

Abstract
Chlamydia trachomatis is an obligate intracellular pathogen with a reduced genome reflecting its host cell dependent life style. However, C. trachomatis has retained all of the genes required for fatty acid and phospholipid synthesis that are present in free-living bacteria. C. trachomatis assembles its cellular membrane using its own biosynthetic machinery utilizing glucose, isoleucine, and serine. This pathway produces disaturated phospholipid molecular species containing a branched-chain 15-carbon fatty acid in the 2-position, which are distinct from the structures of host phospholipids. The enoyl reductase step (FabI) is a target for antimicrobial drug discovery, and the developmental candidate, AFN-1252, blocks the activity of CtFabI. The x-ray crystal structure of the CtFabI•NADH•AFN-1252 ternary complex reveals the interactions between the drug, protein, and cofactor. AFN-1252 treatment of C. trachomatis-infected HeLa cells at any point in the infection cycle reduces infectious titers, and treatment at the time of infection prevents the first cell division. Fatty acid synthesis is essential for C. trachomatis proliferation within its eukaryotic host, and CtFabI is a validated therapeutic target against C. trachomatis.
Keywords: Chlamydia, fatty acid synthesis, phospholipid, enoyl-ACP reductase, antibiotic target
Introduction
Chlamydia trachomatis is an obligate intracellular bacterial parasite that causes a range of human diseases including trachoma and sexually transmitted infections (Belland et al., 2004). In the pre-genomic age, Chlamydia was thought to be an “energy parasite” because they imported ATP from the host cell (Hatch et al., 1982; Iliffe-Lee and McClarty, 1999), and it was unclear which other major metabolic pathways C. trachomatis encoded endogenously and which pathways C. trachomatis depended on the host (Omsland et al., 2014). The publication of the first C. trachomatis genome was reported in 1998, and showed an organism with a reduced genome (ca. 1 million bp) that encoded the major metabolic pathways (glycolysis, DNA synthesis, RNA synthesis, protein synthesis, and phospholipid synthesis) found in free-living bacteria, but lacked many genes necessary to synthesize the precursors to these pathways (Stephens et al., 1998). Instead, C. trachomatis encoded a variety of pumps to acquire these precursors such as amino acids, nucleotides, and cofactors from the host (Braun et al., 2008; Bastidas et al., 2013). The genome sequence of C. trachomatis paints the picture of a free-living bacteria that adapted to intracellular growth by stealing metabolic precursors instead of synthesizing them endogenously, but retained the major metabolic pathways.
Chlamydia trachomatis encodes all the genes necessary to endogenously synthesize phospholipid species typically found in free-living bacteria from acetate, glycerol-3-phosphate, isoleucine, and serine (Yao et al., 2015a) (Figures 1, 2). However, early research into C. trachomatis phospholipid synthesis suggested that C. trachomatis synthesized phospholipids by modifying host phospholipids (Wylie et al., 1997; Su et al., 2004). Because fatty acid and phospholipid synthesis are targets for antibiotic development, extensive efforts were undertaken to characterize the dependence of C. trachomatis on de novo phospholipid synthesis and host fatty acid incorporation (Yao et al., 2014, 2015a,b). In this review, we summarize the experimental evidence that C. trachomatis produces the bulk of their membrane phospholipid via their encoded fatty acid and phospholipid biosynthetic pathways. Although C. trachomatis is capable of activating host fatty acids using an acyl-acyl carrier protein (ACP) synthetase, FASII is essential for Chlamydia proliferation, and we evaluate the potential for targeting this pathway with contemporary FASII therapeutics.
FIGURE 1.

Schematic comparison of phospholipid synthesis in Escherichia coli (free living bacteria) and Chlamydia trachomatis (obligate intracellular bacteria). Modified from Yao and Rock (2017b). (A) Phospholipid synthesis pathway in E. coli. (B) Phospholipid synthesis in C. trachomatis. C. trachomatis encodes all the genes necessary for type II fatty acid (FASII) and phospholipid synthesis from acetate, glycerol-3-phosphate, serine, and isoleucine typically found in free-living bacteria. Fatty acids are made in the type II fatty acid synthesis system (FASII). Two acyltransferases (PlsE and PlsC in C. trachomatis vs. PlsB and PlsC in E. coli) make phosphatidic acid (PA). Phosphatidic acid is the precursor to the various phospholipid species, and C. trachomatis encode for the genes to synthesize phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL) like E. coli. Exogenous fatty acids are converted into acyl-CoA in E. coli. Acyl-CoA cannot be elongated by FASII, and can only be used by the acyltransferases or broken down by β-oxidation. In contrast, exogenous fatty acids are converted into acyl-ACP in C. trachomatis, which can be elongated by the FASII or used by the acyltransferases. C. trachomatis is not predicted to encode for β-oxidation genes.
Fatty Acid and Phospholipid Synthesis in C. trachomatis
Chlamydia trachomatis has a dimorphic life cycle (Abdelrahman and Belland, 2005). The infectious elementary body is responsible for the spread of the infection by attaching and invading susceptible cells. The elementary body is internalized in membrane bound vacuoles, called chlamydial inclusions, and differentiates into the metabolically active reticulate body. The reticulate body undergoes logarithmic growth within the chlamydial inclusions, which expands as the number of reticulate bodies increase. Reticulate bodies directly associate and interact with the chlamydial inclusion using the inclusion membrane proteins (Mirrashidi et al., 2015; Weber et al., 2017). Reticulate bodies differentiate back into elementary bodies, and the lysis of the infected host cell releases the elementary bodies into the environment for further spread and infections.
A major challenge in understanding C. trachomatis phospholipid synthesis is deciphering the contribution of bacterial phospholipid synthesis vs. host phospholipid acquisition in the replication and the life cycle of C. trachomatis. Research has shown that both host and bacterial lipid synthesis is essential for the life cycle of C. trachomatis. C. trachomatis encodes for a fully functional fatty acid and phospholipid synthesis system that is analogous to free living bacteria such as Escherichia coli (Figure 1), and this review will start with the endogenous synthesis of fatty acids and phospholipids.
Type II Fatty Acid Synthesis in C. trachomatis
The genome of C. trachomatis is predicted to encode for a complete type II bacterial fatty acid synthesis (FASII) system capable of producing branched-chain fatty acids (Figure 2, Initiation and Elongation) (Bidawid et al., 1989; Stephens et al., 1998; Yao et al., 2014). C. trachomatis produces branched-chain fatty acids like Staphylococcus aureus (Parsons et al., 2011). Isoleucine is converted to 2-methylbutyryl-CoA by the branched-chain α-ketoacid dehydrogenase complex (Bkd). 3-Ketoacyl-ACP synthase III (FabH) is predicted to prime fatty acid synthesis using 2-methylbutyryl-CoA, as well as acetyl-CoA. Malonyl-CoA is generated from acetyl-CoA by the acetyl-CoA carboxylase complex (AccABCD), and converted into malonyl-ACP by malonyl-CoA:ACP transacylase (FabD). Malonyl-ACP is used for the two-carbon elongation of the growing fatty acid by FabH to initiate FASII, and by 3-ketoacyl-ACP synthase II (FabF) in subsequent elongation steps to make 3-ketoacyl-ACP. The 3-ketoacyl-ACP is reduced by 3-ketoacyl-ACP reductase (FabG) to 3-hydroxyacyl-ACP, which is then dehydrated by 3-hydroxyacyl-ACP dehydratase (FabZ) into trans-2-enoyl-ACP. C. trachomatis is a Gram-negative bacterium, and long-chain 3-hydroxyacyl-ACP is extracted from the FASII pathway for lipo-oligosaccharide synthesis (Rund et al., 1999; Heine et al., 2003, 2007). The trans-2-enoyl-ACP is reduced by enoyl-ACP reductase (FabI) in the last step of each elongation cycle to make acyl-ACP. All the enzymes in C. trachomatis FASII are highly similar (e-value < 1e−40) to their orthologs found in other free-living bacteria such as E. coli and S. aureus. The one exception is the predicted enoyl-ACP reductase FabI (CtFabI), which is more closely related to the FabI found in the plastids of plants and Apicomplexan parasites like malaria (Yao et al., 2014).
FIGURE 2.

Detailed diagram for fatty acid and phospholipid synthesis in C. trachomatis. From Yao et al. (Yao et al., 2015a). This figure shows the protein name designations along with the C. trachomatis gene locus number for each reaction. The biosynthetic pathway is divided into four modules. The initiation module consists of the enzymes that supply the precursors to initiate FASII. The elongation module sequentially elongates the acyl chain. The acyltransfer module extracts acyl-ACP from the elongation cycle to acylate glycerol-phosphate. The phospholipid module diversifies the headgroups to produce PE, PG, and CL. Despite a few minor quirks such as the plastidial related enoyl-ACP reductase (FabI) and 1-acyl-sn-glycerol-3-phosphate acyltransferase (PlsE), fatty acid and phospholipid synthesis in C. trachomatis is the same as other free-living bacteria such as E. coli and Staphylococcus aureus.
Initiation of Phospholipid Assembly by C. trachomatis
Bacterial phospholipid synthesis is initiated by the extraction of acyl-ACP of the appropriate length from the elongation cycle by two acyltransferases to synthesize phosphatidic acid, the key precursor to all phospholipids (Stephens et al., 1998; Yao et al., 2015a). The genome of C. trachomatis encodes two acyltransferases responsible for the successive acylation of sn-glycerol-3-phosphate (G3P) (Figure 2, Acyltransfer). The G3P acyltransferase in C. trachomatis (PlsE) is more related to the soluble G3P acyltranferase found in plant plastids (Turnbull et al., 2001) than the characterized bacterial membrane-bound G3P acyltransferases, PlsY and PlsB (Lightner et al., 1983; Lu et al., 2006; Yao and Rock, 2013). PlsE is established as a bone fide G3P acyltransferase by the biochemical characterization of purified PlsE. These experiments show that PlsE utilizes acyl-ACP as the acyl donor (Yao et al., 2015a).
The second step in the pathway is catalyzed by PlsC, 1-acyl-G3P acyltransferase (Figure 2). CtPlsC is homologous to the well-characterized bacterial PlsCs, and complements E. coli plsC(Ts) mutants (Yao et al., 2015a). These enzymes are known to function as molecular rulers that select a specific acyl-ACP chain length for incorporation into the 2-position (Robertson et al., 2017). CtPlsC exhibits this same property, and has a substrate selectivity like SaPlsC in preferring 15:0-ACP. In complemented plsC(Ts) E. coli strains, CtPlsC inserts 14:0 into the 2-position (like SaPlsC), consistent with C. trachomatis phospholipids possessing 15:0 almost exclusively in the 2-position (Yao et al., 2015a). The presence of 15:0 provides a distinct molecular signature for all phospholipid molecular species produced by the de novo C. trachomatis biosynthetic pathway.
Diversification of Phospholipid Headgroups
Chlamydia trachomatis is predicted to encode for the pathways necessary to synthesize phosphatidylglycerol (PG), cardiolipin (CL), and phosphatidylethanolamine (PE) from phosphatidic acid (Figure 2, Phospholipid) (Stephens et al., 1998; Yao et al., 2015a). These genes and phospholipid species are widely-distributed in free-living Gram-negative bacteria such as E. coli (Parsons and Rock, 2013). Phosphatidic acid is converted by CDP-diacylglyerol synthase (CdsA) into CDP-diacylglycerol. CDP-diacylglycerol is converted into phosphatidylserine by phosphatidylserine synthase (PssA), and then decarboxylated by phosphatidylserine decarboxylase (Psd) to make PE, the major phospholipid species in C. trachomatis. CDP-diacylglycerol is also converted into phosphatidylglycerol phosphate by phosphatidylglycerophosphate synthase (PgsA). The terminal phosphate of phosphatidylglycerolphosphate is removed by phosphatidylglycerophosphatase (Pgp) to make PG. Two PGs are ligated together by CL synthase to make CL.
Phospholipid compositional analysis of C. trachomatis infected HeLa cells showed significant increases in PE, PG, and CL composition compared to uninfected cells (Yao et al., 2015a). All the phospholipid molecular species of the new PE, PG, and CL produced in Chlamydia-infected cells contain 15:0, meaning that they were derived from phosphatidic acid produced by PlsE/PlsC (Yao et al., 2015a). C. trachomatis extracted and purified with the nonionic, non-denaturing detergent Nonidet P-40 was composed of ca. 85% PE/PG/cardiolipin containing branched-chain fatty acids and 15% host PC/sphingomyelin (Yao et al., 2015a). The PE molecular species contain 15:0, while the host PC molecular species did not (Yao et al., 2015a). The results of these experiments show that the bulk of C. trachomatis membrane is composed of chlamydial synthesized phospholipids containing chlamydial synthesized branched-chain fatty acids. The potential importance/function of the associated host lipids is discussed later in this review.
C. trachomatis Does Not Modify Host Phospholipids
Several labs have concluded that host phospholipids such as PE and PC are deacylated and then reacylated with C. trachomatis derived branched-chain fatty acids (Wylie et al., 1997; Su et al., 2004; Soupene et al., 2015; Recuero-Checa et al., 2016; Soupene and Kuypers, 2017). However, there is no evidence that this inferred lipid modification pathway exists in vivo because the phospholipid molecular species analysis of C. trachomatis infected HeLa cells do not detect any of these putative products (Yao et al., 2015a). Branched-chain fatty acids are detected only in the phospholipid species predicted to be synthesized by C. trachomatis, and are not found in phospholipid species synthesized exclusively by the host, such as PC (Yao et al., 2015a). The only potential host phospholipid that may be modified by 15:0 reacylation is PE. Isotopically labeled ethanolamine is incorporated only into host PE species in C. trachomatis-infected HeLa cells because the C. trachomatis biosynthetic pathway produces PE from serine (via PS), not ethanolamine (Dowhan, 1997). The pool of PE labeled by ethanolamine in the C. trachomatis-infected HeLa cells did not contain 15:0, and had the same acyl-chain composition as the PE in uninfected host cells (Yao et al., 2015a). The idea that the host PS is used for PE synthesis (Soupene and Kuypers, 2017) is ruled out because host-derived PE molecular species are not found in purified C. trachomatis (Yao et al., 2015a). Phospholipid molecular species analysis clearly demonstrates that host PE is not deacylated/reacylated with branched-chain fatty acids.
C. trachomatis 2-Acyl-GPE Acyltransferase/Acyl-ACP Synthetase System
Fatty acid synthesis is an energy and material intensive process, and bacteria encode the ability to incorporate exogenous fatty acids into their phospholipids (Yao and Rock, 2015, 2017b). Two mechanisms for incorporating exogenous fatty acids have been characterized in Gram-negative bacteria (Figure 1). First, exogenous fatty acids are converted into acyl-ACP by an acyl-ACP synthetase. The resulting acyl-ACP can be used by the acyltransferases for phosphatidic acid synthesis or further elongated by FASII (Jiang et al., 2006, 2010; Yao et al., 2016a). Second, exogenous fatty acids are converted into acyl-CoA by an acyl-CoA synthetase (Black et al., 1992; Yao et al., 2016a). The resulting acyl-CoA cannot be elongated by FASII, but can be used for phosphatidic acid synthesis (if the bacteria encode an acyltransferase capable of using acyl-CoA) or broken down via β-oxidation (if the bacteria encode for a β-oxidation pathway) (Yao and Rock, 2013). C. trachomatis lacks a β-oxidation pathway and uses acyl-ACP as the acyl donor for its acyltransferases. A third mechanism is fatty acid kinase that exists in Gram-positive bacteria that have a PlsY acyltransferase (Lu et al., 2006; Parsons et al., 2014), and is not present in C. trachomatis.
Chlamydia trachomatis is predicted to encode for the two domains of the E. coli acyl-ACP synthetase/2-acylglycerolphosphoethanolamine (GPE) acyltransferase bifunctional protein as two consecutive, but separate genes (Figure 3). In E. coli, this bifunctional gene is designated aas, and functions to reacylate 2-acyl-GPE formed by Lnt (apo-lipoprotein:phospholipid transacylase) during lipoprotein biogenesis with an acyl-ACP (Jackowski and Rock, 1986). The lipoprotein synthesis pathway is widely distributed in Gram-negative bacteria, and C. trachomatis lipoproteins have the same lipid modifications as observed in E. coli (Everett et al., 1994; Neff et al., 2007). The genes encoding the lipid modification pathway are present (lgt, CT252; lsp, CT408; lnt, CT534). Thus, it is reasonable to conclude that the split acyl-ACP synthetase (AasC) and 2-acyl-GPE acyltransferase (LpaT) system of C. trachomatis also supports phospholipid re-cycling during lipoprotein maturation.
FIGURE 3.
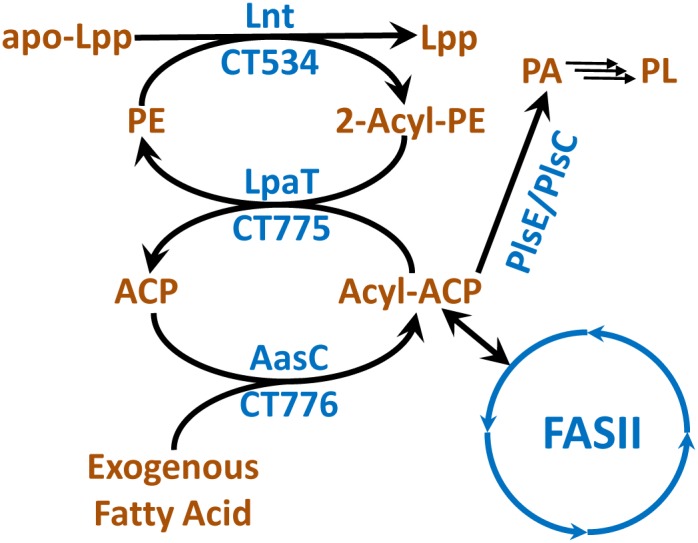
Fatty acid activation and lysophospholipid recycling in C. trachomatis. The amino-terminus of bacterial lipoproteins (apo-Lpp) are acylated by the transfer of fatty acids from the 1-position of PE to generate 2-acyl-glycerophosphoethanolamine (2-acyl-GPE) and acylated lipoproteins (Lpp). Other phospholipid species can also be used in the reaction. The 2-acyl-GPE is reacylated by LpaT using acyl-ACP. Acyl-ACP is derived from FASII or by the activation of exogenous fatty acids by AasC.
There are differences between the E. coli and C. trachomatis systems that expand the function of this system in Chlamydia. In E. coli, the carboxy-terminal acyl-ACP synthetase activity is physically linked to the amino-terminal 2-acyl-GPE acyltransferase activity (Figure 1). Acyl-ACP generated by the E. coli acyl-ACP synthetase domain can only be used by the amino-terminal domain 2-acyl-GPE acyltransferase, and cannot enter FASII or be used by the other cellular acyltransferases (Rock and Cronan, 1979; Cooper et al., 1989; Hsu et al., 1991). In C. trachomatis, the two domains are present as two separate genes (Figure 3). This alternate genetic structure separates C. trachomatis acyl-ACP synthetase (AasC) from the acyltransferase (LpaT) suggesting that AasC may release acyl-ACP that could be used by FASII and all cellular acyltransferases, instead of being metabolically channeled exclusively to LpaT. Biochemical characterization of purified AasC shows that it converts fatty acid only to acyl-ACP (not acyl-CoA) with the same preference for 1-position fatty acids as the E. coli system (Yao et al., 2015b). The heterologous expression of AasC enables the incorporation of exogenous fatty acids into the phospholipids in an E. coli strain deficient in exogenous fatty acid activation showing that AasC generates acyl-ACPs from exogenous fatty acids that is available to cellular acyltransferases (Yao et al., 2015b). LpaT is biochemically characterized as a 2-acyl-lysophosholipid acyltransferase, and CtLpaT also complements E. coli mutants lacking the 2-acyl-GPE acyltransferase (Yao et al., 2015b). Lysophospholipids rapidly undergo acyl chain migration, and these substrates are always present as a mixture of 1-acyl and 2-acyl lysophospholipids (Pluckthun and Dennis, 1982). Thus, one must analyze the positional distribution of acyl chains in the phospholipid product to be certain about the structure of the lysophospholipid substrate used by the enzyme.
The analysis of C. trachomatis-infected HeLa cells showed that about 8% of the chlamydial PE correspond to a PE molecular species containing one 15:0 and one 18:1 unsaturated fatty acid (Yao et al., 2015a). Polyunsaturated fatty acids that are abundant in mammals are not detected in C. trachomatis phospholipids. Because C. trachomatis does not encode for the ability to synthesize unsaturated fatty acid (Stephens et al., 1998), this result suggests that AasC activates host fatty acids in the cell infection model. This hypothesis is tested by labeling C. trachomatis infected HeLa cells with isotopically labeled, medium chain fatty acids (12:0 and 14:0), and observing whether these fatty acids are elongated and incorporated into the C. trachomatis phospholipids (Yao et al., 2015b). C. trachomatis could elongate and incorporate these exogenous, isotopically labeled fatty acids into its phospholipid species, showing that the C. trachomatis AasC is activating exogenous fatty acids during the infection cycle that were utilized by FASII or acyltransferases. Polyunsaturated fatty acids are abundant in the host cell, but are not found in C. trachomatis phospholipids consistent with the observed substrate selectivity for saturated fatty acids, and some reactivity with oleate by the AasC.
Host Phospholipids in the Chlamydial Life Cycle
Host Lipid Trafficking Is Essential for the Growth and Maintenance of the Chlamydial Inclusion
The chlamydial inclusion grows significantly in size during the course of the infection, from holding the single internalized EB at the beginning of the infection cycle to taking up the majority of the internal volume of the host cell at the end of the infection cycle (Elwell and Engel, 2012). The significant increase in the surface area of this chlamydial inclusion membrane during the chlamydial cell cycle necessitates some way to grow this membrane. It has not been possible to define the membrane composition of the inclusion directly because it is challenging to isolate this compartment and to separate the associated C. trachomatis from the membrane. Immunofluorescence studies demonstrated the presence of host lipid species in inclusion membrane (Hackstadt et al., 1995; Carabeo et al., 2003; Beatty, 2008; Elwell et al., 2011), and there is emerging evidence that host and chlamydial proteins are found in the inclusion membrane (Beatty, 2008; Mital et al., 2013; Weber et al., 2017). The landmark paper by Hackstadt et al. (1995) demonstrates that trafficking of the host lipids is essential for the growth of the chlamydial inclusion. Fluorescently labeled sphingolipids synthesized by the host cell from C6-NBD-ceramide co-localized with the inclusion membrane and C. trachomatis, showing that sphingolipids and potentially other host lipids are trafficked into the inclusion. Adding Brefeldin A, an inhibitor of vesicular trafficking, stops this co-localization. The disruption of this trafficking by Brefeldin A led to smaller chlamydial inclusions and premature lysis of chlamydial inclusion (Elwell et al., 2011), demonstrating that the trafficking of host lipids is essential for the growth and maintenance of the chlamydial inclusion. Further research shows that several chlamydial inclusion membrane proteins are essential for the trafficking of host lipids, and the depletion of these proteins also leads to the premature lysis of the chlamydial inclusion (Weber et al., 2017). These experiments show that the chlamydial inclusion membrane proteins recruit host phospholipids via vesicular trafficking pathways to the chlamydial inclusion.
There are extensive investigations in literature demonstrating the transport of other host phospholipids, cholesterol, lipid droplets, high-density lipoproteins, and even host organelles into the chlamydial inclusion as well as the role these transport plays in the infection cycle (Cocchiaro et al., 2008; Elwell et al., 2011; Cox et al., 2012, 2016; Bastidas et al., 2013; Agaisse and Derre, 2014; Al-Zeer et al., 2014; Boncompain et al., 2014; Saka et al., 2015). Inhibition of these pathways usually lead to decreased C. trachomatis infectious titers. These trafficked lipids might be other components of the chlamydial inclusion membrane. C. trachomatis also actively incorporates host fatty acids for de novo phospholipid synthesis, and some of these pathways, such as the lipid droplet trafficking pathway, might deliver the host fatty acids through the chlamydial inclusion membrane. Given that C. trachomatis is auxotrophic for a variety of metabolic precursors, it is very clear that there must be many mechanisms to traffic these metabolic precursors into the chlamydial inclusion. Detailed discussion of host and pathogen interactions can be found in literature (Elwell and Engel, 2012; Bastidas et al., 2013) and is beyond the scope of this review.
Are Host Lipids an Essential Component of the C. trachomatis Membrane?
The fluorescently labeled lipid analogs co-localized with the replicating reticulate bodies (Hackstadt et al., 1995), suggesting that trafficked host lipids are potentially essential constituents of the C. trachomatis membrane. One challenge in interpreting experiments using fluorescently labeled lipids is recognizing that fluorescent groups such as NBD replacing the fatty acid in the fluorescently labeled lipid analogs are significantly bulkier and more hydrophilic. Therefore, these probes have increased solubility compared to the actual lipid, making fluorescently labeled lipid analogs capable of diffusing to places where the actual lipids would not. A variety of different analysis confirms that host phosphatidylcholine and sphingolipids are indeed co-purified at low abundance with C. trachomatis even under stringent purification (Yao et al., 2015a; Cox et al., 2016). Some authors have concluded that this proves that these lipids are essential structural components of the C. trachomatis membrane (Cox et al., 2016). However, these same host lipids are observed using the same purification procedures in the absence of C. trachomatis infection, raising genuine concern about whether the low amounts of host lipids in the purified C. trachomatis preparation are actually components of the bacterial membrane (Yao et al., 2015a).
Whether host phospholipids are essential constituents of the C. trachomatis membrane or just associated with C. trachomatis could be definitely settled by determining if C. trachomatis can replicate in absence of these host lipids. This experiment is impossible to execute in practice because C. trachomatis grows intracellularly with direct contact to the host derived chlamydial inclusion (Mirrashidi et al., 2015; Weber et al., 2017). The low titers observed through inhibiting the various host lipid trafficking pathways do not address whether host lipids are an essential component of the C. trachomatis membrane because the growth of the chlamydial inclusion is also required for the continued replication of the reticulate body (Elwell et al., 2011; Cox et al., 2016). Regardless, inhibition of host lipid tracking pathways is an effective method of disrupting the C. trachomatis life cycle and reducing the generation of infectious elementary bodies.
Inhibition of Fatty Acid Synthesis in C. trachomatis
Fatty acid and phospholipid synthesis is one of the four major conserved biosynthetic pathways in living organisms, and enzymes in the pathway have a long history of being investigated as novel antibiotic targets (Yao and Rock, 2016, 2017a). FabI and FabH were identified and validated as essential gene targets in bacteria by target-based discovery campaigns conducted by GlaxoSmithKline in the 1990s (Payne et al., 2007). FabI have received extensive research as an antibiotic target, with the FabI inhibitor AFN-1252 having recently passed phase 2 clinical trials as a narrow spectrum, S. aureus specific antibiotic (Kaplan et al., 2012, 2013; Flamm et al., 2015; Hunt et al., 2015; Hafkin et al., 2016).
FASII Is Essential in C. trachomatis
Because FabI is the rating-controlling step in fatty acid elongation (Heath and Rock, 1995), CtFabI was biochemically characterized and structurally elucidated. CtFabI catalyzed the reduction of trans-2-enoyl-ACP and complements an E. coli fabI(Ts) strain, validating its function as an enoyl-ACP reductase (Yao et al., 2014). The CtFabI was inhibited by triclosan and AFN-1252, two well-described FabI inhibitors, at low micromolar affinity (Yao et al., 2014). The CtFabI•NAD(H)•AFN-1252 ternary complex was elucidated using X-ray crystallography, and shows that the CtFabI forms a similar ternary complex with AFN-1252 as the previously characterized ternary complex structure with S. aureus FabI (Yao et al., 2014). AFN-1252 has cleared two Phase II clinical efficacy trials, and lacks off-target effects against human cells (Hunt et al., 2015; Hafkin et al., 2016). Therefore, the C. trachomatis-infected HeLa cell model was treated with AFN-1252 to understand how the inhibition of CtFabI and FASII effects the intracellular growth of C. trachomatis. AFN-1252 caused a dose dependent inhibition of the replication of C. trachomatis correlating with selective inhibition of C. trachomatis fatty acid synthesis, demonstrating that the inhibition of chlamydial FASII inhibits the growth and replication of C. trachomatis (Yao et al., 2014). AFN-1252 blocked the transition of C. trachomatis at a reticulate body like state when AFN-1252 was added at infection and prevented the generation of any infectious units (Yao et al., 2014). AFN-1252 also caused decreased infectious titers when added post infection over the entire replication cycle (Yao et al., 2014). These experiments show that FASII is required for the transition of elementary bodies into the metabolically active reticulate bodies as well as the replication of the reticulate bodies. These data also directly validate CtFabI as an anti-chlamydial drug discovery target, and suggest that targeting other enzymes in FASII would be effective as well.
FabI inhibition by AFN-1252 blocked the replication of C. trachomatis despite the abundance of exogenous host fatty acids in the C. trachomatis infected HeLa cell model and the ability to activate exogenous fatty acids with AasC (Yao et al., 2015b). This result appears counterintuitive at first glance, but it is common for FASII inhibitors to block cell growth in the presence of ample extracellular fatty acids (Parsons et al., 2011). The explanation for this finding is that blockage of the elongation cycle leads to the accumulation of the acyl-ACP intermediate used by the inhibited FASII step. This results in free ACP being sequestered as the short-chain thioester biosynthetic intermediate, making it unavailable for exogenous fatty acid activation. The depletion of free ACP prevents the incorporation of exogenous fatty acids by acyl-ACP synthetase, making it impossible to bypass FASII inhibition. This result is observed with many other bacterial species that also encode for exogenous fatty acid incorporation pathways, but cannot bypass FASII inhibition (Yao and Rock, 2015; Yao et al., 2016a).
There has been debate in literature about whether environmental fatty acids can enable the emergence of fatty acid auxotrophic versions of common pathogens like S. aureus (Morvan et al., 2016), how fit these auxotrophic bacteria are (Morvan et al., 2016), whether these auxotrophic bacteria are present in nature (Gloux et al., 2017), and what all this means for FASII targeting antibiotics (Morvan et al., 2017; Yao and Rock, 2017b). Loss of function in the fatty acid initiation genes leads to fatty acid auxotrophic S. aureus (Parsons et al., 2011, 2013). These mutants bypass FASII inhibition by utilizing exogenous fatty acids for phospholipid synthesis instead. These mutants occur at high frequency (1 × 10−6) from any mutation that causes loss of function in the initiation module (Parsons et al., 2011, 2013). High levels of exogenous fatty acids are required for the growth of these mutants, and their growth rate in liquid culture significantly lags their wild type counterpart (Parsons et al., 2011, 2013). The fatty acid auxotrophic mutants appear to have a significant growth defect in vivo as well. The ΔaccD mutant does not proliferate at all in the bacteremia model (Parsons et al., 2013). The fabD gene truncation mutant has decreased growth in the tail vein infection model compared to wild type (Morvan et al., 2016). Certain point mutations to FASII genes can enable S. aureus to become more resistant to FASII therapeutics, but without becoming fatty acid auxotrophic and not able to bypass FASII inhibition. These mutants have minimal growth defects (Morvan et al., 2016) as they are able to synthesize fatty acids endogenously. The issue of resistance in antibiotic discovery will be discussed in the next section. The environmental prevalence of these auxotrophic S. aureus mutants was estimated by screening S. aureus on media containing the FASII inhibitor triclosan and fatty acid supplementation (Gloux et al., 2017). Screening found that 7% of clinical and veterinary samples contained colonies that grew on the selective media and requires fatty acid supplementation. This result was interpreted as a wide prevalence of fatty acid auxotrophic S. aureus in the environment (Gloux et al., 2017). Whether this “prevalence” represents a persistent population of fatty acid auxotrophic S. aureus in the environment, or transient mutants that arise from the high frequency loss of function mutations is unclear. Regardless, the evidence is clear that fatty acid auxotrophic versions of S. aureus can arise frequently under selection pressure even if they are not in the environment already. Environment prevalence or fast emergence of S. aureus capable of bypassing FASII inhibition is predicted to cause significant failures to FASII therapeutics if these mutants don’t have significant fitness defects (Morvan et al., 2016). However, AFN-1252 was highly successful in clinical trials even in patients with significant comorbidities (Hafkin et al., 2016), consistent with the observations that fatty acid auxotrophic S. aureus has significant growth defects (Parsons et al., 2011, 2013). We have noted that the endogenous synthesis of branched-chain fatty acids appear essential for the proper growth and proliferation of bacterial species containing anteiso15:0 (Yao and Rock, 2015, 2017a,b).
Bypassing FASII inhibition through fatty acid auxotrophy is not predicted to be a viable mechanism in C. trachomatis, although more experiments are needed. First, C. trachomatis could not bypass FASII inhibition even when AFN-1252 is added 8 h post infection when there is a large population of growing C. trachomatis and host cell protein synthesis is inhibited (Yao et al., 2014). This result suggests that fatty acid auxotrophic mutants must have fitness defects given the high frequency that mutations causing fatty acid auxotrophy is predicted to occur. Second, C. trachomatis encodes for a fully functional fatty acid synthesis system (Stephens et al., 1998; Yao et al., 2014) despite being in an exogenous fatty acid rich environment and actively trafficking/incorporating host fatty acids over the growth phase (Yao et al., 2015b). The conservation of FASII suggests an essential role given that C. trachomatis has a reduced genome that has replaced biosynthetic capabilities with host trafficking in other essential pathways (Stephens et al., 1998). The essentiality of FASII may be related to branched-chains fatty acids or lipooligosaccharides (Yao and Rock, 2015).
FabI as a Pathogen Selective Antibacterial Target
The well-reasoned “multi-target” hypothesis proposes that broad-spectrum, monotherapeutic antibiotics in clinical use today target multiple gene targets (Silver, 2007, 2011; Yao and Rock, 2017a) because point mutations in the gene target allows the fast development of resistance against single-target antibiotics in bacteria. Because broad-spectrum antibiotics usually only have micromolar affinity as a compromise to achieve the broad-spectrum, a small increase in resistance is sufficient to render the antibiotic ineffective in the clinic. Multi-targeting overcomes this hurdle because the probability of developing resistance against several gene targets is multiplicative (i.e., 1 × 10−9 vs. one target, 1 × 10−18 vs. two targets, 1 × 10−27 for three targets, etc) (Yao and Rock, 2017a). Unfortunately, only three known sets of gene targets are structurally close enough to allow for efficient multi-targeting: penicillin binding proteins, ribosomes, and DNA gyrases (Silver, 2007).
The FabI inhibitor, AFN-1252, is currently under clinical trials as an example of a pathogen selectively antibiotic. Rather than compromising affinity to achieve a broad spectrum of activity, AFN-1252 is designed to have low nanomolar affinity against S. aureus FabI (Yao et al., 2013). Missense mutations to the S. aureus FabI can significantly increase resistance to AFN-1252, from 4 ng/ml against FabI(WT) to 250 ng/ml against FabI(M99T) and 500 ng/ml against FabI(Y147H) (Yao et al., 2013). However, additional rounds of selection did not produce mutants with higher levels of resistance. The FabI(M99T, Y147H) double mutant was nonviable. These results show that even the most resistant mutant found via the selection experiment is still susceptible to therapeutically achievable doses of AFN-1252 (Kaplan et al., 2013), suggesting that high affinity, pathogen selective inhibitors might prove to be therapeutically effective antibiotics. AFN-1252 is the only antibiotic following this paradigm in clinical trials, having recently passed phase II trials (Hafkin et al., 2016). Its development will validate or disprove the pathogen selective inhibitor idea.
A narrow spectrum is a feature of FabI inhibitors because many bacteria, such as Streptococcus pneumonia, encode for a different structural class of enoyl-ACP reductases called FabK that is resistant to FabI inhibitors (Heath and Rock, 2000; Marrakchi et al., 2003). Although pharmaceutical companies traditionally strived for broad spectrum antibiotics due to economic incentives, narrow spectrum antibiotics have gained increasing interest (Maxson and Mitchell, 2016; Spaulding et al., 2017). Recent advances in microbiome research demonstrates that broad spectrum antibiotic treatment decimates the commensal microbiota, and is linked to adverse effects such as increased secondary infections in the short term (Bohnhoff and Miller, 1962; Buffie et al., 2012) and increased chances of metabolic diseases in the long term (Blaser and Falkow, 2009; Cho et al., 2012; Modi et al., 2014; Boursi et al., 2015). The normal treatment for C. trachomatis infection is high-dosage Azithromycin, which is known to significantly alter the microbiome (Million et al., 2013; Parker et al., 2017). The narrow spectrum FabI inhibitor, AFN-1252, has been shown to minimize collateral damage to the microbiome, and would decrease these potential negative side effects (Yao et al., 2016b). A chlamydial selective FabI inhibitor is particularly alluring for novel anti-chlamydial therapeutics, because FabI is a validated target and minimizing collateral damage to the microbiome would be an improvement over existing therapeutics. Furthermore, AFN-1252 had reasonable affinity against the CtFabI in the cellular model, suggesting that AFN-1252 is a valid starting structure for additional rounds of structural optimization to generate a high affinity C. trachomatis selective inhibitor and potentially speeding up the discovery process (Yao et al., 2014).
Author Contributions
JY and CR conceived, drafted, and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by NIGMS grant GM034496 and the American Syrian Associated Charities.
References
- Abdelrahman Y. M., Belland R. J. (2005). The chlamydial developmental cycle. FEMS Microbiol. Rev. 29 949–959. 10.1016/j.femsre.2005.03.002 [DOI] [PubMed] [Google Scholar]
- Agaisse H., Derre I. (2014). Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect. Immun. 82 2037–2047. 10.1128/IAI.01530-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zeer M. A., Al-Younes H. M., Kerr M., Abu-Lubad M., Gonzalez E., Brinkmann V., et al. (2014). Chlamydia trachomatis remodels stable microtubules to coordinate Golgi stack recruitment to the chlamydial inclusion surface. Mol. Microbiol. 94 1285–1297. 10.1111/mmi.12829 [DOI] [PubMed] [Google Scholar]
- Bastidas R. J., Elwell C. A., Engel J. N., Valdivia R. H. (2013). Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 3:a010256. 10.1101/cshperspect.a010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty W. L. (2008). Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect. Immun. 76 2872–2881. 10.1128/IAI.00129-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belland R., Ojcius D. M., Byrne G. I. (2004). Focus: chlamydia. Nat. Rev. Microbiol. 2 530–531. 10.1038/nrmicro931 [DOI] [PubMed] [Google Scholar]
- Bidawid S., Chou S., Ng C. W., Perry E., Kasatiya S. (1989). Fatty acid profiles of Chlamydia using capillary gas chromatography. Antonie Van Leeuwenhoek 55 123–131. 10.1007/Bf00404752 [DOI] [PubMed] [Google Scholar]
- Black P. N., Dirusso C. C., Metzger A. K., Heimert T. L. (1992). Cloning, sequencing, and expression of the fadD gene of Escherichia coli encoding acyl coenzyme A synthetase. J. Biol. Chem. 267 25513–25520. [PubMed] [Google Scholar]
- Blaser M. J., Falkow S. (2009). What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 7 887–894. 10.1038/nrmicro2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnhoff M., Miller C. P. (1962). Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J. Infect. Dis. 111 117–127. 10.1093/infdis/111.2.117 [DOI] [PubMed] [Google Scholar]
- Boncompain G., Muller C., Meas-Yedid V., Schmitt-Kopplin P., Lazarow P. B., Subtil A. (2014). The intracellular bacteria Chlamydia hijack peroxisomes and utilize their enzymatic capacity to produce bacteria-specific phospholipids. PLoS One 9:e86196. 10.1371/journal.pone.0086196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursi B., Mamtani R., Haynes K., Yang Y. X. (2015). The effect of past antibiotic exposure on diabetes risk. Eur. J. Endocrinol. 172 639–648. 10.1530/EJE-14-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun P. R., Al-Younes H., Gussmann J., Klein J., Schneider E., Meyer T. F. (2008). Competitive inhibition of amino acid uptake suppresses Chlamydial growth: involvement of the Chlamydial amino acid transporter BrnQ. J. Bacteriol. 190 1822–1830. 10.1128/JB.01240-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie C. G., Jarchum I., Equinda M., Lipuma L., Gobourne A., Viale A., et al. (2012). Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 80 62–73. 10.1128/IAI.05496-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo R. A., Mead D. J., Hackstadt T. (2003). Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. U.S.A. 100 6771–6776. 10.1073/pnas.1131289100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Yamanishi S., Cox L., Methe B. A., Zavadil J., Li K., et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488 621–626. 10.1038/nature11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro J. L., Kumar Y., Fischer E. R., Hackstadt T., Valdivia R. H. (2008). Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U.S.A. 105 9379–9384. 10.1073/pnas.0712241105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper C. L., Hsu L., Jackowski S., Rock C. O. (1989). 2-Acylglycerolphosphoethanolamine acyltransferase/acyl-acyl carrier protein synthetase is a membrane-associated acyl carrier protein binding protein. J. Biol. Chem. 264 7384–7389. [PubMed] [Google Scholar]
- Cox J. V., Abdelrahman Y. M., Peters J., Naher N., Belland R. J. (2016). Chlamydia trachomatis utilizes the mammalian CLA1 lipid transporter to acquire host phosphatidylcholine essential for growth. Cell. Microbiol. 18 305–318. 10.1111/cmi.12523 [DOI] [PubMed] [Google Scholar]
- Cox J. V., Naher N., Abdelrahman Y. M., Belland R. J. (2012). Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cell. Microbiol. 14 1497–1512. 10.1111/j.1462-5822.2012.01823.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowhan W. (1997). Phosphatidylserine decarboxylases: pyruvoyl-dependent enzymes from bacteria to mammals. Method Enzymol. 280 81–88. 10.1016/S0076-6879(97)80104-8 [DOI] [PubMed] [Google Scholar]
- Elwell C. A., Engel J. N. (2012). Lipid acquisition by intracellular Chlamydiae. Cell. Microbiol. 14 1010–1018. 10.1111/j.1462-5822.2012.01794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C. A., Jiang S., Kim J. H., Lee A., Wittmann T., Hanada K., et al. (2011). Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathog. 7:e1002198. 10.1371/journal.ppat.1002198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett K. D., Desiderio D. M., Hatch T. P. (1994). Characterization of lipoprotein EnvA in Chlamydia psittaci 6BC. J. Bacteriol. 176 6082–6087. 10.1128/jb.176.19.6082-6087.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamm R. K., Rhomberg P. R., Kaplan N., Jones R. N., Farrell D. J. (2015). Activity of debio1452, a FabI inhibitor with potent activity against Staphylococcus aureus and coagulase-negative Staphylococcus spp., including multidrug-resistant strains. Antimicrob. Agents Chemother. 59 2583–2587. 10.1128/AAC.05119-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloux K., Guillemet M., Soler C., Morvan C., Halpern D., Pourcel C., et al. (2017). Clinical relevance of type II fatty acid synthesis bypass in Staphylococcus aureus. Antimicrob. Agents Chemother. 61:e02515-16. 10.1128/AAC.02515-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T., Scidmore M. A., Rockey D. D. (1995). Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc. Natl. Acad. Sci. U.S.A. 92 4877–4881. 10.1073/pnas.92.11.4877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafkin B., Kaplan N., Murphy B. (2016). Efficacy and safety of AFN-1252, the first Staphylococcus-specific antibacterial agent, in the treatment of acute bacterial skin and skin structure infections, including those in patients with significant comorbidities. Antimicrob. Agents Chemother. 60 1695–1701. 10.1128/Aac.01741-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch T. P., Al-Hossainy E., Silverman J. A. (1982). Adenine nucleotide and lysine transport in Chlamydia psittaci. J. Bacteriol. 150 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath R. J., Rock C. O. (1995). Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 270 26538–26542. 10.1074/jbc.270.44.26538 [DOI] [PubMed] [Google Scholar]
- Heath R. J., Rock C. O. (2000). A triclosan-resistant bacterial enzyme. Nature (London) 406 145–146. 10.1038/35018162 [DOI] [PubMed] [Google Scholar]
- Heine H., Gronow S., Zamyatina A., Kosma P., Brade H. (2007). Investigation on the agonistic and antagonistic biological activities of synthetic Chlamydia lipid A and its use in in vitro enzymatic assays. J. Endotoxin. Res. 13 126–132. 10.1177/0968051907079122 [DOI] [PubMed] [Google Scholar]
- Heine H., Muller-Loennies S., Brade L., Lindner B., Brade H. (2003). Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur. J. Biochem. 270 440–450. 10.1046/j.1432-1033.2003.03392.x [DOI] [PubMed] [Google Scholar]
- Hsu L., Jackowski S., Rock C. O. (1991). Isolation and characterization of Escherichia coli K-12 mutants lacking both 2-acyl-glycerophosphoethanolamine acyltransferase and acyl-acyl carrier protein synthetase activity. J. Biol. Chem. 266 13783–13788. [PubMed] [Google Scholar]
- Hunt T., Kaplan N., Hafkin B. (2015). Safety, tolerability and pharmacokinetics of multiple oral doses of AFN-1252 administered as immediate release (IR) tablets in healthy subjects. J. Chemother. 28 164–171. 10.1179/1973947815Y.0000000075 [DOI] [PubMed] [Google Scholar]
- Iliffe-Lee E. R., McClarty G. (1999). Glucose metabolism in Chlamydia trachomatis: the ‘energy parasite’ hypothesis revisited. Mol. Microbiol. 33 177–187. 10.1046/j.1365-2958.1999.01464.x [DOI] [PubMed] [Google Scholar]
- Jackowski S., Rock C. O. (1986). Transfer of fatty acids from the 1-position of phosphatidylethanolamine to the major outer membrane lipoprotein of Escherichia coli. J. Biol. Chem. 261 11328–11333. [PubMed] [Google Scholar]
- Jiang Y., Chan C. H., Cronan J. E. (2006). The soluble acyl-acyl carrier protein synthetase of Vibrio harveyi B392 is a member of the medium chain acyl-CoA synthetase family. Biochemistry 45 10008–10019. 10.1021/bi060842w [DOI] [PubMed] [Google Scholar]
- Jiang Y., Morgan-Kiss R. M., Campbell J. W., Chan C. H., Cronan J. E. (2010). Expression of Vibrio harveyi acyl-ACP synthetase allows efficient entry of exogenous fatty acids into the Escherichia coli fatty acid and lipid A synthetic pathways. Biochemistry 49 718–726. 10.1021/bi901890a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Albert M., Awrey D., Bardouniotis E., Berman J., Clarke T., et al. (2012). Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 56 5865–5874. 10.1128/AAC.01411-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Garner C., Hafkin B. (2013). AFN-1252 in vitro absorption studies and pharmacokinetics following microdosing in healthy subjects. Eur. J. Pharm. Sci. 50 440–446. 10.1016/j.ejps.2013.08.019 [DOI] [PubMed] [Google Scholar]
- Lightner V. A., Bell R. M., Modrich P. (1983). The DNA sequences encoding plsB and dgk loci of Escherichia coli. J. Biol. Chem. 258 10856–10861. [PubMed] [Google Scholar]
- Lu Y.-J., Zhang Y.-M., Grimes K. D., Qi J., Lee R. E., Rock C. O. (2006). Acyl-phosphates initiate membrane phospholipid synthesis in gram-positive pathogens. Mol. Cell 23 765–772. 10.1016/j.molcel.2006.06.030 [DOI] [PubMed] [Google Scholar]
- Marrakchi H., Dewolf W. E., Jr., Quinn C., West J., Polizzi B. J., So C. Y., et al. (2003). Characterization of Streptococcus pneumoniae enoyl-[acyl carrier protein] reductase (FabK). Biochem. J. 370 1055–1062. 10.1042/BJ20021699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxson T., Mitchell D. A. (2016). Targeted treatment for bacterial infections: prospects for pathogen-specific antibiotics coupled with rapid diagnostics. Tetrahedron 72 3609–3624. 10.1016/j.tet.2015.09.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Million M., Lagier J. C., Yahav D., Paul M. (2013). Gut bacterial microbiota and obesity. Clin. Microbiol. Infect. 19 305–313. 10.1111/1469-0691.12172 [DOI] [PubMed] [Google Scholar]
- Mirrashidi K. M., Elwell C. A., Verschueren E., Johnson J. R., Frando A., Von Dollen J., et al. (2015). Global mapping of the inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 18 109–121. 10.1016/j.chom.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J., Miller N. J., Dorward D. W., Dooley C. A., Hackstadt T. (2013). Role for chlamydial inclusion membrane proteins in inclusion membrane structure and biogenesis. PLoS One 8:e63426. 10.1371/journal.pone.0063426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S. R., Collins J. J., Relman D. A. (2014). Antibiotics and the gut microbiota. J. Clin. Invest. 124 4212–4218. 10.1172/JCI72333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan C., Halpern D., Kenanian G., Hays C., Anba-Mondoloni J., Brinster S., et al. (2016). Environmental fatty acids enable emergence of infectious Staphylococcus aureus resistant to FASII-targeted antimicrobials. Nat. Commun. 7:12944. 10.1038/ncomms12944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan C., Halpern D., Kenanian G., Pathania A., Anba-Mondoloni J., Lamberet G., et al. (2017). The Staphylococcus aureus FASII bypass escape route from FASII inhibitors. Biochimie 141 40–46. 10.1016/j.biochi.2017.07.004 [DOI] [PubMed] [Google Scholar]
- Neff L., Daher S., Muzzin P., Spenato U., Gulacar F., Gabay C., et al. (2007). Molecular characterization and subcellular localization of macrophage infectivity potentiator, a Chlamydia trachomatis lipoprotein. J. Bacteriol. 189 4739–4748. 10.1128/JB.01889-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A., Sixt B. S., Horn M., Hackstadt T. (2014). Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol. Rev. 38 779–801. 10.1111/1574-6976.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker E. P. K., Praharaj I., John J., Kaliappan S. P., Kampmann B., Kang G., et al. (2017). Changes in the intestinal microbiota following the administration of azithromycin in a randomised placebo-controlled trial among infants in south India. Sci. Rep. 7:9168. 10.1038/s41598-017-06862-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B., Broussard T. C., Bose J. L., Rosch J. W., Jackson P., Subramanian C., et al. (2014). Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 111 10532–10537. 10.1073/pnas.1408797111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B., Frank M. W., Rosch J. W., Rock C. O. (2013). Staphylococcus aureus fatty acid auxotrophs do not proliferate in mice. Antimicrob. Agents Chemother. 57 5729–5732. 10.1128/AAC.01038-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B., Frank M. W., Subramanian C., Saenkham P., Rock C. O. (2011). Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108 15378–15383. 10.1073/pnas.1109208108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B., Rock C. O. (2013). Bacterial lipids: metabolism and membrane homeostasis. Prog. Lipid Res. 52 249–276. 10.1016/j.plipres.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J., Gwynn M. N., Holmes D. J., Pompliano D. L. (2007). Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6 29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- Pluckthun A., Dennis E. A. (1982). Acyl and phosphoryl migration in lysophospholipids: importance in phospholipid synthesis and phospholipase specificity. Biochemistry 21 l743–l1750. 10.1021/bi00537a007 [DOI] [PubMed] [Google Scholar]
- Recuero-Checa M. A., Sharma M., Lau C., Watkins P. A., Gaydos C. A., Dean D. (2016). Chlamydia trachomatis growth and development requires the activity of host Long-chain Acyl-CoA Synthetases (ACSLs). Sci. Rep. 6:23148. 10.1038/srep23148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson R. M., Yao J., Gajewski S., Kumar G., Martin E. W., Rock C. O., et al. (2017). A two-helix motif positions the lysophosphatidic acid acyltransferase active site for catalysis within the membrane bilayer. Nat. Struct. Mol. Biol. 24 666–671. 10.1038/nsmb.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock C. O., Cronan J. E., Jr. (1979). Solubilization, purification, and salt activation of acyl-acyl carrier protein synthetase from Escherichia coli. J. Biol. Chem. 254 7116–7122. [PubMed] [Google Scholar]
- Rund S., Lindner B., Brade H., Holst O. (1999). Structural analysis of the lipopolysaccharide from Chlamydia trachomatis serotype L2. J. Biol. Chem. 274 16819–16824. 10.1074/jbc.274.24.16819 [DOI] [PubMed] [Google Scholar]
- Saka H. A., Thompson J. W., Chen Y. S., Dubois L. G., Haas J. T., Moseley A., et al. (2015). Chlamydia trachomatis infection leads to defined alterations to the lipid droplet proteome in epithelial cells. PLoS One 10:e0124630. 10.1371/journal.pone.0124630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L. L. (2007). Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6 41–55. 10.1038/nrd2202 [DOI] [PubMed] [Google Scholar]
- Silver L. L. (2011). Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24 71–109. 10.1128/CMR.00030-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupene E., Kuypers F. A. (2017). Phosphatidylserine decarboxylase CT699, lysophospholipid acyltransferase CT775, and acyl-ACP synthase CT776 provide membrane lipid diversity to Chlamydia trachomatis. Sci. Rep. 7:15767. 10.1038/s41598-017-16116-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupene E., Wang D., Kuypers F. A. (2015). Remodeling of host phosphatidylcholine by Chlamydia acyltransferase is regulated by acyl-CoA binding protein ACBD6 associated with lipid droplets. MicrobiologyOpen 4 235–251. 10.1002/mbo3.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding C. N., Klein R. D., Ruer S., Kau A. L., Schreiber H. L., Cusumano Z. T., et al. (2017). Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature 546 528–532. 10.1038/nature22972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens R. S., Kalman S., Lammel C., Fan J., Marathe R., Aravind L., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282 754–759. 10.1126/science.282.5389.754 [DOI] [PubMed] [Google Scholar]
- Su H., Mcclarty G., Dong F., Hatch G. M., Pan Z. K., Zhong G. (2004). Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 279 9409–9416. 10.1074/jbc.M312008200 [DOI] [PubMed] [Google Scholar]
- Turnbull A. P., Rafferty J. B., Sedelnikova S. E., Slabas A. R., Schierer T. P., Kroon J. T., et al. (2001). Analysis of the structure, substrate specificity, and mechanism of squash glycerol-3-phosphate (1)-acyltransferase. Structure 9 347–353. 10.1016/S0969-2126(01)00595-0 [DOI] [PubMed] [Google Scholar]
- Weber M. M., Lam J. L., Dooley C. A., Noriea N. F., Hansen B. T., Hoyt F. H., et al. (2017). Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep. 19 1406–1417. 10.1016/j.celrep.2017.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie J. L., Hatch G. M., Mcclarty G. (1997). Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 179 7233–7242. 10.1128/jb.179.23.7233-7242.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Abdelrahman Y. M., Robertson R. M., Cox J. V., Belland R. J., White S. W., et al. (2014). Type II fatty acid synthesis is essential for the replication of Chlamydia trachomatis. J. Biol. Chem. 289 22365–22376. 10.1074/jbc.M114.584185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Bruhn D. F., Frank M. W., Lee R. E., Rock C. O. (2016a). Activation of exogenous fatty acids to acyl-acyl carrier protein cannot bypass FabI inhibition in Neisseria. J. Biol. Chem. 291 171–181. 10.1074/jbc.M115.699462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Carter R. A., Vuagniaux G., Barbier M., Rosch J. W., Rock C. O. (2016b). A pathogen-selective antibiotic minimizes disturbance to the microbiome. Antimicrob. Agents Chemother. 60 4264–4273. 10.1128/AAC.00535-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Cherian P. T., Frank M. W., Rock C. O. (2015a). Chlamydia trachomatis relies on autonomous phospholipid synthesis for membrane biogenesis. J. Biol. Chem. 290 18874–18888. 10.1074/jbc.M115.657148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Dodson V. J., Frank M. W., Rock C. O. (2015b). Chlamydia trachomatis scavenges host fatty acids for phospholipid synthesis via an acyl-acyl carrier protein synthetase. J. Biol. Chem. 290 22163–22173. 10.1074/jbc.M115.671008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Maxwell J. B., Rock C. O. (2013). Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem. 288 36261–36271. 10.1074/jbc.M113.512905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rock C. O. (2013). Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta 1831 495–502. 10.1016/j.bbalip.2012.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rock C. O. (2015). How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J. Biol. Chem. 290 5940–5946. 10.1074/jbc.R114.636241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rock C. O. (2016). Resistance mechanisms and the future of bacterial enoyl-acyl carrier protein reductase (FabI) antibiotics. Cold Spring Harb. Perspect. Med. 6:a027045. 10.1101/cshperspect.a027045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rock C. O. (2017a). Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim. Biophys. Acta 1862 1300–1309. 10.1016/j.bbalip.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rock C. O. (2017b). Exogenous fatty acid metabolism in bacteria. Biochimie 141 30–39. 10.1016/j.biochi.2017.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]