induced pluripotent stem cells. Development. 2017;144:1056–1064. doi: 10.1242/dev.142794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–484. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 56.Willenbring H, Soto-Gutierrez A. Transplantable liver organoids made from only three ingredients. Cell Stem Cell. 2013;13:139–140. doi: 10.1016/j.stem.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu BR, Brindley SM, Tucker RM, Lambert CL, Mack CL. alpha-enolase autoantibodies cross-reactive to viral proteins in a mouse model of biliary atresia. Gastroenterology. 2010;139:1753–1761. doi: 10.1053/j.gastro.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mack CL, Tucker RM, Lu BR, Sokol RJ, Fontenot AP, Ueno Y, Gill RG. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44:1231–1239. doi: 10.1002/hep.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, Caldwell C, et al. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133:268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mack CL, Tucker RM, Sokol RJ, Karrer FM, Kotzin BL, Whitington PF, Miller SD. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr Res. 2004;56:79–87. doi: 10.1203/01.PDR.0000130480.51066.FB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohya T, Fujimoto T, Shimomura H, Miyano T. Degeneration of intrahepatic bile duct with lymphocyte infiltration into biliary epithelial cells in biliary atresia. Journal of Pediatric Surgery. 1995;30:515–518. doi: 10.1016/0022-3468(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 62.Feng J, Li M, Gu W, Tang H, Yu S. The aberrant expression of HLA-DR in intrahepatic bile ducts in patients with biliary atresia: an immunohistochemistry and immune electron microscopy study. J Pediatr Surg. 2004;39:1658–1662. doi: 10.1016/j.jpedsurg.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 63.Mack CL, Anderson KM, Aubrey MT, Rosenthal P, Sokol RJ, Freed BM. Lack of HLA predominance and HLA shared epitopes in biliary Atresia. Springerplus. 2013;2:42. doi: 10.1186/2193-1801-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuasa T, Tsuji H, Kimura S, Niwa N, Yurugi K, Egawa H, Tanaka K, et al. Human leukocyte antigens in Japanese patients with biliary atresia: retrospective analysis of patients who underwent living donor liver transplantation. Hum Immunol. 2005;66:295–300. doi: 10.1016/j.humimm.2004.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.