

Abstract
Human milk oligosaccharides (HMOs) are the third largest macromolecular component of breast milk and offer infants numerous health benefits, most of which stem from the development of a healthy microbiome. Characterization, quantification, and chemical derivatization of HMOs remains a frontier issue in glycobiology due to the challenge of isolating appreciable quantities of homogenous HMOs from breast milk. Herein, we report the synthesis of the human milk tetrasaccharide lacto-N-tetraose (LNT). LNT is ubiquitous in human breast milk as it is a core structure common to longer-chain HMOs and many glycolipids.
Graphical Abstract
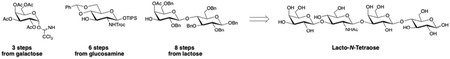
1. Introduction
Human milk is among the earliest vehicles for intestinal bacterial colonization.1–3 In addition to labor and delivery, human milk assists in establishing a microbiota that provides several health benefits for the breastfed infant. There are an estimated 700 species of bacteria present in human milk at any given time.1,4–7 The majority of these bacteria are symbiotic or commensal with roles varying from metabolic functioning to immune enhancing.4 These protective measures are due, in part, to the human milk oligosaccharides (HMOs), a family of unconjugated glycans unique to human milk.8–13
HMOs are prebiotics that stimulate the growth of symbiotic bacteria whose colonization in the infant gut competes with pathogenic colonization.14 This activity is critical to infant health as symbiotic species, such as bifidobacteria, aid in digestion, metabolism, immunity, and the production of neurotransmitters that affect behavior and cognition.1,15,16 HMOs also function as anti-adhesive antimicrobial agents by serving as free flowing decoy receptors. This role allows HMOs to inhibit pathogen adhesion to host cell ligands thus preventing the first stage of infection. Although the general functionings of pooled HMOs have been explored, the functional roles of individual HMOs are far less clear due to a limited access to large quantities of single-entity HMOs.8,11
HMOs are the third most abundant macromolecule in human milk after lactose and fat. Colostrum, or the first milk, is produced during the perinatal period and contains ca. 25 g/L of HMOs.17,18 As nursing progresses and milk matures, the concentration of HMOs decreases. Interestingly, premature milk features a greater concentration of HMOs than term milk.18 In general, the concentration and molecular architecture of HMOs vary depending on stage of lactation and Lewis (Le) blood group and secretor status of the mother.19
To date, the structures of nearly 200 HMOs have been elucidated.20–22 However, of these, only a handful of short-chain HMOs can be produced. Moreover, their production is only possible on a multi-milligram scale. As short-chain HMOs serve as building blocks for long-chain HMOs featuring branching, fucosylation, and/or sialylation, insufficient access to shorter HMOs has largely limited the access to long-chain HMOs. Not surprisingly, a number of strategies have been developed to synthesize structurally defined single-entity HMOs.23–30
We recently initiated a program aimed at studying the glycobiology of human milk. As shown in Figure 1, donor milk from two mothers was fingerprinted using MALDI FT ICR in positive ion mode, to reveal the structures of HMOs in a mass range of 500–2000 m/z. A mass of 730.241 was observed, corresponding to the two isomeric tetrasaccharides, lacto-N-tetraose (LNT) and lacto-N-neotetraose (LNnT). LNT is more common in human milk being present at 8 times the level of LNnT. In contrast, LNnT is more common in bovine milk and by extension more common in infant formula.28 The relative intensities of this peak will vary from sample to sample as LNT and/or LNnT undergoes enzymatic functionalization to larger, fucosylated derivatives. Unlike LNnT, LNT is not currently available at large scale through chemo-enzymatic synthesis.28 We therefore selected this molecule as a target for chemical synthesis. Chemical syntheses of several LNT derivatives and a chemical synthesis of LNT have been previously reported.30–35 Additionally, an elegant chemoenzymatic approach was recently reported which yielded ca. 20 mg of LNT.36 These preparations, however, are on scales smaller than that featured in the present work. Herein, we describe the chemical synthesis of homogeneous LNT using an approach that produces ca. 1 gram of protected tetrasaccharide in a single glycosylation event.
Figure 1.
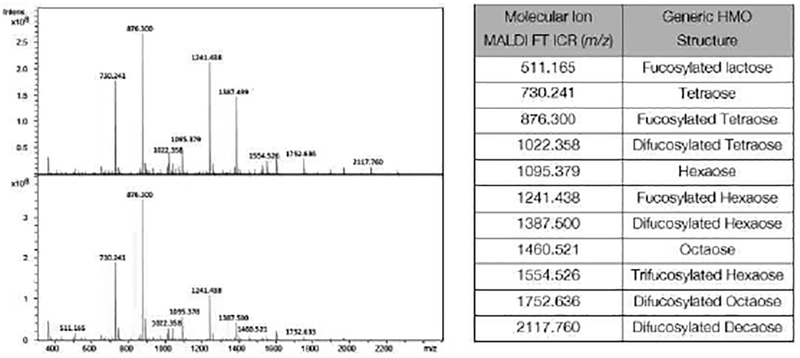
HMO mass fingerprinting of two human milk samples using MALDI FT ICR. A generic structural description is given for each molecular ion observed.
As depicted in Figure 1, LNT (1) is a linear tetrasaccharide in which all glycosidic bonds are b-configured. More specifically, LNT contains lactose (Galb1–4Glc) at the reducing end, which is elon-gated by b-1,3 linked lacto-N-biose (Galb1–3GlcNAc). Our general synthetic strategy envisioned a twoglycosylation approach wherein the tetrasaccharide could be assembled from galactose-, glucosamine-, and lactose-derived building blocks 2, 3, and 4 respectively.
2. Results and discussion
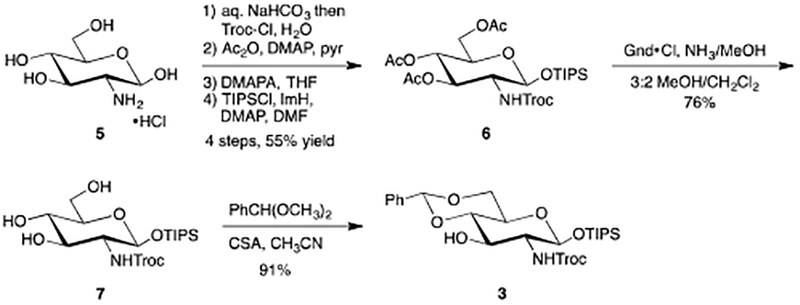
In the opening stages of the program, we synthesized the requisite glucosamine residue 3, which is related to a previously described building block reported by the Boons laboratory.37 Starting from glucosamine hydrochloride 5, the C-2 amine was protected as its trichloroethyl (Troc) carbamate after treatment with saturated aqueous sodium bicarbonate solution followed by addition of 2,2,2-trichloroethyl chloroformate (Troc-Cl). Following peracetylation, selective removal of the anomeric acetate was accomplished using 3-(dimethylamino)-1-propylamine (DMAPA).38 Finally, the anomeric hemiacetal was converted to its triisopropyl silyl ether using standard Corey conditions (imidazole and DMF)39 to provide b-silyl ether 6 as a single anomer in 54% yield for the 4-step sequence. We initially attempted to advance substrate 6 to its triol 7 using sodium methoxide in methanol. Unfortunately, in our hands, we observed compatibility issues with the Troc carbamate. After screening several sets of conditions, we realized acetate removal could be accomplished cleanly using guanidinium chloride in the presence of ammonia to arrive at triol 7 in 76% yield. Finally, 7 was converted to the 4,6-benzylidene acetal upon reaction with benzaldehyde dimethyl acetal and catalytic 10-camphorsulfonic acid (CSA) to yield glucosamine acceptor 3 in 91% yield.
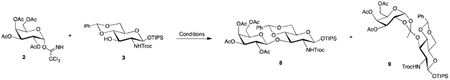
We next turned our attention to the synthesis of lacto-N-biose disaccharide 8, which required uniting a galactose donor with glucosamine acceptor 3. Known galactose-based a-trichloroacetimidate donor 2240 was prepared in two steps from commercially available galactose pentaacetate and reacted with acceptor 3. Interestingly, the glycosylation was not as straightforward as anticipated as significant amounts of orthoester were observed, depending on reaction conditions. As shown in Table 1, our initial strategy used acetonitrile as the solvent in the hope that it would assist in providing the desired bglycoside. Unfortunately, use of acetonitrile and colder reaction temperatures resulted in significant formation of orthoacetate 9. Ultimately, we discovered formation of the orthoacetate could be alleviated by using dichloromethane as the solvent alongside catalytic TMSOTf as an activator with warmer reaction temperatures. Using these conditions, the desired, protected lacto-N-biose disaccharide 8 could be obtained in 85% yield.
Table 1.
Synthesis of Lacto-N-Biose Disaccharide 8 and Unexpected Orthoacetate 9.
| Entry | Activator | Solvent | Temperature (°C) | %Yield 9 | %Yield 10 |
|---|---|---|---|---|---|
| 1 | TMSOTf | CH3CN | −40 | 18 | 50 |
| 2 | BF3⋅OEt2 | CH3CN | −40 | 0 | 23 |
| 3 | TMSOTf | CH3CN | −10 | 28 | 40 |
| 4 | TMSOTf | CH3CN | 0 | 36 | 44 |
| 5 | TMSOTf | CH3CN | r.t. | 49 | 0 |
| 6 | TMSOTf | CH2Cl2 | −78 | 0 | 22 |
| 7 | TMSOTf | CH2Cl2 | 0 | 85 | 0 |
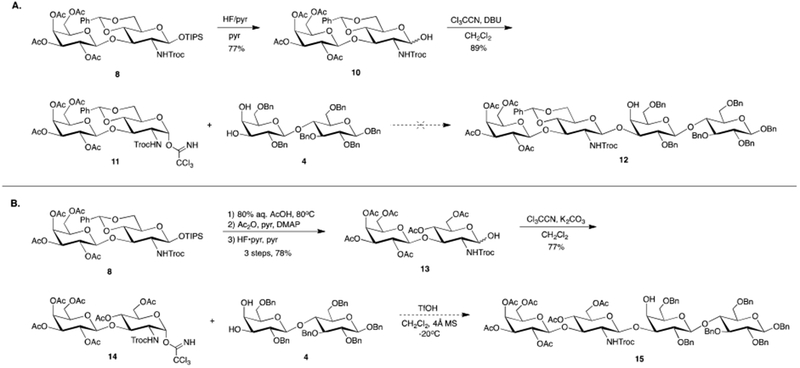
After obtaining gram quantities of 8, the β-silyl ether was removed via treatment with HF in pyridine to yield anomeric hemiacetal 10 in 77% yield. 10 was then converted to α-trichloroacetimidate donor 11 in 89% yield. With this donor in hand, the campaign to produce protected tetrasaccharide 12 began. Using known lactose acceptor 441, prepared in six steps from lactose octaacetate,42 we attempted to form the final glycosidic linkage. Unfortunately, we were unable to successfully generate this linkage despite screening a variety of activators, including TMSOTf, BF3⋅OEt2, AgOTf, TfOH, and MeOTf, as well as a number of reaction solvents and reaction temperatures. Ultimately, we concluded our inability to form the desired bond was due to the rigidity of the benzylidene acetal of the lacto-N-biose based donor and its prohibitive effect on the generation of the requisite oxonium ion. In retrospect, the poor reactivity of glycosyl donors featuring benzylidene acetals is well precedented.43–47
To circumvent the poor reactivity of donor 11, we first removed the benzylidene acetal by heating 11 to 80oC and treating with 80% aqueous acetic acid. The resulting diol was then acetylated and the β-silyl ether removed to yield anomeric hemiacetal 13 in 78% yield over 3 steps. With 13 in hand, we moved to form a-trichloroacetimidate donor 14. Initial attempts relied on treatment of anomeric hemiacetal 13 with 1,8-diazabicylco[5.4.0]undec-7-ene (DBU) and trichloroacetonitirle (Cl3CCN). Surprisingly, these conditions proved unreliable as frequent decomposition was observed. In an effort to lessen this degradation, potassium carbonate (K2CO3) and prolonged reaction times were used to generate atrichloroacetimidate donor 14 in 77% yield.
With the new lacto-N-biose donor 14 in hand, we once again turned our attention to forming the final glycosidic linkage. Unfortunately, glycosylation with donor 14 and acceptor 4 did not proceed as smoothly as expected. While mass spectral data suggested we had formed the desired tetrasaccharide 15, we were unable to fully characterize the compound as multiple byproducts were observed. One of the suspected byproducts was the hexasaccharide resulting from glycosylation at both the C-3’ and C-4’ hydroxyls of acceptor 4. In an effort to help the glycosylation event proceed more smoothly, we attempted to better exploit the increased reactivity of the equatorial C-3’ hydroxyl over the C-4’ axial hydroxyl by adding the donor dropwise into the reaction vessel containing the acceptor and TfOH. Much to our disappointment, multiple side products were once again observed.
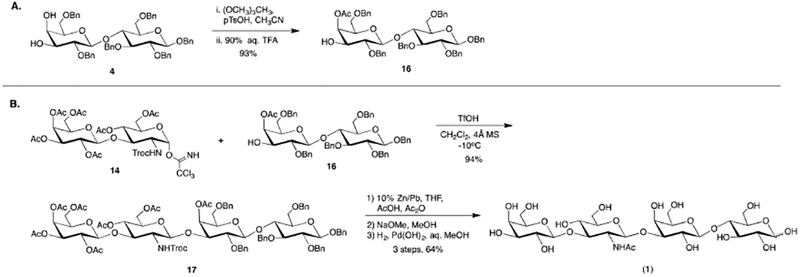
Although we were unable to confirm formation of the hexasaccharide, we hypothesized the glycosylation would be improved by protecting the C-4’ hydroxyl of diol acceptor 4. To do this, we employed a simple two-step, one-pot procedure to selectively acetylate the axial C-4’ hydroxyl. Treatment of diol 4 with trimethylorthoacetate followed by treatment with 90% aqueous trifluoroacetic acid furnished known axially acetylated acceptor 16 in a satisfactory 93% yield.48,49 Much to our delight, TfOH promoted glycosylation of donor 14 with acceptor 16 resulted in formation of the desired fully protected tetrasaccharide 17 in 94% yield.
With 17 in hand, global deprotection to afford lacto-N-tetraose (1) commenced with Troc removal accompanied by in situ acetamide formation. To this end, it is important to highlight that the use of activated Zn dust in a mixed solvent system of acetic acid and acetic anhydride to reductively convert the N-Troc-protecting group to its corresponding acetamide resulted in partial decomposition and undesirably low yields. To overcome this shortcoming, we moved to less conventional reducing agents, including 10% Zn/Pb couple and 10% Cd/Pb couple.50,51 Of these, 10% Zn/Pb couple performed best and reductively cleaved the Troc carbamate without any accompanying decomposition. Following successful acetamide formation, deacetylation followed by hydrogenolysis furnished lacto-N-tetraose (1) in 64% yield over the three step deprotection sequence.
In summary, the total synthesis of lacto-N-tetraose has been completed using a linear approach from three readily synthesized building blocks. Of note is that the key, final glycosylation event was conducted to produce ca. 1 gram of protected LNT. With this molecule now available at large scale, future directions in our lab concern studying the conversion of this molecule to other complex HMOs. Progress in this regard, will be reported in due course.
3. Experimental Section
3.1. General materials and methods
Commercial reagents were used as received. Anhydrous solvents were taken from an MBRAUN solvent purification system (MB SPS) and stored over 4 Å or 3 Å molecular sieves. All moisture-sensitive reactions were performed in flame- or oven-dried round bottom flasks under an argon atmosphere. All airor moisture-sensitive liquids were transferred via oven-dried stainless steel syringes or cannula. Reaction temperatures were monitored and controlled via thermocouple thermometer and corresponding hot plate stirrer. Flash column chromatography was performed as described by Still et. al. using silica gel 230–400 mesh. Analytical thin-layer chromatography (TLC) was performed on glass-backed Silica gel 60 F254 plates (EMD/Merck KGaA) and visualized using UV, cerium ammonium molybdate stain, and anisaldehyde stain. 1H NMR spectra were obtained on a Bruker 400 or 600 MHz spectrometer with reporting relative to residual solvent signals (CDCl3, 7.26 ppm; CH3OD, 3.31 ppm; D2O, 4.79 ppm). 1H NMR spectral data are presented as follows: chemical shifts (δ ppm), multiplicity (s=singlet, d=doublet, dd=doublet of doublets, t=triplet, q=quartet, m=multiplet, br=broad), coupling constants (Hz), integration, proton assignment. 13C NMR spectra were obtained on a Bruker 100 MHz spectrometer with reporting relative to residual solvent signals (CDCl3, 77.16 ppm; CH3OD, 49.0 ppm). 13C NMR spectral data are presented as follows: chemical shifts (δ ppm), carbon assignment. Proton and carbon assignments were made with the aid of 2D NMR techniques (COSY, HSQC, and HMBC). High resolution mass spectra were recorded on a high resolution Thermo Electron Corporation MAT 95XP-Trap by use of electro-spray ionization (ESI) by the Indiana University Mass Spectrometry facility and a SYNAPT G2 or SYNAPT G2-S spectrometer (Waters, for TOF-MS) by the McLean lab of Vanderbilt University. Low resolution mass spectra were recorded on a Thermo Scientific Dionex Ultimate 3000 HPLC system with MSQ Plus Mass Detector. Optical rotations were obtained using a Perkin Elmer 341 polarimeter.
3.2. Synthetic procedures
3.2.1. (2R,3S,4R,5R,6S)-2-(acetoxymethyl)-5-(((2,2,2-trichloroethoxy)carbonyl)amino)-6-((triisopropylsilyl)oxy)tetrahydro-2H-pyran-3,4-diyl diacetate (6)
To a suspension of D-(+)-glucosamine hydrochloride 5 (1.0 eq, 3.0 g, 14 mmol) in H2O (30 mL) was added NaHCO3 (3.0 eq, 3.5 g, 42 mmol) at room temperature and the resulting mixture was stirred 1 H. 2,2,2-trichloroethyl chloroformate (5.0 eq, 9.4 mL, 70 mmol) was then added dropwise over 20 minutes. The reaction was stirred an additional 3 H after which a white solid had formed. The reaction was filtered, washed with additional H2O (250 mL), and allowed to dry overnight. The crude Troc-protected glucosamine was coevaporated with benzene (3x) then dissolved in pyridine (130 mL) and acetic anhydride (5.0 eq, 6.6 mL, 70 mol) was added dropwise over 5 minutes. The resulting solution was stirred 6 H then was diluted with EtOAc (250 mL), washed with 2 N HCl (6 × 90 mL), brine (1 × 90 mL), dried (MgSO4), filtered, and concentrated in vacuo to yield a pale yellow foam. The crude tetraacetate was dissolved in THF (70 mL) and 3-(dimethylamino)-1-propylamine (DMAPA) (5.0 eq, 8.8 mL, 70 mmol) was added. The reaction was stirred 1 H and 15 minutes then was diluted with CH2Cl2 (130 mL), washed with 2 N HCl (2 × 60 mL), dried (MgSO4), filtered, and concentrated in vacuo to yield a white foam. The crude anomeric hemiacetal was dissolved in DMF (90 mL) and imidazole (2.0 eq, 1.9 g, 28 mmol), DMAP (cat), and chlorotriisopropylsilane (1.2 eq, 3.6 mL, 17 mmol) were added sequentially. The resulting solution was stirred 16 H then additional chlorotriisopropylsilane was added (1.5 mL). The reaction stirred an additional 22 H then was diluted with EtOAc (180 mL), washed with H2O (3 × 80 mL), 2N HCl (1 × 80 mL), brine (1 × 80 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (2:1 hexanes/EtOAc) to yield anomeric silyl ether 6 (4.87 g, 7.65 mmol, 55% over 4 steps) as a white solid: Rf 0.48 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 5.23 (t, J=10.1 Hz , 1H, H-3), 5.06 (d, J=8.6 Hz, 1H, NH), 5.04 (t, J=9.6 Hz, 1H, H-4), 4.89 (d, J=7.8 Hz, 1H, H-1), 4.75 (d, J=12.0 Hz, 1H, Troc CH), 4.61 (d, J=12.0 Hz, 1H, Troc CH), 4.15 (d, J=4.2 Hz, 2H, H-6a, H-6b), 3.72–3.61 (m, 2H, H-2, H-5), 2.06 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 1.12–1.01 (m, 21H, TIPS), 1.60 (s, H2O); 13C (100 MHz, CDCl3) δ 170.9 (COCH3), 170.8 (COCH3), 169.7 (COCH3), 154.1 (Troc CO), 96.1 (C-1), 95.4 (Troc CCl3), 74.8 (Troc CH2), 72.1 (C-3), 71.9 (C-5), 69.1 (C-4), 62.6 (C-6), 58.7 (C-2), 20.8 (COCH3), 17.9 (TIPS), 17.8 (TIPS), 12.3 (TIPS). HRMS (ESI) calcd for C24H40Cl3NO10Si [M+Na]+ 658.1385, found 658.1356.
3.2.2. 2,2,2-trichloroethyl ((2S,3R,4R,5S,6R)-4,5-dihydroxy-6-(hydroxymethyl)-2-((triisopropylsilyl)oxy)tetrahydro-2H-pyran-3-yl)carbamate (7)
To a solution of 6 (1.0 eq, 4.0 g, 6.3 mmol) in CH2Cl2 (60 mL) and MeOH (60 mL), was added guanidine hydrochloride (5.0 eq, 3.0 g, 31 mmol) and 7 N NH3/MeOH (12.0 eq, 8.4 mL, 75 mmol) sequentially. The resulting solution was stirred 21 H then was neutralized with AcOH and concentrated in vacuo. The crude residue was purified via flash column chromatography (10:1 CHCl3/MeOH) to yield triol 7 (2.45 g, 4.80 mmol, 76%) as a white solid: Rf 0.41 (10:1 CH2 Cl2/MeOH); 1H NMR (400 MHz, MeOD) δ 4.73 (d, J=7.7 Hz, 1H, H-1), 4.70 (s, 2H, Troc CH, Troc CH), 3.84 (dd, J=2.6, 11.8 Hz, 1H, H-6a), 3.72 (dd, J=5.0, 11.8 Hz, 1H, H-6b), 3.47–3.33 (m, 3H, H-2, H-3, H-4), 3.23 (ddd, J=2.6, 5.0, 9.0 Hz, 1H, H-5), 1.16–1.07 (m, 21 H, TIPS), 1.98 (s, AcOH); 13C (100 MHz, MeOD) δ 156.9 (Troc CO), 97.7 (C-1), 97.0 (Troc CCl3), 77.7 (C-5), 75.8 (Troc CH2), 75.7 (C-3), 72.3 (C-4), 62.8 (C-6), 61.5 (C-2), 18.4 (TIPS), 18.4 (TIPS), 13.5 (TIPS). HRMS (ESI) calcd for C18 H34Cl3NO7Si [M+Na]+ 532.1068, found 532.1059.
3.2.3. 2,2,2-trichloroethyl((2R,4aR,6S,7R,8R,8aS)-8-hydroxy-2-phenyl-6-((triisopropylsilyl)oxy)hexahydropyrano[3,2-d][1,3]dioxin-7-yl)carbamate (3)
To a solution of 7 (1.0 eq, 1.5 g, 2.9 mmol) in CH3CN (30 mL) was added benzaldehyde dime-thyl acetal (2.0 eq, 0.88 mL, 5.9 mmol). The reaction pH was adjusted between 2–4 using DL-10-camphorsulfonic acid and the reaction heated to 60oC. The reaction was stirred 2 H then was neutralized with Et3N, diluted with EtOAc (70 mL), washed with water (2 × 30 mL), brine (1 × 30 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (6:1 hexanes/EtOAc) to yield benzylidene acetal 3 (1.6 g, 3.0 mmol, 91%) as a white foam: Rf 0.60 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.50–7.48 (m, 2H, aromatic), 7.40–7.36 (m, 3H, aromatic), 5.55 (s, 1H, benzylidene CH), 5.15 (d, J=5.9 Hz, 1H, NH), 4.92 (d, J=7.5 Hz, 1H, H-1) 4.74 (d, J=12.0 Hz, 1H, Troc CH), 4.66 (d, J=11.9 Hz, 1H, Troc CH), 4.29 (dd, J=10.5, 4.9 Hz, 1H, H-6a), 4.06–4.02 (m, 1H, H-3), 3.78 (t, J=10.2 Hz, 1H, H-6b), 3.58 (t, J=9.2 Hz, 1H, H-4), 3.49–3.36 (m, 2H, H-5, H-2), 1.15–1.03 (m, 21H, TIPS); 13C (100 MHz, CDCl3) δ 154.6 (Troc CO), 137.2 (aromatic), 129.5 (aromatic), 128.5 (aromatic), 126.5 (aromatic), 102.1 (benzylidene CH), 96.2 (C-1), 95.3 (Troc CCl3), 81.7 (C-4), 75.0 (Troc CH2), 71.0 (C-3), 68.7 (C-6), 66.3 (C-5), 61.3 (C-2), 17.9 (TIPS), 12.3 (TIPS). HRMS (ESI) calcd C25H38Cl3NO7Si [M+Na]+ 620.1381, found 620.1367.
3.2.4. (2R,3S,4S,5R,6R)-2-(acetoxymethyl)-6-(((2R,4aR,6S,7R,8R,8aS)-2-phenyl-7-(((2,2,2-trichloroethoxy)carbonyl)amino)-6-((triisopropylsilyl)oxy)hexahydropyrano[3,2-d][1,3]dioxin-8-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (8)
Donor 2 (1.4 eq, 1.46 g, 3.0 mmol) and acceptor 3 (1.0 eq, 1.27 g, 2.1 mmol) were coevaporated with benzene (2 × 8 mL) and placed in a vacuum desiccator containing P2O5 overnight. The donor/acceptor mixture was dissolved in CH2Cl2 (14 mL) and the resulting solution was cannulated into a reaction flask containing 4Å powdered molecular sieves (2.7 g). The mixture was stirred under argon 1 H then cooled to −10oC and TMSOTf (0.1 eq, 0.038 mL in 0.2 mL CH2Cl2) was added. The reaction was stirred 10 minutes then quenched with Et3N. The reaction was diluted with CH2Cl2, filtered through celite, dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (2:1 hexanes/EtOAc) to yield diasaccharide 8 (1.59 g, 1.71 mmol, 81%) as a white foam: Rf 0.35 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.49–7.45 (m, 2H, aromatic), 7.40–7.34 (m, 3H, aromatic), 5.54 (s, 1H, benzylidene CH), 5.29 (d, J=2.8 Hz, 1H, H-4’), 5.19 (dd, J=8.4, 10.4 Hz, 1H, H-2’), 5.18 (d, J=8.0 Hz, NH), 5.11 (d, J=7.8 Hz, H-1), 4.92 (dd, J=3.4, 10.4 Hz, 1H, H-3’), 4.69 (s, 2H, Troc CH, Troc CH), 4.68 (d, J=8.6 Hz, H-1’), 4.32 (t, J=9.2 Hz, H-3), 4.27 (dd, J=5.0, 10.6 Hz, 1H, H-6a), 4.11 (q, EtOAc), 4.06 (dd, J=8.4, 10.6 Hz, 1H, H-6’a), 3.83 (dd, J=5.4, 10.7 Hz, 1H, H-6’b), 3.78 (t, J=10.4 Hz, H-6b), 3.72 (t, J=9.2 Hz, 1H, H-4), 3.64 (t, J=6.7 Hz, 1H, H-5’), 3.50–3.44 (ddd, J=4.9, 9.7, 9.7 Hz, 1H, H-5), 3.28 (q, J=8.0 Hz, 1H, H-2), 2.11 (s, 3H, COCH3), 2.04 (s, EtOAc), 2.03 (s, 3H, COCH3), 1.95 (s, 3H, COCH3), 1.93 (s, 3H, COCH3), 1.62 (s, H2O), 1.25 (t, EtOAc), 1.12–1.00 (m, 21H, TIPS), 0.06 (s, silicone grease); 13C (100 MHz, CDCl3) δ 170.4 (COCH3), 170.3 (COCH3), 170.2 (COCH3), 169.6 (COCH3), 153.9 (Troc CO), 137.2 (aromatic), 129.4 (aromatic), 128.5 (aromatic), 126.2 (aromatic), 101.5 (benzylidene CH), 101.0 (C-1’), 95.3 (C-1), 95.2 (Troc CCl3), 80.4 (C-4), 78.2 (C-3), 74.9 (Troc CH2), 71.1 (C-3’), 70.6 (C-5’), 69.4 (C-2’), 68.8 (C-6), 67.0 (C-4’), 66.2 (C-5), 61.0 (C-6’), 60.7 (C-2), 20.9 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3), 17.9 (TIPS), 17.9 (TIPS), 12.3 (TIPS). HRMS (ESI) calcd for C39H56Cl3NO16Si [M+Na]+ 950.2332, found 950.2311.
3.2.5. (3aR,5R,6S,7S,7aR)-5-(acetoxymethyl)-2-methyl-2-(((2R,4aR,6S,7R,8R,8aS)-2-phenyl-7-(((2,2,2-trichloroethoxy)carbonyl)amino)-6-((triisopropylsilyl)oxy)hexahydropyrano[3,2-d][1,3]dioxin-8-yl)oxy)tetrahydro-5H-[1,3]dioxolo[4,5-b]pyran-6,7-diyl diacetate (9)
Donor 2 (1.3 eq, 0.16 g, 0.33 mmol) and acceptor 3 (1.0 eq, 0.15 g, 0.25 mmol) were coevapo-rated with benzene (2 × 4 mL) and placed in a vacuum desicator containing P2O5 overnight. The donor/acceptor mixture was dissolved in CH3CN (1.25 mL) and the resulting solution was cannulated into a reaction flask containing 4Å powdered molecular sieves (0.4 g). The mixture was stirred under argon 1 H then cooled to −40oC and TMSOTf (1 drop) was added. The reaction was stirred 1 H then quenched with Et3N. The reaction was diluted with EtOAc (25 mL), filtered through celite, washed with water (3 × 10 mL), brine (1 × 10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromotography (2:1 hexanes/EtOAc) to yield orthoacetate 9 (0.115 g, 0.124 mmol, 50%) as a white foam: R 1f 0.48 (2:1 hexanes/EtOAc);1H NMR (400 MHz, CDCl3) δ 7.45–7.43 (m, 2H, aromatic), 7.35–7.33 (m, 3H, aromatic), 5.82 (d, J=4.8 Hz, 1H, H-1’), 5.51 (s, 1H, benzylidene CH), 5.33 (dd, J=1.5, 3.2 Hz, 1H, H-4’), 5.26 (d, J=8.7 Hz, 1H, NH), 4.94 (d, J=7.2 Hz, 1H, H-3’), 4.93 (d, J=7.1 Hz, 1H, H-1), 4.83 (d, J=11.9 Hz, 1H, Troc CH), 4.55 (d, J=12.0 Hz, 1H, Troc CH), 4.30–4.23 (m, 3H, H-6a, H-2’, H-5’), 4.11–4.05 (m, 3H, H-6’a, H-3, H-6’b), 3.76 (t, J=10.2 Hz, 1H, H-6b), 3.60 (t, J=9.3 Hz, 1H, H-4), 3.45–3.36 (m, 2H, H-5, H-2), 2.08 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.65 (orthoacetate CH3), 1.25 (m, grease), 0.06 (s, silcone grease), 1.11–1.02 (m, 21H, TIPS); 13C (100 MHz, CDCl3) δ 170.6 (COCH3), 170.1 (COCH3), 170.0 (COCH3), 154.0 (Troc CO), 137.2 (aromatic), 129.2 (aromatic), 128.3 (aromatic), 126.2 (aromatic), 120.5 (orthoacetate C), 101.7 (benzylidene CH), 98.4 (C-1’), 96.6 (C-1), 95.2 (Troc CCl3), 80.5 (C-4), 75.0 (Troc CH2), 72.5 (C-2’), 71.6 (C-3’), 71.6 (C-3), 69.0 (C-5’), 68.7 (C-6), 66.5 (C-5), 65.8 (C-4’), 61.6 (C-6’), 60.3 (C-2), 25.1 (orthoacetate CH3), 20.9 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 17.9 (TIPS), 17.8 (TIPS), 12.3 (TIPS). HRMS (TOF) calcd for C H39H56Cl3NO16Si [M+Na]+ 950.2326, found 950.2365.
3.2.6. (2R,3S,4S,5R,6R)-2-(acetoxymethyl)-6-(((2R,4aR,6R,7R,8R,8aS)-6-hydroxy-2-phenyl-7-(((2,2,2-trichloroethoxy)carbonyl)amino)hexahydropyrano[3,2-d][1,3]dioxin-8-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (10)
To a solution 8 (1.0 eq, 1.0 g, 1.1 mmol) in pyridine (11 mL) cooled to 0oC was added 70% HF in pyridine (5.4 mL) dropwise over 5 minutes. The solution was stirred 30 minutes then was allowed to warm to room temperature and stir an additional 4.5 H. The reaction was diluted with water (40 mL) and extracted with EtOAc (4 × 15 mL). The combined organics were quenched with solid NaHCO3, and saturated NaHCO3 solution, washed with 2 N HCl (3 × 20 mL), brine (1 × 20 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (2:3 hexanes/EtOAc) to yield anomeric hemiacetal 10 (0.64 g, 0.83 mmol, 77%) (α/β 2.2:1) as a white foam: Rf 0.38 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ (α anomer) 7.48–7.45 (m, 2H, aromatic), 7.38–7.35 (m, 3H, aromatic), 5.55 (s, 1H, benzylidene CH), 5.54 (s, 0.45 H, minor anomer), 5.30–5.25 (m, 2H, H-1, H-4’), 5.19 (dd, J=8.0, 10.4 Hz, minor anomer), 5.17 (dd, J=8.0, 10.4 Hz, H-2’), 4.96 (d, J=12.0 Hz, 1H, Troc CH), 4.92 (dd, J=3.4, 10.5 Hz, 1H, H-3’), 4.80 (d, J=11.9 Hz, 0.45 H, minor anomer), 4.71 (m, 0.45 H, minor anomer), 4.69 (d, J=8.0 Hz, 1H, H-1’), 4.59 (d, J=12.1 Hz, 1H, Troc CH), 4.43 (m, 0.45 H, minor anomer), 4.33 (dd, J=4.9, 10.5 Hz, 0.45 H, minor anomer), 4.24 (dd, J=4.8, 10.3 Hz, 1H, H-6a), 4.14–3.98 (m, 4H, H-5, H-2, H-3, H-6’a), 3.83–3.71 (m, 3H, H-6’b, H-6b, H-4), 3.57 (t, J=6.7 Hz, 1H, H-5’), 3.52 (d, J=3.2 Hz, 1H, OH), 3.53–3.47 (m, 0.9 H, minor anomer), 2.11 (s, 1.35 H, minor anomer), 2.10 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 1.97 (s, 1.35 H, minor anomer), 1.95 (s, 1.35 H, minor anomer), 1.95 (s, 3H, COCH3), 1.91 (s, 3H, COCH3), 1.75 (br s, 0.45 H, minor anomer OH), 1.25 (m, grease), 0.06 (s, silicone grease); 13C (100 MHz, CDCl3) d 170.5 (COCH3), 170.4 (minor anomer), 170.4 (COCH3), 170.3 (COCH3), 169.7 (COCH3), 154.2 (Troc CO), 137.2 (aromatic), 137.0 (minor anomer), 129.5 (minor anomer), 129.4 (aromatic), 128.5 (aromatic), 126.2 (minor anomer), 126.1 (aromatic), 126.1 (aromatic), 101.6 (minor anomer), 101.6 (benzylidene CH), 101.2 (C-1’), 95.5 (Troc CCl3), 95.3 (minor anomer), 92.5 (C-1), 80.8 (C-4), 77.3 (C-3), 75.0 (Troc CH2), 71.1 (C-3’), 71.0 (minor anomer), 70.8 (minor anomer), 70.6 (C-5’), 69.6 (C-2’), 69.4 (minor anomer), 69.0 (C-6), 68.7 (minor anomer), 66.9 (C-4’), 66.5 (minor anomer), 62.7 (C-5), 60.9 (C-6’), 54.8 (C-2), 20.8 (COCH3), 20.8 (COCH3), 20.7 (COCH3). HRMS (ESI) calcd for C30H36Cl3NO16 [M+Na]+ 794.0997, found 794.1010.
3.2.7. (2R,3S,4S,5R,6R)-2-(acetoxymethyl)-6-(((2R,4aR,6R,7R,8R,8aS)-2-phenyl-6-(2,2,2-trichloro-1-iminoethoxy)-7-(((2,2,2-trichloroethoxy)carbonyl)amino)hexahydropyrano[3,2-d][1,3]dioxin-8-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (11)
To a solution of 10 (1.0 eq, 0.35 g, 0.45 mmol) in CH2Cl2 (6.2 mL) cooled to 0oC was added Cl3CCN (10 eq, 0.45 mL, 4.5 mmol) followed by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (cat.). The reaction was stirred 1 H then was allowed to warm to room temperature and stir an additional 1 H. The reaction was concentrated and the crude residue purified via flash column chromatography (1:1 hexanes/ethyl acetate) to yield a-trichloroacetimidate 11 (0.36 g, 0.40 mmol, 89%) as a pale yellow powder: Rf 0.71 (1:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H, imidate NH), 7.52–7.45 (m, 2H, aromatic), 7.40–7.35 (m, 3H, aromatic), 6.40 (d, J=3.8 Hz, H-1), 5.60 (s, 1H, benzylidene CH), 5.32 (d, J=3.0 Hz, 1H, H-4’), 5.21 (dd, J=7.9, 10.4 Hz, H-2’), 5.12 (d, J=8.6 Hz, 1H, NH), 4.97 (dd, J=3.4, 10.5 Hz, H-3’), 4.94 (d, J=11.9 Hz, 1H, Troc CH), 4.80 (d, J=7.9 Hz, 1H, H-1’), 4.56 (d, J=12.0 Hz, 1H, Troc CH), 4.34 (dd, J=4.6, 10.3 Hz, 1H, H-6a), 4.25 (ddd, J=3.8, 10.2, 10.2 Hz, 1H, H-2), 4.12–4.04 (m, 2H, H-3, H-6’a), 3.98 (ddd, J=4.7, 9.7, 9.7 Hz, H-5), 3.92–3.84 (m, 2H, H-6’b, H-4), 3.82 (t, J=10.3 Hz, 1H, H-6b), 3.72 (t, J=6.7 Hz, H-5’), 2.12 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.96 (s, 3H, COCH3), 1.95 (s, 3H, COCH3), 1.56 (s, H O), 1.25 (m, grease); 13C (100 MHz, CDCl3) δ 170.4 (COCH3), 170.3 (COCH3), 169.6 (COCH3), 160.5 (imidate CNH), 154.1 (Troc CO), 136.9 (aromatic), 129.5 (aromatic), 128.5 (aromatic), 126.1 (aromatic), 101.6 (benzylidene CH), 100.9 (C-1’), 95.4 (Troc CCl3), 95.3 (C-1), 90.9 (imidate CCl3), 79.8 (C-4), 76.8 (C-3), 74.9 (Troc CH2), 71.0 (C-3’), 70.9 (C-5’), 69.8 (C-2’), 68.6 (C-6), 66.8 (C-4’), 65.2 (C-5), 61.0 (C-6’), 54.4 (C-2), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3). HRMS (ESI) calcd for C32H36Cl6N2O16 [M+Na]+ 937.0094, found 937.0067.
3.2.8. (2R,3S,4S,5R,6R)-2-(acetoxymethyl)-6-(((2R,4aR,6R,7R,8R,8aS)-6-hydroxy-2-phenyl-7-(((2,2,2trichloroethoxy)carbonyl)amino)hexahydropyrano[3,2-d][1,3]dioxin-8-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (13)
A solution of 8 (1.0 eq, 1.45 g, 1.56 mmol) in 80% aqueous acetic acid (15 mL) was heated to 80oC and stirred 5 H and 15 minutes. The reaction was diluted with water (40 mL) and extracted with EtOAc (4 × 15 mL) while solid NaHCO3 was added. The combined organics were washed with saturated NaHCO3 solution (4 × 20 mL), brine (1 × 20 mL), dried (MgSO4), filtered, and concentrated in vacuo to yield a white solid. The crude diol was dissolved in pyridine (16 mL) and acetic anhydride (2.5 eq, 0.37 mL, 3.9 mmol) and DMAP (cat.) were added. The reaction was stirred 1 H and 15 minutes then diluted with EtOAc (50 mL), washed with 2 N HCl (4 × 20 mL), brine (1 × 20 mL), dried (MgSO4), filtered, and concentrated in vacuo to yield a white foam. The crude diacetate was dissolved in pyridine (15 mL) and the resulting solution cooled to 0oC. 70% HF in pyridine (8 mL) was added dropwise over 5 minutes. The reaction was stirred 1 H then allowed to warm to room temperature and stir an additional 5 H. The reaction was diluted with water (40 mL) and extracted with EtOAc (4 × 15 mL) while solid NaHCO3 was added. The combined organics were quenched with saturated NaHCO3 solution, washed with 2 N HCl (4 × 20 mL), brine (1 × 20 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (2:3 hexanes/ethyl acetate) to yield anomeric hemiacetal 13 (0.94 g, 1.2 mmol, 78% over 3 steps) (α/β 8:1) as a white foam: Rf 0.20 (1:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ (α anomer) 5.36 (m, 1H, NH), 5.35 (d, J=3.2 Hz, 1H, H-4’), 5.25 (t, J=2.9 Hz, 1H, H-1), 5.05 (dd, J=7.8, 10.4 Hz, 1H, H-2’), 4.99 (t, J=8.8 Hz, H-4), 4.92 (dd, J=3.4, 10.5 Hz, 1H, H-3’), 4.74 (s, 2H, Troc CH, Troc CH), 4.65 (d, J=7.8 Hz, H-1’), 4.24–4.15 (m, 4H, H-6’a, H-5, H-6a, H-6b), 4.07–4.00 (m, 3H, H-3, H-6’b, H-2), 3.90 (dd, J=7.0, 6.7 Hz, 1H, H-5’), 3.35 (d, J=3.0, 1H, OH), 2.14 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 1.95 (s, 3H, COCH3); 13C (100 MHz, CDCl3) δ (α anomer) 171.1 (COCH3), 170.6 (COCH3), 170.3 (COCH3), 170.3 (COCH3), 169.8 (COCH3), 169.5 (COCH3), 154.1 (Troc CO), 100.7 (C-1’), 95.3 (Troc CCl3), 92.1 (C-1), 75.4 (C-3), 74.9 (Troc CH2), 70.9 (C-3’), 70.6 (C-5’), 69.1 (C-2’), 68.8 (C-4), 67.9 (C-5), 67.0 (C-4’), 62.5 (C-6), 61.0 (C-6’), 55.2 (C-2), 21.0 (COCH3), 20.9 (COCH3), 20.9 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 20.7 (COCH3). HRMS (ESI) calcd for C27H36Cl3NO18 [M+Na]+ 790.0896, found 790.0905.
3.2.9. (2R,3R,4S,5S,6R)-2-(((2R,3S,4R,5R,6R)-3-acetoxy-2-(acetoxymethyl)-6-(2,2,2-trichloro-1-iminoethoxy)-5-(((2,2,2-trichloroethoxy)carbonyl)amino)tetrahydro-2H-pyran-4-yl)oxy)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (14)
To a solution of 13 (1.0 eq, 0.30 g, 0.39 mmol) in CH2Cl2 (5.6 mL) was added potassium carbonate (3.0 eq, 1.6 g, 1.2 mmol) and trichloroacetonitirle (10 eq, 0.39 mL, 3.9 mmol) sequentially. The reaction was stirred 22 H then diluted with CH2Cl2, filtered through celite, and concentrated in vacuo. The crude residue was purified via flash column chromatography (1:1 hexanes/ethyl acetate) to yield atrichloroacetimidate 14 (0.27 g, 0.30 mmol, 77%) as a white foam: Rf 0.38 (1:1 hexanes/ethyl acetate); 1H (400 MHz, CDCl3) d 8.83 (s, 1H, imidate NH), 6.31 (d, J=3.7 Hz, 1H, H-1), 5.37 (dd, J=0.8, 3.5 Hz, 1H, H-4’), 5.14–5.05 (m, 3H, H-2’, H-4, NH), 4.96 (dd, J=3.4, 10.4 Hz, 1H, H-3’), 4.78 (d, J=12.0 Hz, 1H, Troc CH), 4.70 (d, J=7.5 Hz, 1H, H-1’), 4.69 (d, 12.2 Hz, 1H, Troc CH), 4.29 (td, J=3.8, 10.1, 10.1 Hz, 1H, H-2), 4.23–4.17 (m, 2H, H-6’a, H-6a), 4.14–4.02 (m, 4H, H-6b, H-5, H-3, H-6’b), 3.96 (t, J=6.7 Hz, 1H, H-5’), 2.15 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 1.96 (s, 3H, COCH), 1.66 (s, H2O), 1.25 (m, grease), 0.06 (s, silicon grease); C (100 MHz, CDCl3) δ 170.9 (COCH3), 170.5 (COCH3), 170.2 (COCH3), 170.2 (COCH3), 169.5 (COCH3), 169.5 (COCH3), 160.4 (imidate CNH), 154.0 (Troc CO), 100.6 (C-1’), 95.2 (Troc CCl3), 90.9 (imidate CCl3), 75.8 (C-3), 75.0 (Troc CH2), 70.9 (C-5’), 70.8 (C-3’), 70.3 (C-5), 69.3 (C-2’), 68.0 (C-4), 67.0 (C-4’), 61.8 (C-6), 61.2 (C-6’), 54.7 (C-2), 20.9 (COCH3), 20.9 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 20.7 (COCH3). HRMS (ESI) calcd for C29H36Cl6N2O18 [M+Na]+ 932.9992, found 932.9962.
3.2.10. (2R,3R,4S,5R,6S)-5-(benzyloxy)-2-((benzyloxy)methyl)-4-hydroxy-6-(((2R,3R,4S,5R,6R)-4,5,6-tris(benzyloxy)-2-((benzyloxy)methyl)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-3-yl acetate (16)48,49
To a solution of 4 (1.0 eq, 0.9 g, 1.0 mmol) in CH3CN (10 mL) was added trimethylorthoacetate (3.0 eq, 0.38 mL, 3.0 mmol) and p-toluene sulfonic acid (0.1 eq, 0.02 g, 0.1 mmol). The reaction was stirred 25 minutes then 90% trifluroacetic acid (0.36 mL) was added. The resulting solution was stirred 20 minutes then diluted with water (15 mL) and extracted with EtOAc (4 × 5 mL). The combined organics were washed with a saturated NaHCO3 solution (2 × 7 mL), brine (1 × 7 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (2:1 hexanes/ethyl acetate) to yield axial acetate 16 (0.87 g, 0.94 mmol, 93%) as a white foam: Rf 0.32 (2:1 hexanes/ethyl acetate); 1H (600 MHz, CDCl3) δ 7.34–7.13 (m, 30H, aromatic), 5.30 (d, J=3.2 Hz, 1H, H-4’), 4.93 (d, J=10.5 Hz, 1H, PhCH), 4.91 (d, J=11.9 Hz, 1H, PhCH), 4.87 (d, J=10.9 Hz, 1H, PhCH), 4.76 (d, J=11.4 Hz, 1H, PhCH), 4.71 (d, J=10.6 Hz, 1H, PhCH), 4.69 (d, J=10.9 Hz, 1H, PhCH), 4.63 (d, J=11.3 Hz, 1H, PhCH), 4.62 (d, J=12.1 Hz, 1H, PhCH), 4.59 (d, J=12.1 Hz, 1H, PhCH), 4.45 (d, J=7.8 Hz, 1H, H-1), 4.44 (d, J=7.7 Hz, 1H, H-1’), 4.42 (d, J=11.4 Hz, 1H, PhCH), 4.41 (d, J=12.1 Hz, 1H, PhCH), 4.20 (d, J=12.0 Hz, 1H, PhCH), 3.99 (t, J=9.2 Hz, 1H, H-4), 3.77 (dd, J=4.0, 11.0 Hz, 1H, H-6a), 3.71 (dd, J=1.2, 10.7 Hz, H-6b), 3.60 (dd, J=3.4, 9.6 Hz, 1H, H-3’), 3.53 (t, J=9.1 Hz, 1H, H-3), 3.48 (t, J=6.6 Hz, 1H, H-5’), 3.45 (dd, J=7.9, 8.9 Hz, 1H, H-2), 3.36 (dd, J=7.9, 9.4 Hz, 1H, H-2’), 3.33 (ddd, J=1.6, 3.8, 9.5 Hz, 1H, H-5), 3.30 (d, J=6.7 Hz, 2H, H-6’a, H-6’b), 2.20 (br s, 1H, OH), 1.99 (s, 3H, COCH3), 1.53 (s, H2O); 13C (100 MHz, CDCl3) δ 171.1 (COCH3), 139.2 (aromatic), 138.7 (aromatic), 128.4 (aromatic), 138.3 (aromatic), 138.1 (aromatic), 137.6 (aromatic), 128.6 (aromatic), 128.5 (aromatic), 128.5 (aromatic), 128.4 (aromatic), 128.2 (aromatic), 128.1 (aromatic), 128.0 (aromatic), 128.0 (aromatic), 127.9 (aromatic), 127.8 (aromatic), 127.8 (aromatic), 127.8 (aromatic), 127.7 (aromatic), 127.5 (aromatic), 102.7 (C-1), 102.5 (C-1’), 82.9 (C-3), 81.9 (C-2), 80.3 (C-2’), 76.5 (C-4), 75.4 (PhCH2), 75.2 (PhCH2), 75.2 (C-5), 73.6 (PhCH2), 73.4 (PhCH2), 72.6 (C-3’), 72.1 (C-5’), 71.1 (PhCH2), 69.7 (C-4’), 68.3 (C-6), 67.4 (C-6’), 20.9 (COCH3). LRMS calcd for C56H60 O12 [M+NH4]+ 942.44, found 942.56.
3.2.11. (2R,3R,4S,5S,6R)-2-(((2R,3S,4R,5R,6S)-3-acetoxy-6-(((2R,3S,4S,5R,6S)-3-acetoxy-5-(benzyloxy)-2-((benzyloxy)methyl)-6-(((2R,3R,4S,5R,6R)-4,5,6-tris(benzyloxy)-2-((benzyloxy)methyl)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-4-yl)oxy)-2-(acetoxymethyl)-5-(((2,2,2-trichloroethoxy)carbonyl)amino)tetrahydro-2H-pyran-4-yl)oxy)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (17)
Donor 14 (1.1 eq, 0.55 g, 0.60 mmol) and acceptor 16 (1.0 eq, 0.50 g, 0.54 mmol) were coevaporated with benzene (2 × 6 mL) and placed in a vacuum desiccator containing P2O5 overnight. The donor/acceptor mixture was dissolved in CH2Cl2 (18 mL) and the resulting solution was cannulated into a reaction flask containing 4Å powdered molecular sieves (1.1 g). The mixture was stirred under argon 1 H then cooled to −10oC and TfOH (cat.) was added. The reaction was stirred 12 minutes then quenched with Et3N. The reaction was diluted with CH2Cl2, filtered through celite, dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified via flash column chromatography (1:1 hexanes/ethyl acetate) to yield tetrasaccharide 17 (0.85 g, 0.51 mol, 94%) as a white foam: Rf 0.35 (1:1 hexanes/ethyl acetate); 1H (400 MHz, CDCl3) δ 7.48–7.44 (m, 2H, aromatic), 7.38–7.25 (m, 25H, aromatic), 7.23–7.16 (m, 3H, aromatic), 5.42 (d, J=3.6 Hz, 1H, H-4’), 5.33 (d, J=3.0 Hz, H-4’’’), 5.01–4.82 (m, 7H, H-3’’’, H-2’’’, Troc CH, PhCH, PhCH, PhCH, PhCH), 4.77–4.69 (m, 5H, PhCH, PhCH, PhCH, H-1”, H-4”), 4.64 (d, J=12.3 Hz, 1H, PhCH), 4.61 (d, J=13.2 Hz, 1H, PhCH), 4.49–4.42 (m, 4H, PhCH, PhCH, H-1’, H-1), 4.32 (d, J=12.1 Hz, 1H, Troc CH), 4.28 (d, J=11.8 Hz, PhCH), 4.23–4.14 (m, 4H, H-6’’’a, H-1’’’, H-6”a, H-6”b), 4.10–4.03 (m, 3H, H-6’’’b, H-4, NH), 3.81–3.77 (m, 2H, H-5’’’, H-6a), 3.71–3.60 (m, 3H, H-5”, H-3’, H-6b), 3.58–3.52 (m, 3H, H-5’, H-2’, H-3), 3.49–3.42 (m, 3H, H-3”, H-2”, H-2), 3.36–3.31 (m, 3H, H-6a, H-6b, H-5), 2.13 (s, 3H, COCH3), 2.12 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 1.99 (s, 3H, COCH3), 1.96 (s, 3H, COCH3), 1.95 (s, 3H, COCH3), 1.64 (s, H2O), 1.26 (m, grease); 13C (100 MHz, CDCl3) δ 171.0 (COCH3), 170.5 (COCH3), 170.3 (COCH3), 170.3 (COCH3), 169.8 (COCH3), 169.3 (COCH3), 169.3 (COCH3), 153.9 (Troc CO), 139.3 (aromatic), 139.1 (aromatic), 138.7 (aromatic), 138.2 (aromatic), 138.1 (aromatic), 137.6 (aromatic), 128.9 (aromatic), 128.6 (aromatic), 128.5 (aromatic), 128.4 (aromatic), 128.3 (aromatic), 128.2 (aromatic), 128.2 (aromatic), 128.1 (aromatic), 128.0 (aromatic), 128.0 (aromatic), 127.9 (aromatic), 127.9 (aromatic), 127.8 (aromatic), 127.7 (aromatic), 127.5 (aromatic), 126.9 (aromatic), 102.6 (C-1), 102.0 (C-1’), 100.9 (C-1’’’), 100.7 (C-1”), 95.6 (Troc CCl3), 82.8 (C-3), 81.9 (C-2’), 81.7 (C-2), 77.2 (C-3”), 76.7 (C-3’), 76.0 (C-4), 75.5 (PhCH2), 75.2 (PhCH2), 75.1 (C-5), 74.6 (PhCH2), 74.3 (Troc CH2), 73.7 (PhCH2), 73.7 (PhCH2), 72.8 (C-5’), 71.7 (C-5”), 71.0 (PhCH2), 70.9 (C-3’’’), 70.5 (C-5’’’), 69.4 (C-4’), 69.4 (C-4”), 68.8 (C-2’’’), 68.1 (C-6), 68.0 (C-6’), 66.9 (C-4’’’), 62.5 (C-6”), 61.1 (C-6’’’), 57.5 (C-2”), 21.0 (COCH3), 21.0 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3). HRMS (ESI) calcd for C83H94Cl3NO29 [M+Na]+ 1696.4875, found 1696.4839.
3.2.12. N-((2S,3R,4R,5S,6R)-2-(((2R,3S,4S,5R,6S)-3,5-dihydroxy-2-(hydroxymethyl)-6-(((2R,3S,4S,5R,6R)-4,5,6-trihydroxy-2-(hydroxymethyl)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-
To a solution of 17 (1.0 eq, 0.10 g, 0.06 mmol) in THF (1.5 mL) was added acetic acid (1.0 mL) and acetic anhydride (0.5 mL) followed by 10% Zn/Pb couple solid (0.24 g).50,51 The resulting mixture was stirred 6 H and 15 min then was diluted with CH2Cl2 (20 mL), filtered through celite, washed with saturated NaHCO3 solution (3 × 7 mL), brine (1 × 7 mL), dried (MgSO4), filtered, concentrated in vacuo, and coevaprotated with toluene to yield a white foam. The crude acetamide was suspended in MeOH (2 mL) and a concentrated NaOMe solution was added. The reaction was stirred 2 H then neutralized with Dowex 50Wx8, filtered, and concentrated in vacuo to yield a white solid. The crude heptanol was suspended in MeOH (2 mL) and H2O (1 mL) and Pd(OH)2 was added (2.0 eq, 0.083 g, 0.119 mmol). The reaction was stirred under H2 for 3 days then was diluted with H2O and MeOH, filtered through celite, concentrated in vacuo, coevaporated with toluene, and lyophilized to yield lacto-N-tetraose (1) (0.027 g, 0.038 mmol, 64% over 3 steps) as a white solid: [α]20D +13.5 (c 0.26, DMSO); 1H and 13C spectroscopy data were in accordance with literature data.52 HRMS (TOF) calcd for C26H45NO21 [M+Na]+ 730.2382, found 730.3188.
Supplementary Material
Figure 2.
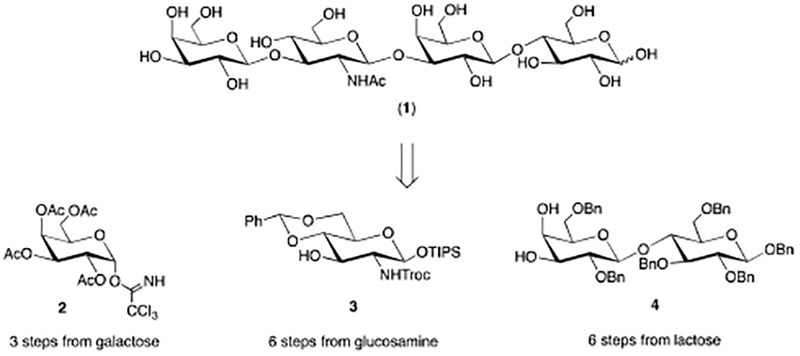
Retrosynthetic analysis of 1.
Scheme 1.
Synthesis of Glucosamine Acceptor 8.
Scheme 2.
Use of Lactose Acceptor 4 in Lacto-N-Tetraose Glycosylation.
Scheme 3.
Synthesis of Lacto-N-Tetraose (1).
Highlights.
MALDI FT ICR analysis of human milk
Gram Scale synthesis of protected Lacto-N-Tetraose (LNT)
Interesting trapping of acyloxonium ion, resulting from participation of a C2 acetate during a glycosylation event
Acknowledgements
The authors would like to acknowledge Vanderbilt University and the Institute of Chemical Biology for financial support. KMC acknowledges support from the Vanderbilt Chemical Biology Interface (CBI) training program (T32 GM065086) and the Vanderbilt Pre3 (Preventing adverse Pregnancy outcomes & Prematurity) Initiative for a travel grant. Dr. Michelle Reyzer and Prof. Richard Caprioli are acknowledged for conducting HRMS analysis of human milk oligosaccharide samples. James C. Poland and Prof. John McLean are acknowledged for conducting compound HRMS. Jade A. Bing is acknowledged for her meaningful contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information
Experimental procedures and characterization data (including spectra for new compounds). This material is available free of charge via the internet at http://pubs.acs.org
References
- (1).German JB, Freeman SL, Lebrilla CB, Mills DA, Nestle Nutr. Workshop Ser. Pediatr. Program 62 (2008) 205–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Martin R, Langa S, Reviriego C, Jiminez E, Marin ML, Xaus J, Fernandez L, Rodriguez JM, J. Pediatr 143 (2003) 754–758. [DOI] [PubMed] [Google Scholar]
- (3).Heikkila MP, Saris PE, J. Appl. Microbiol 95 (2003) 471–478. [DOI] [PubMed] [Google Scholar]
- (4).Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, Mira A, Am. J. Clin. Nutr 96 (2012) 544–551. [DOI] [PubMed] [Google Scholar]
- (5).Bashiardes S, Thaiss CA, Elinav E, Cell Metab 23 (2016) 393–394. [DOI] [PubMed] [Google Scholar]
- (6).Praveen P, Jordan F, Priami C, Morine MJ, Microbiome 3 (2015) 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Meropol SB, Edwards A, Birth Defects Res. C Embryo Today 105 (2015) 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu H, Lau K, Thon V, Autran CA, Jantscher-Krenn E, Xue M, Li Y, Sugiarto G, Qu J, Mu S, Ding L, Bode L, Chen X, Angew. Chem 53 (2014) 6687–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Newburg DS, Grave G, Pediatr. Res 75 (2014) 675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Musilova S, Rada V, Vlkova E, Bunesova V, Benef. Microbes 5 (2014) 273–283. [DOI] [PubMed] [Google Scholar]
- (11).He Y, Liu S, Kling DE, Leone S, Lawlor NT, Huang Y, Feinberg SB, Hill DR, Newburg DS, Gut 65 (2014) 33–46. [DOI] [PubMed] [Google Scholar]
- (12).Newburg DS, Biochemistry (Mosc) 78 (2013) 771–785. [DOI] [PubMed] [Google Scholar]
- (13).Bode L, Glycobiology 22 (2012) 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Newburg DS, Ruiz-Palacios GM, Morrow AL, Annu. Rev. Nutr 25 (2005) 37–58. [DOI] [PubMed] [Google Scholar]
- (15).Ruiz-Moyano S, Totten SM, Garrido DA, Smilowitz JT, German JB, Lebrilla CB, A Mills DA, Appl. Environ. Microbiol 79 (2013) 6040–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Marcobal A, Barboza M, W Froehlich JW, Block DE, German JB, Lebrilla CB, Mills DA, J. Agric. Food. Chem 58 (2010) 5334–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bode L, Kuhn L, Kim HY, Hsiao L, Nissan C, Sinkala M, Kankasa C, Mwiya M, Thea DM, Aldrovandi GM, Am. J. Clin. Nutr 96 (2012) 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Morrow AL, Chen C, Cline A, Newburg DS, FASEB J 30 (2016) Supplement 673.10. [Google Scholar]
- (19).Stahl B, Thurl S, Henker J, Siegel M, Finke B, Sawatzki G, Adv. Exp. Med. Biol 501 (2001) 299–306. [DOI] [PubMed] [Google Scholar]
- (20).Wu S, Grimm R, German JB, Lebrilla CB, J. Proteome Res 10 (2011) 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ruhaak LR, Lebrilla CB, Adv. Nutr 3 (2012) 406S–414S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pfenninger A, Karas M, Finke B, Stahl B, J. Am. Soc. Mass Spectrom 13 (2002) 1341–1348. [DOI] [PubMed] [Google Scholar]
- (23).Xiao Z, Guo Y, Liu Y, Li L, Zhang Q, Wen L, Wang X, Kondengaden SM, Wu Z, Zhou J, Cao X, Li X, Ma C, Wang PG, J. Org. Chem 81 (2016) 5851–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Love KR, Seeberger PH, J. Org. Chem 70 (2005) 3168–3177. [DOI] [PubMed] [Google Scholar]
- (25).Roussel F, Takhi M, e Schmidt RR, J. Org. Chem 66 (2001) 8540–8548. [DOI] [PubMed] [Google Scholar]
- (26).Roussel F, Knerr L, Schmidt RR, Eur. J. Org. Chem 11 (2001) 2067–2073. [Google Scholar]
- (27).Knuhr P, Castro-Palomino J, Grathwohl M, Schmidt RR, Eur. J. Org. Chem 22 (2001) 4239–4246. [Google Scholar]
- (28).Han NS, Kim TJ, Park YC, Kim J, Seo JH, Biotechnol. Adv 30 (2012) 1268–1278. [DOI] [PubMed] [Google Scholar]
- (29).La Ferla B, Prosperi D, Lay L, Russo G, Panza L, Carbohydr. Res 337 (2002) 1333–1342. [DOI] [PubMed] [Google Scholar]
- (30).Jennum CA, Fenger TH, Bruun LM, Madsen R, Eur. J. Org. Chem 2014 (2014) 3232–3241 [Google Scholar]
- (31).Aly MRE, Ibrahim ESI, El Ashry ESH, Schmidt RR, Carbohydr. Res 316 (1999) 121–132. [DOI] [PubMed] [Google Scholar]
- (32).Hahm HS, Liang CF, Lai CH, Fair RJ, Schuhmacher F, Seeberger PH, J. Org. Chem 81 (2016) 5866–5877. [DOI] [PubMed] [Google Scholar]
- (33).Hsu Y, Lu XA, Zulueta MM, Tsai CM, Lin KI, Hung SC, Wong CH, J. Am. Chem. Soc 134 (2012) 4549–4552. [DOI] [PubMed] [Google Scholar]
- (34).Shengshu H, Hai Y, Xi C, Sci. China Chem 54 (2011) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sherman AA, Yudina ON, Mironov YV, Sukhova EV, Shashkov AS, Menshov VM, Nifantiev NE, Carbohydr. Res 336 (2001) 13–46. [DOI] [PubMed] [Google Scholar]
- (36).Yao W, Yan J, Chen X, Wang F, Cao H, Carbohydr. Res 401 (2015) 5–10. [DOI] [PubMed] [Google Scholar]
- (37).Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons GJ, Science 341 (2013) 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Andersen SM, Heuckendorff M, Jensen HH, Org. Lett 17 (2015) 944–947. [DOI] [PubMed] [Google Scholar]
- (39).Corey EJ, Venkateswarlu A, J. Am. Chem. Soc 94 (1972) 6190–6191. [Google Scholar]
- (40).Schmidt RR, Stumpp M, Liebigs Ann. Chem 1983 (1983) 1249–1256. [Google Scholar]
- (41).Jung KH, Hoch M, Schmidt RR, Liebigs Ann. Chem 1989 (1989) 1099–1106. [Google Scholar]
- (42).Allen JR, Danishefsky SJ, J. Am. Chem. Soc 121 (1999) 10875–10882. [Google Scholar]
- (43).Nigudkar SS, Demchenko AV, Chem. Sci 6 (2015) 2687–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Crich D, Sharma I, J. Org. Chem 75 (2010) 8383–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Crich D, de la Mora M, Vinod AU, J. Org. Chem 68 (2003) 8142–8148. [DOI] [PubMed] [Google Scholar]
- (46).Fraserreid B, Wu ZF, Andrews CW, Skowronski E, Bowen JP, J. Am. Chem. Soc 113 (1991) 1434–1435. [Google Scholar]
- (47).Jensen HH, Nordstrom LU, Bols M, J. Am. Chem. Soc 126 (2004) 9205–9213. [DOI] [PubMed] [Google Scholar]
- (48).Lay L, Manzoni L, Schmidt Carbohydr RR. Res 310 (1998) 157–171. [DOI] [PubMed] [Google Scholar]
- (49).Nicolaou KC, Bockovich NJ, Carcanague DR, J. Am. Chem. Soc 115 (1993) 8843–8844. [Google Scholar]
- (50).Dong Q, Anderson CE, Ciufolini MA, Tetrahedron Lett 36 (1995) 5681–5682. [Google Scholar]
- (51).Boger DL, Kim SH, Mori Y, Weng JH, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc 123 (2001)1862–1871. [DOI] [PubMed] [Google Scholar]
- (52).Strecker G, Wieruszeski JM, Michalski JC, Montreuil J Glycoconjugate J 6 (1989) 67–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.