

Abstract
Plazomicin is a next-generation, semisynthetic aminoglycoside antibiotic currently under development for the treatment of infections due to multidrug-resistant Enterobacteriaceae. The compound was designed by chemical modification of the natural product sisomicin to provide protection from common aminoglycoside modifying enzymes that chemically alter these drugs via N-acetylation, O-adenylylation, or O-phosphorylation. In this study, plazomicin was profiled against a panel of isogenic strains of Escherichia coli individually expressing twenty-one aminoglycoside resistance enzymes. Plazomicin retained antibacterial activity against 15 of the 17 modifying enzyme-expressing strains tested. Expression of only two of the modifying enzymes, aac(2′)-Ia and aph(2″)-IVa, decreased plazomicin potency. On the other hand, expression of 16S rRNA ribosomal methyltransferases results in a complete lack of plazomicin potency. In vitro enzymatic assessment confirmed that AAC(2′)-Ia and APH(2′′)-IVa (aminoglycoside acetyltransferase, AAC; aminoglycoside phosphotransferase, APH) were able to utilize plazomicin as a substrate. AAC(2′)-Ia and APH(2′′)-IVa are limited in their distribution to Providencia stuartii and Enterococci, respectively. These data demonstrate that plazomicin is not modified by a broad spectrum of common aminoglycoside modifying enzymes including those commonly found in Enterobacteriaceae. However, plazomicin is inactive in the presence of 16S rRNA ribosomal methyltransferases, which should be monitored in future surveillance programs.
Keywords: plazomicin, antibiotic resistance, aminoglycoside-modifying enzymes, Enterobacteriaceae
Graphical abstract
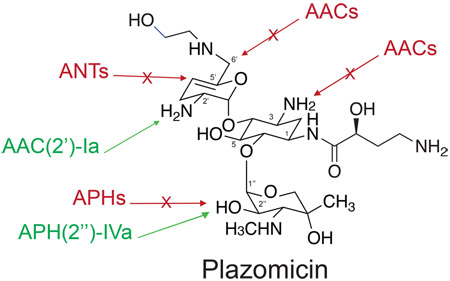
The need for new antibacterial agents that target multidrug resistant (MDR) Gram-negative pathogenic bacteria is critical.1 A well-established approach to address this demand involves the chemical modification of existing antibiotic scaffolds to circumvent resistance mechanisms.2 The aminoglycosides are a class of naturally produced antibiotics extensively used in the clinic for over 70 years. However, their utility has been eroded due to the availability of newer agents with similar antibacterial spectra as well as the emergence of aminoglycoside-modifying enzymes (AMEs), which chemically alter the antibiotic, reducing affinity for the bacterial ribosome.3
Enzyme-mediated chemical modification can occur via N-acetylation (aminoglycoside acetyltransferases, AACs), O-adenylylation (aminoglycoside nucleotidyltransferases, ANTs), and O-phosphorylation (aminoglycoside phosphotransferases, APHs). These enzymes have diverse regiospecificities that can modify many sites in the aminoglycoside molecule, thereby generating a daunting challenge to aminoglycoside use and new drug design.4–6 The emergence of plasmid-mediated AMEs spurred the development of the semisynthetic aminoglycosides amikacin, isepamicin, netilmicin, and arbekacin that avoid the activity of some AMEs.4 Inevitably, AMEs have emerged that mediate resistance to each of these aminoglycosides.6–8Finally, resistance mechanisms involving modification of the drug target, the bacterial ribosome, have also appeared in recent years. These enzymes are 16S rRNA methyltransferases (16S RMTases) that result in reduced affinity of aminoglycosides to the ribosomal target. Indeed, these enzymes mediate resistance to all 4,6-disubstituted aminoglycosides due to N7 methylation of G14059,10 (e.g., ArmA and RmtB) or to all 4,6- and 4,5-disubstituted aminoglycosides through N1 methylation of A140811–13 (e.g., NpmA) of the 16S rRNA, which corresponds to the aminoglycoside binding site.11
As the proportion of aminoglycoside resistant isolates increased in the clinic, approaches were taken to rationally develop an aminoglycoside that evades the action of AMEs but retains activity against Gram-negative pathogens.4 The result of such efforts led the development of the next-generation aminoglycoside plazomicin (previously known as ACHN-4904,14). The plazomicin scaffold is derived from the naturally produced aminoglycoside sisomicin15 (Figure 1), a molecule that does not possess 3′ and 4′-OH groups thereby protecting the drug from the common APH(3′) and ANT(4′) modifying enzymes that are responsible for resistance to the semisynthetic aminoglycoside amikacin.6,8 Through thoughtful design, plazomicin demonstrates promising activity against strains harboring a variety of aminoglycoside resistance enzymes.4,14 The antibiotic is thought to be recalcitrant to modification by the ANT(2′′), APH(2′′), and AAC(3) enzymes due to the addition of a 2(S)-hydroxy aminobutyryl substituent at N14,14 (Figure 1). Finally, plazomicin was rendered impervious to modification by the common AAC(6′)6 AME by the addition of a hydroxyethyl substituent at the 6′ position4,14 (Figure 1). These chemical modifications do not compromise antibacterial potency but instead allow for increased activity against isolates harboring aminoglycoside resistance enzymes.14 Despite studies exhibiting promising plazomicin activity toward aminoglycoside resistant isolates, a systematic study investigating activity in the presence of well-characterized individual resistance determinants has not been undertaken. Here, we perform a rigorous cell-based and in vitro biochemical study of plazomicin activity in the presence of a panel of aminoglycoside resistance enzymes individually expressed in an isogenic strain of Escherichia coli, allowing for the generation of a comprehensive plazomicin potency map.
Figure 1.
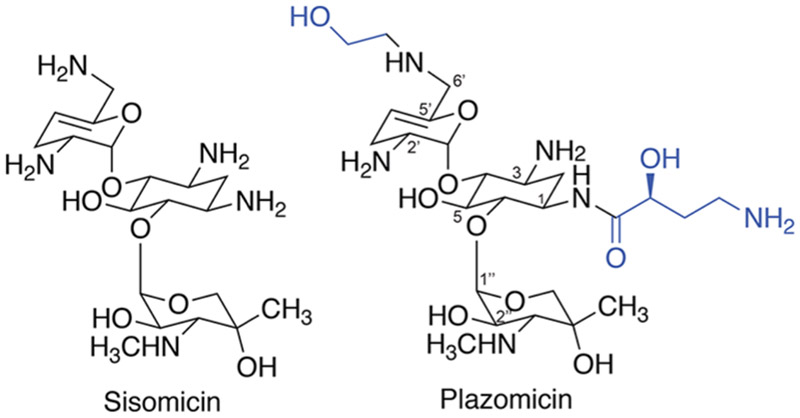
Chemical structures of sisomicin and plazomicin. The next-generation aminoglycoside plazomicin is derived from the naturally produced sisomicin scaffold by the addition of a N1 2(S)-hydroxy aminobutyryl and a hydroxyethyl substituent at the 6′ position (shown in blue).
■RESULTS
Antibacterial Activity of Plazomicin against a Panel of Isogenic E. coli Strains Expressing Aminoglycoside Resistance Enzymes.
Plazomicin susceptibility testing was performed utilizing strains derived from our recently developed antibiotic resistance platform (ARP).16 This platform is a cell-based array of individual resistance genes present in wild-type E. coli BW25113 and a hyper-permeable, efflux deficient mutant (E. coli BW25113 ΔbamB ΔtolC) of this strain that has increased sensitivity to many antibiotics.16 In this survey, gene expression levels were under control of the constitutive Pbla promoter, which results in high-level constitutive resistance to reference aminoglycosides (Table 1). To assess potency, plazomicin was tested against control strains containing only an empty vector (i.e., the wild-type E. coli BW25113 and the hyper-permeable, efflux deficient mutant), which revealed that plazomicin has a comparable potency to the reference aminoglycosides (gentamicin and kanamycin). Furthermore, plazomicin potency was comparable between wild-type E. coli BW25113 and E. coli BW25113 ΔbamB ΔtolC, suggesting that plazomicin is not significantly hindered by the outer membrane and is not a substrate for efflux in E. coli. Overall, plazomicin was profiled against twenty-one aminoglycoside resistance elements, comprising eight AACs, two ANTs, seven APHs, and four 16S RMTases. The majority of AMEs did not impact plazomicin potency, with the exception of AAC(2′)-Ia (16-fold increase in minimum inhibitory concentration, MIC) and APH(2′′)-IVa (4- to 8-fold increase in MIC) (Table 1). Expression of 16S RMTases that methylate either G1405 (ArmA, RmtB) or A1408 (NpmA, KamB) led to highly elevated plazomicin MICs (MICs ranging from 64 to >512 μg/mL; Table 1).
Table 1.
Plazomicin Susceptibility Testing of E. coli BW25113 and E. coli BW25113 ΔbamB ΔtolC Expressing Aminoglycoside Resistance Enzymesa
| aminoglycoside resistance gene |
E. coli BW25113 plazomicin MICs (μg/mL) |
E. coli BW25113 ΔbamB ΔtolC plazomicin MICs (μg/mL) |
E. coli BW25113 reference aminoglycoside MICs (μg/mL) |
E. coli BW25113 ΔbamB ΔtolC reference aminoglycoside MICs(μg/mL) |
|---|---|---|---|---|
| empty vector | 2 | 1 | 0.5 (GENT) | 0.25 (GENT) |
| 1 (KAN) | 1 (KAN) | |||
| 2 (STREP) | 2 (STREP) | |||
| 32 (HYGRO) | 16 (HYGRO) | |||
| 16 (SPEC) | 8 (SPEC) | |||
| aac(2′)-Ia | 32 | 16 | 4 (GENT) | 4 (GENT) |
| aac(3)-Ia | 2 | 1 | 128 (KAN) | 128 (KAN) |
| aac(3)-II | 4 | 2 | 128 (KAN) | 64 (KAN) |
| aac(3)-IV | 2 | 2 | 128 (KAN) | 128 (KAN) |
| aac(6′)-Ib | 2 | 1 | 128 (KAN) | 128 (KAN) |
| aac(6′)-Ib-cr | 2 | 2 | 32 (KAN) | 64 (KAN) |
| aac(6′)-Ie-aph(2′′)-Ia | 2 | 2 | 128 (KAN) | 128 (KAN |
| aac(6′)-Ii | 2 | 1 | 256 (KAN) | 128 (KAN) |
| ant(2′′)-Ia | 2 | 2 | 128 (KAN) | 128 (KAN) |
| ant(4′)-Ia | 2 | 2 | >256 (KAN) | >256 (KAN) |
| aph(2′′)-IIa (-Ib) | 2 | 1 | 8 (KAN) | 4 (KAN) |
| aph(2′′)-IVa (-Id) | 8 | 8 | 18 (KAN) | 8 (KAN) |
| aph(3′)-IIIa | 2 | 1 | >256 (KAN) | >256 (KAN) |
| aph(3′′)-Ia | 2 | 1 | 16 (STREP) | 16 (STREP) |
| aph(4)-Ia | 2 | 1 | >256 (HYGRO) | >256 (HYGRO) |
| aph(6)-Ia | 2 | 2 | 256 (STREP) | 256 (STREP) |
| aph(9)-Ia | 2 | 1 | >256 (SPEC) | >256 (SPEC) |
| armA | 256 | 64 | 128 (KAN) | 256 (KAN) |
| rmtB | >512 | >512 | >1024 (KAN) | >1024 (KAN) |
| npmA | 256 | 256 | >512 (KAN) | >512 (KAN) |
| kamB | 256 | 256 | 512 (KAN) | 512 (KAN) |
Control aminoglycoside MICs are included for reference (kanamycin, KAN; gentamicin, GENT; spectinomycin, SPEC; hygromycin, HYGRO; streptomycin, STREP).
In Vitro Enzymatic Assessment of Plazomicin Modification by Aminoglycoside Modifying Enzymes.
Following whole cell-based assessment of plazomicin activity, the antibiotic was next profiled against a panel of purified AMEs utilizing steady-state kinetic enzyme assays. Assays were performed with purified AAC(2′)-Ia, AAC(3′)-Ia, AAC(3)-II, AAC(3)-IV, AAC(6′)-Ii, AAC(6′)-Ie-APH(2′′)-Ia, APH(2′′)-IIa, APH(2′′)-IVa, APH(3′′)-Ia, APH(3′)-IIIa, APH(4)-Ia, APH(6)-Ia, APH(9)-Ia, and ANT(2′′)-Ia. Enzyme activity was confirmed by varying concentrations of reference substrate aminoglycosides and determination of steady-state kinetic parameters. The enzymes were then examined for their ability to utilize plazomicin (≥250 μM) as a substrate. Consistent with the susceptibility testing, both AAC(2′)-Ia and APH(2′′)-IVa (-Id) were able to modify plazomicin. The bifunctional AAC(6′)Ie-APH(2′′)-Ia AME that is found in some Grampositive cocci was unexpectedly able to modify plazomicin in the phosphotransferase mode; however, the presence of this gene does not confer significant resistance in MRSA, possibly reflecting differences in resistance gene expression between this model system engineered to provide high level resistance and clinical isolates and enzyme efficiency (see below)17 (Table 1). Full steady-state kinetic parameters were determined for all three enzymes with both plazomicin and comparator substrate aminoglycosides (Table 2). The remaining enzymes were not able to utilize plazomicin (250–300 ¼M) as a substrate under the conditions described in the Methods section. Finally, we hypothesized that plazomicin may act to inhibit the activity of AMEs, since the molecule does not possess the functional moieties for modification but may still interact with the enzyme active sites. Utilizing the same enzyme assays, plazomicin was profiled at 300 μM in the presence of reference aminoglycosides at their determined Km concentration. We detected no inhibition by plazomicin at 300 μM of the activity of any of the enzymes profiled demonstrating no observable affinity for AMEs even in nonproductive modification roles.
Table 2.
Steady-State Kinetic Parameters at 25°C of APH(2′′)-IVa, AAC(2′)-Ia, and AAC(6′)-Ie-APH(2′′)-Ia Activity
| enzyme | substrate | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| APH(2′′)-IVa | kanamycin B | 11 ± 3 | 0.27 ± 0.01 | 2.5 × 104 |
| APH(2′′)-IVa | plazomicin | 34 ± 5 | 0.20 ± 0.01 | 5.9 × 103 |
| AAC(2′)-Ia | gentamicin | 22 ± 8 | 0.12 ± 0.04 | 5.5 × 103 |
| AAC(2′)-Ia | plazomicin | 370 ± 90 | 0.9 ± 0.1 | 2.4 × 103 |
| AAC(6′)-Ie-APH(2′′)-Ia | amikacin | 25 ± 6 | 0.58 ± 0.04 | 2.3 × 104 |
| AAC(6′)-Ie-APH(2′′)-Ia | plazomicin | 69 ± 7 | 0.126 ± 0.004 | 1.8 × 103 |
Confirmation of Plazomicin Modification by AAC(2′)-Ia, APH(2′′)-IVa, and AAC(6′)-Ie-APH(2′′)-Ia.
To further test enzyme-mediated modification of the antibiotic, high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) was performed with plazomicin and reference aminoglycosides. Reactions were performed with the purified recombinant enzymes utilized in the kinetic assays. AAC(2′)-Ia acetylation (plazomicin mass increase of 42.0 Da) and phosphorylation (plazomicin mass increase of 79.9 Da) mediated by APH(2′′)-IVa and AAC(6′)-Ie-APH(2′′)-Ia was confirmed by HR-ESI-MS (Table 3). Of note, for AAC(6′)-Ie-APH(2′′)-Ia, the quantities of phosphorylated plazomicin (plazomicin-P) were significantly lower (3.5% turnover) than the reference phosphorylated aminoglycoside, indicating that the antibiotic is a poor substrate for the enzyme.
Table 3.
HR-ESI-MS Analysis of Acetylated (AAC(2′)-Ia) and Phosphorylated (APH(2′′)-IVa and AAC(6′)-Ie-APH(2′′)-Ia) Plazomicin in Positive Ion Mode
| aminoglycoside modifying enzyme |
molecular formula |
exact mass [M + H]+
m/z |
|
|---|---|---|---|
| calculated | observed | ||
| no enzyme control | C25H49N6O10+ | 593.3507 | 593.3520 |
| AAC(2′)-Ia | C27H51N6O11+ | 635.3610 | 635.3628 |
| AAC(6′)-Ie-APH(2′′)-Ia | C25H50N6O13P+ | 673.3168 | 673.3164 |
| APH(2′′)-IVa | C25H50N6O13P+ | 673.3168 | 673.3169 |
Addition of the 2(S)-hydroxy aminobutyryl substituent at the N1 of plazomicin (Figure 1) was speculated to avoid modification by APH(2′′) enzymes;4,14 however, in this study, we observed that APH(2′′)-IVa can utilize plazomicin as a substrate both in the cell-based assays and in vitro. In order to determine the site of phosphorylation mediated by APH(2′′)-IVa, a combination of one- and two-dimensional nuclear magnetic resonance spectroscopy (NMR) techniques was employed (Tables S1 and S2). Complete NMR assignments for both plazomicin and plazomicin-phosphate were performed. The overall appearance of the 1H spectra of plazomicin-phosphate was similar to that of plazomicin, with the exception of a few distinct chemical shift changes. One of the multiplets at 4.01–4.03 ppm, corresponding to the 2′′ proton in the plazomicin spectra, is deshielded in the phosphorylated product and appears at 4.35 ppm as a doublet. In the 13C NMR spectra of plazomicin-phosphate, the carbon chemical shift assigned for 2′′ is deshielded from 66.05 to 68.42 ppm. Moreover, the carbon resonance mentioned above, as well as the carbon resonance for the 1″, are both split by a 2-and 3-bond 31P spin coupling, with coupling constants of 4.23 and 8.47 Hz, respectively. A 31P–H decoupled experiment revealed a 31P chemical shift at 4.78 ppm. On the basis of this analysis, we can unequivocally assign the site of phosphorylation locating to the 2′′-OH.
Crystal Structures of the APH(2′′)-IVa–Plazomicin Complex and the AAC(2′)-Ia Enzyme.
To gain insight into the interaction between plazomicin and APH(2′′)-IVa, we determined the crystal structure of the antibiotic bound to the APH(2′′)-IVa enzyme (at a resolution of 1.51 A, see crystallographic statistics in Table S3). The crystals were isomorphous with our previous crystal structure of APH(2′′)-IVa in complex with kanamycin A18 and contained two chains in the asymmetric unit. Residual difference density was observed in the aminoglycoside binding site of one of the two chains corresponding to the plazomicin molecule (Figure 2A). Overall, the plazomicin molecule adopted an unexpected conformation than what was previously observed for the binding of aminoglycosides to this enzyme, and the binding pocket underwent some conformational changes. The plane bisecting the three rings of plazomicin was rotated relative to the binding position of kanamycin A (Figure 2B). Furthermore, aminoglycosides have been observed to pack against aromatic residues in the APH binding pocket; the rotated binding mode of plazomicin altered its packing against aromatic residues in the APH(2′′)-IVa active site. Plazomicin’s central desosasmine packed against the Trp271 and Trp287 side chains from the C-terminal region of the enzyme, which dramatically contrasted with the binding position of this ring in kanamycin A, which did not pack against these residues and instead was anchored through hydrogen bonds with Asp197, Ser199, and Glu239 (Figure 2B). Notably, the 1-N-2-hydroxybutyrlamine substituent of the central ring occupied the position of the double prime ring of kanamycin A. Instead, the double-prime ring of plazomicin was positioned to interact with the C-terminal region, packing against Trp287, Tyr284, and Tyr278, the latter two of which underwent a rotation of their side chain rotamers. The 2′′-OH and 3″-N of plazomicin interacted with Asn32 from the nucleotide positioning loop of the N-terminal domain of the APH, but the 2′′-OH was too far (8 Å) to interact with the catalytic base, Asp197. Finally, plazomicin’s prime ring did not pack against Trp271 as was observed for this ring of kanamycin A; instead, this ring was more solvent exposed and only formed an interaction between the (2-hydroxyethylamino)methyl substituent at the 6′ position. This structural analysis reveals that APH(2′′)-IVa is able to accommodate plazomicin by conformational rearrangement of active site residues.
Figure 2.
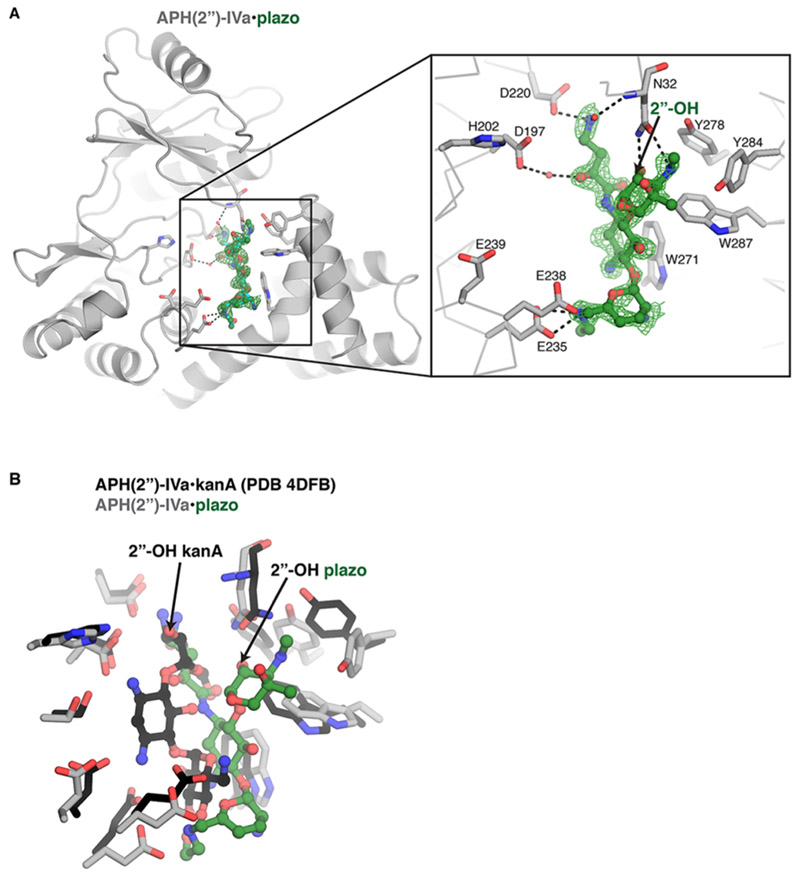
Crystal structure of APH(2′′)-IVa bound to plazomicin. (A) APH(2′′)-IVa, in gray; plazomicin (plazo), in green. Interacting residue side chains are highlighted and shown as sticks. The site of modification on plazomicin (2′′-OH group) is labeled. The electron density Fo–Fc map for plazomicin is contoured at 2.0 σ after refinement. (B) Overlay of APH(2′′)-IVa bound to plazo (colored green) and kanamycin A (kanA, colored black). The 2′′-OH of each molecule is labeled.
Despite extensive screening of crystallization conditions, we were not successful in crystallizing the AAC(2′)-Ia/plazomicin complex. However, we did determine the first crystal structure of this enzyme bound to gentamicin C1A (Table S3), which provides insight into how this enzyme can accommodate plazomicin. The AAC(2′)-Ia enzyme crystallized with 6 dimers in the asymmetric unit. We modeled four coenzyme A/acetylcoenzyme A molecules and 10 2′-N-acetylgentamicin molecules; one dimer contained a coenzyme A bound to each active site plus one 2′-N-acetylgentamicin molecule (Figure 3). The acetylated gentamicin was bound to the enzyme via only four hydrogen bonds: two from the C-terminal carboxylate of Trp178 to the 3-N of the drug; an interaction between Glu149 and the 1-N; an interaction between the backbone carbonyl of Ser114 and the 2′-N group. Stacking interactions from the side chain of Phe29 with the gentamicin prime ring also contributes to the interaction. Using this structure, we modeled plazomicin into the active site (Figure 3). From this modeling, the enzyme’s hydrogen bonds and the stacking interaction with gentamicin could be maintained when binding plazomicin, with the addition of a further two hydrogen bonds potentially forming between Asp176 and the hydroxyl group of the 2(S)-hydroxy aminobutyryl substituent at N1 and between Asp37 and the hydroxyl group of the hydroxyethyl substituent at the 6′-N. Experimental structure determination of this complex will validate this binding mode.
Figure 3.
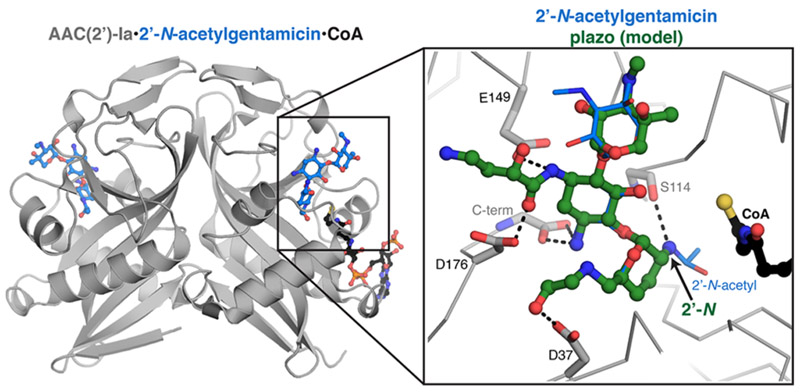
Crystal structure of AAC(2′)-Ia bound to acetylated gentamicin. AAC(2′)-Ia dimer, in gray; 2′-N-acetylgentamicin, in blue; coenzyme A (CoA), in black. The active site of one of the AAC(2′)-Ia molecules is highlighted, and plazomicin (plazo, colored green) has been modeled into the gentamicin binding site. Plazomicin interacting residue side chains are highlighted and shown as sticks.
■DISCUSSION
MDR Enterobacteriaceae represent an increasing global health threat, largely due to the production of extended-spectrum β-lactamases and carbapenemases that are frequently coexpressed with elements that confer resistance to other antibiotics such as aminoglycosides, macrolides, and fluoroquinolones.19 To combat this global health threat, alternative treatment options are required. Plazomicin is a next-generation aminoglycoside currently in development for the treatment of serious infections caused by MDR Enterobacteriaceae.
Although plazomicin has been shown to exhibit promising activity against clinical isolates harboring aminoglycoside resistance enzymes,14,19–21 a comprehensive study of plazomicin activity in the presence of individual resistance elements has not previously been reported. Since clinical isolates often harbor more than one resistance element,4 interpretation of antibiograms becomes complex. In this study, we profiled plazomicin against a highly curated panel of individual aminoglycoside resistance enzymes. As anticipated due to the observed ability of 16S RMTases to mediate resistance to all known 4,6-disubstituted aminoglycosides,9 the methyltransferases profiled in this study resulted in highly elevated plazomicin MICs via target protection. In contrast, plazomicin was unaffected by nearly all clinically relevant AMEs. Since plazomicin possesses a 2′-amino group, the antibiotic is not active against some isolates of Providencia stuartii14 that contain an inducible, chromosomally located AAC(2′)-Ia enzyme.22,23 Indeed, we confirm that this enzyme is capable of utilizing plazomicin as a substrate (Table 1–3). Furthermore, we determined the first crystal structure of AAC(2′)-Ia and modeled plazomicin into the active site. On the basis of our findings, it seems likely that the enzyme would be able to accommodate and modify the antibiotic. AAC(2′)-Ia plays a peptidoglycan housekeeping role in this species, which may explain why this gene has not been mobilized or detected in other species and is therefore unlikely to reduce the intended clinical efficacy of plazomicin.4,22,24
An unexpected outcome of this study was the ability of a subset of APH(2′′) enzymes to modify plazomicin. Crystal structure analysis of APH(2′′)-IVa and ANT(2′′)-Ia bound to kanamycin (PDB codes 4DFB and 4WQL, respectively) suggests that the N1 2(S)-hydroxy aminobutyryl modification of plazomicin would preclude modification by these enzymes, since the unprotected N1 of kanamycin is in close proximity to the enzymes. While ANT(2′′)-Ia and APH(2′′)-IIa (there is no available crystal structure for APH(2′′)-IIa) were not able to modify the antibiotic, we show that APH(2′′)-IVa (formerly designated APH(2′′)-Id)25 is capable of phosphorylating the 2′′-OH, with comparable steady-state kinetic parameters to kanamycin (Table 2). To gain insight into this unexpected result, we determined the crystal structure of the enzyme bound to plazomicin (Figure 2), which revealed that the antibiotic adopts a different conformation than that observed for the binding of other aminoglycosides. Indeed, this dramatically different binding position illustrates the versatility of this APH to accommodate and modify plazomicin. The 2′′-OH phosphorylation site of plazomicin is not present in the position normally adopted for modification by APH(2′′)-IVa (Figure 2B). For plazomicin modification to occur, conformational mobility of the relative positions of the N- and C-terminal domains, as well as flexibility of the nucleotide positioning loop,26,27 must play key roles in the versatility of this enzyme. Finally, the bifunctional AAC(6′)-Ie-APH(2′′)-Ia enzyme was also able to utilize plazomicin as a substrate in the phosphorylation enzyme assay (Table 2). Similar to APH(2′′)-IVa and ANT(2′′)-Ia, analysis of the APH(2′′)-Ia/kanamycin crystal structure (PDB code: 5IQB) suggests that plazomicin would be precluded from the aminoglycoside binding pocket. Indeed, antimicrobial potency reinforced this observation, and previous studies have reported that plazomicin remains active against Staphylococcus aureus harboring AAC(6′)-Ie-APH(2′′)-Ia, consistent with the relatively high Km (Table 3).20
Overall, since aph(2′′)-IVa distribution has thus far been limited to enterococci,28 it is unlikely that this enzyme will significantly impact plazomicin clinical utility. Plazomicin’s potency against Enterobacteriaceae, including those producing the majority of common AMEs, makes it a promising candidate for the intended treatment of infections caused by MDR Enterobacteriaceae.
■METHODS
Aminoglycoside Susceptibility Testing.
The MICs of aminoglycosides were determined in triplicate, in E. coli BW25113 (wild-type strain) and BW25113 ΔbamB ΔtolC16 expressing 21 different aminoglycoside resistance genes ligated into the pGDP316 vector. As a control, MICs were also determined for both strains carrying pGDP3 alone (no insert present). MICs were performed according to Clinical & Laboratory Standards Institute (CLSI) protocols29 in cation adjusted Mueller Hinton broth (CAMHB) with inoculum prepared using the colony resuspension method. Plates were incubated for 18 h at 37 °C in a stationary incubator.
Aminoglycoside Acetyltransferase Enzyme Assays.
Kinetic assays were performed with gentamicin and acetyl coenzyme A (AcCoA) as substrates, the thiol reagent 5,5-dithio-bis(2-nitrobenzoic acid) (DTNB) as an indicator, and five different AACs. All assays were carried out in 96-well plates with a final reaction volume of 100 μL and 25 mM MES (pH 6), 150 μM AcCoA, 500 μM DTNB, and varying concentrations of aminoglycoside. Assay components, except for AcCoA, were incubated for 10 min at room temperature. The reaction was initiated by the addition of AcCoA and followed for 10 min at 412 nm. Varying concentrations of gentamicin (4–62 μM) were used to determine the Km of each enzyme. The enzymes were then examined for their ability to use plazomicin as a substrate. Assays were conducted under the same conditions, substituting 300 μM gentamicin for plazomicin. If the enzyme utilized plazomicin as a substrate, the Km was determined (31–500 μM plazomicin). A final concentration of 0.5 μM AAC(2′)-Ia, a path length of 0.249 cm, and an extinction coefficient (TNB) of 14150 M−1 cm−1 were used. If plazomicin was not a substrate, the antibiotic was assessed for its ability to inhibit the enzyme using the same assay, with gentamicin at the respective Km and plazomicin at 300 μM. For all enzyme data analysis, Grafit 4 software (Erithacus Software, Staines, UK) was utilized.
Aminoglycoside Nucelotidyltransferase Enzyme Assay.
Adenylylation of aminoglycosides was monitored using the continuous EnzChek pyrophosphatase assay (Life Technologies). Reactions were carried out in 96-well plates, monitored at 360 nm, and consisted of 50 mM HEPES (pH 7.5), 40 mM KCl, 10 mM MgCl2, 5 μg of ANT(2′′)-Ia, and varying concentrations of aminoglycoside, in a final volume of 100 μL. Reactions were performed in triplicate and incubated at 25 °C for 10 min followed by initiation with ATP (300 μM final concentration); reactions were subsequently monitored for 10 min. Varying concentrations of kanamycin B (Sigma-Aldrich) were used to determine the Km of ANT(2′′)-Ia. The enzyme was then examined for its ability to use plazomicin as a substrate. Assays were conducted utilizing the same conditions, substituting kanamycin B for plazomicin (250 μM). Plazomicin was then assessed for its ability to inhibit ANT(2′′)-Ia using the same assay, with kanamycin B at Km and plazomicin at 300 μM
Aminoglycoside Phosphotransferase Enzyme Assay.
Phosphorylation of aminoglycosides (release of ADP) was detected using a coupled pyruvate kinase/lactate dehydrogenase assay, by monitoring the oxidization of NADH at 340 nm. All assays were carried out in 96-well plates in 50 mM HEPES (pH 7.5), 40 mM KCl, 10 mM MgCl2, 0.3 mM NADH, 3.5 mM phosphoenolpyruvate, 0.00125 units of PK/LDH, and varying concentrations of aminoglycoside in a final volume of 250 μL. Reactions were performed in triplicate and incubated at 25 °C for 10 min followed by initiation with nucleotide; reactions were subsequently monitored for 10 min. All enzymes were initiated with ATP (either 300 or 1000 μM depending upon the Km), with the exception of AAC(6′)-Ie-APH(2′′)-Ia that utilizes GTP (500 μM final concentration). The enzymes were examined for their ability to use plazomicin as a substrate. Assays were conducted utilizing the same conditions, substituting reference aminoglycosides for plazomicin (250 μM).
For determination of steady-state kinetic parameters of APH(2′′)-IVa (final concentration of 0.2 μM enzyme for kanamycin B and 0.9 μM for plazomicin) and AAC(6′)-Ie-APH(2′′)-Ia (final concentration of 0.34 μM enzyme for amikacin and 1.1 μM for plazomicin), varying concentrations of aminoglycoside were utilized to determine the Km of each enzyme. A path length of 0.621 cm and an extinction coefficient of 6220 M−1 cm−1 were used.
HR-ESI-MS and NMR Analysis of Enzyme Mediated Plazomicin Modification.
APH(2′′)-IVa catalyzed phosphorylated plazomicin was generated following incubation for 2 h at 37 °C (100% turnover). The reaction consisted of 50 mg of plazomicin, 138 mg of ATP, 5 mg of APH(2′′)-IVa, 25 mM HEPES (pH7.5), 40 mM KCl, and 10 mM MgCl2, in a final volume of 50 mL. The reaction was quenched with 50 mL of acetonitrile followed by centrifugation to remove precipitant. The supernatant was concentrated and reconstituted in H2O, followed by purification using AG50W-X8 strong cation exchange resin. The resin was pre-equilibrated with 1% NH4OH and washed with H2O until a neutral pH was achieved. Fractions containing phosphorylated plazomicin were identified with LC/ESI-MS using a QTRAP 2000 (Applied Biosystems) system equipped with an Agilent 1100 LC interface. A final purification step involved application of phosphorylated plazomicin to a C18 cartridge followed by analysis using NMR and HR-ESI-MS. HR-ESI-MS was acquired using an Agilent 1290 UPLC separation module and qTOF G6550A mass detector in positive ion mode. Liquid chromatography separation was performed using an Eclipse C18 (3.5 μm, 2.1 × 100 mm) column (Agilent Technologies) and the following pump method: at 0 min 95% solvent A (0.1% vol/vol of formic acid in water) and from 1 to 7 min up to 97% solvent B (0.1% vol/vol of formic acid in acetonitrile), at a flow rate of 0.4 mL/min. NMR data were acquired on a Bruker AVIII 700 MHz instrument in deuterated water as the solvent. Chemical shifts are reported in parts per million (ppm) relative to tetramethyl silane using the residual solvent signal as an internal signal.
For HR-ESI-MS analysis of modified aminoglycosides by AAC(2′)-Ia and AAC(6′)-Ie-APH(2′′)-Ia, 1 mL reactions were performed consisting of 25 mM HEPES (pH7.5), 40 mM KCl, and 10 mM MgCl2, 2 mg of plazomicin or kanamycin B, 0.5 mg of enzyme, and a final concentration of 3 mM ATP (AAC(6′)-Ie-APH(2′′)-Ia) or AcCoA (AAC(2′)-Ia). Reactions were incubated at 37 °C with shaking at 250 rpm for 6 h followed by the addition of 1 mL of MeOH to quench the reaction and analysis by HR-ESI-MS.
Crystallization of APH(2′′)-IVa in Complex with Plazomicin.
For crystallization, APH(2′′)-IVa was purified as previously described18 with a final buffer of 0.3 M NaCl, 10 mM HEPES (pH 7.5), and 0.5 mM TCEP. Crystals were grown using the sitting drop method, and the reservoir buffer was composed of 0.2 M NaCl, 10 mM HEPES (pH 7.5), 25% (w/v) PEG3350, and 10 mM plazomicin. Primarily long, thin crystals were formed, and these were determined to be mainly the apoenzyme in the primitive orthorhombic space group P212121; the APH(2′′)-IVa/plazomicin complex formed a rare, thicker crystal of the primitive monoclinic space group P21. After crystal growth, 10 mM plazomicin was added to the drop and incubated for 30 min before freezing using paratone oil and then transferred to an X-ray goniometer for data collection. X-ray diffraction data was collected at 100 K using a Rigaku FR-E Superbright anode and a Rigaku Saturn A200 CCD detector and then processed using XDS30 and Aimless.31 The structure was solved by Molecular Replacement using the APH(2′′)-IVa/kanamycin A complex18 and Phenix.phaser.32 Structure refinement was completed using Phenix.refine and Coot.33 The presence of one plazomicin molecule was validated using a difference omit map after positioning the protein molecules, and the occupancy of plazomicin was refined to a value of 0.90. B-factors were refined as isotropic, and TLS parametrization was included in the refinement. Average B-factor and bond angle/length RMSD values were calculated using Phenix. All geometry was verified using the Phenix, Coot, and wwPDB validation tools. The structure was deposited in the Protein Databank (PDB) with the accession number 6CD7.
Crystallization of AAC(2′)-Ia in Complex with 2′-N-Acetylgentamicin.
AAC(2′)-Ia was purified as previously described18 with a final buffer of 0.3 M NaCl, 10 mM HEPES (pH 7.5), and 0.5 mM TCEP. Crystals were grown using the sitting drop method; the reservoir buffer was composed of 0.1 M ammonium tartrate (pH 7), 12% (w/v) PEG3350, and 1 mM gentamicin. The crystal was cryoprotected using 8% glycerol, 8% ethylene glycol, and 8% sucrose. X-ray diffraction data was collected at 100 K at the Advanced Photon Source Life beamline 21-ID-G with a Marmosaic 300 mm CCD and processed using XDS and Aimless. The structure was solved by molecular replacement using the structure of AAC(2′)-Ic from Mycobacterium tuberculosis (PDB accession number: 1M44). Structure refinement was completed using Phenix.refine and Coot. The presence of ten 2′-N-acetylgentamicin, one acetyl-coenzyme A, and three coenzyme A molecules was evident in difference density maps after initial molecular replacement (MR) phasing. B-factors were refined as isotropic, and TLS parametrization was included in the refinement. Average B-factor and bond angle/length RMSD values were calculated using Phenix. All geometry was verified using the Phenix, Coot, and wwPDB validation tools. The structure was deposited in the PDB with the accession number 5US1.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by Achaogen, a Canadian Institutes of Health Research grant (FRN-148463), and a Canada Research Chair in Antibiotic Biochemistry (to G.D.W.). We thank Aiping Dong at the Structural Genomics Consortium, Toronto, and Zdzislaw Wawrzak at Life Sciences Collaborative Access Team, Advanced Photon Source, Argonne National Laboratory, for X-ray diffraction data collection. The crystal structure determination in this project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272201200026C and HHSN272201700060C.
ABBREVIATIONS
- MDR
multidrug resistant
- AMEs
aminoglycoside-modifying enzymes
- AACs
aminoglycoside acetyltransferases
- ANTs
aminoglycoside nucleotidyltransferases
- APHs
aminoglycoside phosphotransferases
- 16S
RMTases, 16S rRNA methyltransferases
- ARP
antibiotic resistance platform
- MIC
minimum inhibitory concentration
- HR-ESI-MS
high-resolution electrospray ionization mass spectrometry
- NMR
nuclear magnetic resonance spectroscopy
- CAMHB
cation adjusted Mueller Hinton broth
- AcCoA
acetyl coenzyme A
- DTNB
5,5-dithiobis(2-nitrobenzoic acid)
- PDB
Protein Databank
- MR
molecular replacement
- Plazo
plazomicin
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acsinfecdis.8b00001.
HRMS (ESI) of plazomicin and phosphorylated plazomicin; 1H-NMR spectra of plazomicin-2″-phosphate in D2O; 13C-DEPTQ-NMR spectra of plazomicin-2″-phosphate in D2O; 1D-31P-H decoupling NMR spectra of plazomicin-2″-phosphate in D2O; 1H chemical shifts for plazomicin and plazomicin-2″-phosphate; 13C chemical shifts for plazomicin and plazomicin-2″-phosphate; X-ray diffraction statistics (PDF)
Notes
The authors declare the following competing financial interest(s): A.W.S. and K.M.K. are employees and shareholders of Achaogen.
REFERENCES
- (1).Brown ED, and Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature 529 (7586), 336–343. [DOI] [PubMed] [Google Scholar]
- (2).Pawlowski AC, Johnson JW, and Wright GD (2016) Evolving medicinal chemistry strategies in antibiotic discovery. Curr. Opin. Biotechnol 42, 108–117. [DOI] [PubMed] [Google Scholar]
- (3).Garneau-Tsodikova S, and Labby KJ (2016) Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. MedChemComm 7 (1), 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Armstrong ES, and Miller GH (2010) Combating evolution with intelligent design: the neoglycoside ACHN-490. Curr. Opin. Microbiol 13 (5), 565–573. [DOI] [PubMed] [Google Scholar]
- (5).Bacot-Davis VR, Bassenden AV, and Berghuis AM (2016) Drug-target networks in aminoglycoside resistance: hierarchy of priority in structural drug design. MedChemComm 7 (1), 103–113. [Google Scholar]
- (6).Shaw KJ, Rather PN, Hare RS, and Miller GH (1993) Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev 57 (1), 138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wright GD (1999) Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol 2 (5), 499–503. [DOI] [PubMed] [Google Scholar]
- (8).Ramirez MS, and Tolmasky ME (2010) Aminoglycoside modifying enzymes. Drug Resist. Updates 13 (6), 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liou GF, Yoshizawa S, Courvalin P, and Galimand M (2006) Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens. J. Mol. Biol 359 (2), 358–364. [DOI] [PubMed] [Google Scholar]
- (10).Perichon B, Courvalin P, and Galimand M (2007) Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob. Agents Chemothe 51 (7), 2464–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Doi Y, Wachino JI, and Arakawa Y (2016) Aminoglycoside Resistance: The Emergence of Acquired 16S Ribosomal RNA Methyltransferases. Infect. Dis. Clin. North. Am 30 (2), 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Husain N, Obranic S, Koscinski L, Seetharaman J, Babic F, Bujnicki JM, Maravic-Vlahovicek G, and Sivaraman J (2011) Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res. 39 (5), 1903–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wachino J, Shibayama K, Kurokawa H, Kimura K, Yamane K, Suzuki S, Shibata N, Ike Y, and Arakawa Y (2007) Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob. Agents Chemother 51 (12), 4401–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Aggen JB, Armstrong ES, Goldblum AA, Dozzo P, Linsell MS, Gliedt MJ, Hildebrandt DJ, Feeney LA, Kubo A, Matias RD, Lopez S, Gomez M, Wlasichuk KB, Diokno R, Miller GH, and Moser HE (2010) Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob. Agents Chemother 54 (11), 4636–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Weinstein MJ, Marquez JA, Testa RT, Wagman GH, Oden EM, and Waitz JA (1970) Antibiotic 6640, a new Micromonospora-produced aminoglycoside antibiotic. J. Antibiot 23 (11), 551–554. [DOI] [PubMed] [Google Scholar]
- (16).Cox G, Sieron A, King AM, De Pascale G, Pawlowski AC, Koteva K, and Wright GD (2017) A Common Platform for Antibiotic Dereplication and Adjuvant Discovery. Cell chem. biol 24 (1), 98–109. [DOI] [PubMed] [Google Scholar]
- (17).López MC, Picazo JJ, Simaluiza JR, Rodríguez-Avial I, and Culebras E (April 25–28, 2015) Plazomicin has potent in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA) isolates carrying different aminoglycoside-modifying enzymes In ECCMID Conference Programme, Copenhagen, Denmark. [Google Scholar]
- (18).Shakya T, Stogios PJ, Waglechner N, Evdokimova E, Ejim L, Blanchard JE, McArthur AG, Savchenko A, and Wright GD (2011) A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol 18 (12), 1591–1601. [DOI] [PubMed] [Google Scholar]
- (19).Haidar G, Alkroud A, Cheng S, Churilla TM, Churilla BM, Shields RK, Doi Y, Clancy CJ, and Nguyen MH (2016) Association between the Presence of Aminoglycoside-Modifying Enzymes and In Vitro Activity of Gentamicin, Tobramycin, Amikacin, and Plazomicin against Klebsiella pneumoniae Carbapenemase- and Extended-Spectrum-beta-Lactamase-Producing Enterobacter Species. Antimicrob. Agents Chemother 60 (9), 5208–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lopez Diaz MC, Rios E, Rodriguez-Avial I, Simaluiza RJ, Picazo JJ, and Culebras E (2017) In-vitro activity of several antimicrobial agents against methicillin-resistant Staphylococcus aureus (MRSA) isolates expressing aminoglycoside-modifying enzymes: potency of plazomicin alone and in combination with other agents. Int. J. Antimicrob. Agents 50 (2), 191–196. [DOI] [PubMed] [Google Scholar]
- (21).Lopez-Diaz MD, Culebras E, Rodriguez-Avial I, Rios E, Vinuela-Prieto JM, Picazo JJ, and Rodriguez-Avial C (2017) Plazomicin Activity against 346 Extended-Spectrum-beta-Lactamase/AmpC-Producing Escherichia coli Urinary Isolates in Relation to Aminoglycoside-Modifying Enzymes. Antimicrob. Agents Chemother 61 (2), AAC.02454–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Macinga DR, and Rather PN (1999) The chromosomal 2’-N-acetyltransferase of Providencia stuartii: physiological functions and genetic regulation. Front. Biosci., Landmark Ed 4, D132–140. [DOI] [PubMed] [Google Scholar]
- (23).Swiatlo E, and Kocka FE (1987) Inducible expression of an aminoglycoside-acetylating enzyme in Providencia stuartii. J. Antimicrob. Chemother 19 (1), 27–30. [DOI] [PubMed] [Google Scholar]
- (24).Payie KG, Rather PN, and Clarke AJ (1995) Contribution of gentamicin 2’-N-acetyltransferase to the O-acetylation of peptidoglycan in Providencia stuartii. J. Bacteriol 177 (15), 4303–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kaplan E, Guichou JF, Chaloin L, Kunzelmann S, Leban N, Serpersu EH, and Lionne C (2016) Aminoglycoside binding and catalysis specificity of aminoglycoside 2’-phosphotransferase IVa: A thermodynamic, structural and kinetic study. Biochim. Biophys. Acta, Gen. Subj 1860 (4), 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shi K, Houston DR, and Berghuis AM (2011) Crystal structures of antibiotic-bound complexes of aminoglycoside 2’-phosphotransferase IVa highlight the diversity in substrate binding modes among aminoglycoside kinases. Biochemistry 50 (28), 6237–6244. [DOI] [PubMed] [Google Scholar]
- (27).Thompson PR, Boehr DD, Berghuis AM, and Wright GD (2002) Mechanism of aminoglycoside antibiotic kinase APH(3′)-IIIa: role of the nucleotide positioning loop. Biochemistry 41 (22), 7001–7007. [DOI] [PubMed] [Google Scholar]
- (28).Tsai SF, Zervos MJ, Clewell DB, Donabedian SM, Sahm DF, and Chow JW (1998) A new high-level gentamicin resistance gene, aph(2’)-Id, in Enterococcus spp. Antimicrob. Agents Chemother 42 (5), 1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).CLSI. (2015) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, Tenth ed., Document M07-A10, Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- (30).Kabsch W (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 67 (Pt 4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr, 60 (Pt 12), 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.