

Abstract
Kushneria konosiri X49T is a member of the Halomonadaceae family within the order Oceanospirillales and can be isolated from salt-fermented larval gizzard shad. The genome of K. konosiri X49T reported here provides a genetic basis for its halophilic character. Diverse genes were involved in salt-in and -out strategies enabling adaptation of X49T to hypersaline environments. Due to resistance to high salt concentrations, genome research of K. konosiri X49T will contribute to the improvement of environmental and biotechnological usage by enhancing understanding of the osmotic equilibrium in the cytoplasm. Its genome consists of 3,584,631 bp, with an average G + C content of 59.1%, and 3261 coding sequences, 12 rRNAs, 66 tRNAs, and 8 miscRNAs.
Keywords: Kushneria konosiri, Halomonadaceae, Halophile, Complete genome, Konosirus punctatus
Introduction
The gizzard shad, Konosirus punctatus, is a popular marine fish used as a food source in Northeast Asia and is usually consumed as a grilled dish, sushi, or jeotgal (or jeot). Jeotgal is a traditional Korean fermented food made by adding a substantial amount of solar salt to seafood such as fish, shrimp, or shellfish. During fermentation, jeotgal gains an extra flavor that may be caused by a microorganism derived from the environment, solar salt, or sea organisms. In an analysis of the microbiota of salt-fermented seafood, the strain X49T (= KACC 14623T = JCM 16805T) was isolated from the salt-fermented larval gizzard shad, known as Daemi-jeot in Korean [1]. Blast analysis and phylogenetic analysis using the 16S rRNA sequence revealed that the strain X49T belongs to the genus Kushneria.
The genus Kushneria was first proposed as a novel genus by Sanchez-Porro in 2009, on the basis of phylogenetic analyses of 16S and 23S rRNA gene sequences [2]. It comprises a group of related Gram-negative, aerobic, motile, and rod- or oval-shaped bacteria. Most Kushneria strains have been isolated from saline environments and possess hypersaline resistance [2–7]. Strain X49T also has a halophilic character [1]. At present, there are four sequenced Kushneria strains, but only the genome of Kushneria marisflavi KCCM 80003T has been reported [8]. Thus, the genomic analysis of strain X49T should help us to understand the genetic basis of adaptation to a hypersaline environment. The present study determined the classification and features of strain X49T, as well as its genome sequence and gene annotations.
Organism information
Classification and features
Serially diluted suspensions of Daemi-jeot were plated directly on Marine agar medium and maintained under aerobic condition at 25 ± 1 °C for 14 days. To obtain pure isolates, a single colony was repeatedly transferred to new agar plates. Comparison between the 16S rRNA gene sequence of strain X49T (Accession number: GU198748) and those obtained using NCBI BLASTN [9] with the settings for highly similar sequences produced 100 hits: 47, 36, 1, and 1 from the genera Kushneria, Halomonas, Halomonadaceae, and Chromohalobacter, respectively, and the remaining 15 from uncultured bacteria. The validated species with the maximum sequence similarity was K. marisflavi SW32T (NR_025094), which shared a sequence identity of 98.63%. Phylogenetic analysis using MEGA6 [10] based on 16S rRNA gene sequences of the Kushneria members and related taxa showed that strain X49T was within the cluster comprising the genus Kushneria (Fig. 1). Strain X49T is classified as Proteobacteria, Oceanospirillales, Halomonadaceae, and Kushneria, and is named Kushneria konosiri. Characteristics of K. konosiri X49T are presented in Table 1. The cells were aerobic, Gram-negative, rod- or oval-shaped, and 1.2–3.2 μm in length and 0.5–1.0 μm in width. A flagellum was observed (Fig. 2). The colonies were orange-colored and circular with entire margins on marine agar medium. Growth was observed at 10–37 °C, at pH 4.5–8.5, and in the presence of 0–26% (w/v) NaCl. The physiological characteristics, such as the growth substrates of K. konosiri X49T, have been described in detail previously [1].
Fig. 1.

Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences of K. konosiri X49T and closely related taxa. Numbers at nodes indicate bootstrap values (over 70%, 1000 replicates) for neighbor-joining, maximum-likelihood, and maximum-parsimony. Closed circles indicate the nodes that were also generated by maximum-likelihood and maximum-parsimony. Scale bar, 0.005 accumulated changes per nucleotide
Table 1.
Classification and general features of K. konosiri X49T according to the Minimum Information about a Genome Sequence (MIGS) recommendations
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [28] | |
| Phylum Proteobacteria | TAS [29] | ||
| Class Gammaproteobacteria | TAS [30] | ||
| Order Oceanospirillales | TAS [31] | ||
| Family Halomonadaceae | TAS [32] | ||
| Genus Kushneria | TAS [2] | ||
| Species Kushneria konosiri | TAS [1] | ||
| Type strain X49T (Accession GU198748) | TAS [1] | ||
| Gram stain | Negative | TAS [1] | |
| Cell shape | Rod, oval-shaped | TAS [1] | |
| Motility | Motile | TAS [1] | |
| Sporulation | Not reported | TAS [1] | |
| Temperature range | 10–37 °C | TAS [1] | |
| Optimum temperature | 15–25 °C | TAS [1] | |
| pH range | pH 4.5–8.5 | TAS [1] | |
| Optimum pH range | pH 5.0–7.0 | TAS [1] | |
| Carbon source | Heterotroph | TAS [1] | |
| MIGS-6 | Habitat | Fermented food | TAS [1] |
| MIGS-6.3 | Salinity | 0–26% NaCl (w/v) | TAS [1] |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [1] |
| MIGS-15 | Biotic relationship | Free-living | NAS |
| MIGS-14 | Pathogenicity | Not reported | |
| MIGS-4 | Geographic isolation | South Korea: Goheung | TAS [1] |
| MIGS-5 | Sample collection date | Apr-09 | |
| MIGS-4.1 | Latitude | Not reported | |
| MIGS-4.1 | Longitude | Not reported | |
| MIGS-4.3 | Depth | Not reported | |
| MIGS-4.4 | Altitude | Not reported |
The evidence codes are as follows. TAS: traceable author statement (i.e., a direct report exists in the literature). NAS: non-traceable author statement (i.e., not observed directly in a living, isolated sample, but based on a generally accepted property of the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [13]
Fig. 2.
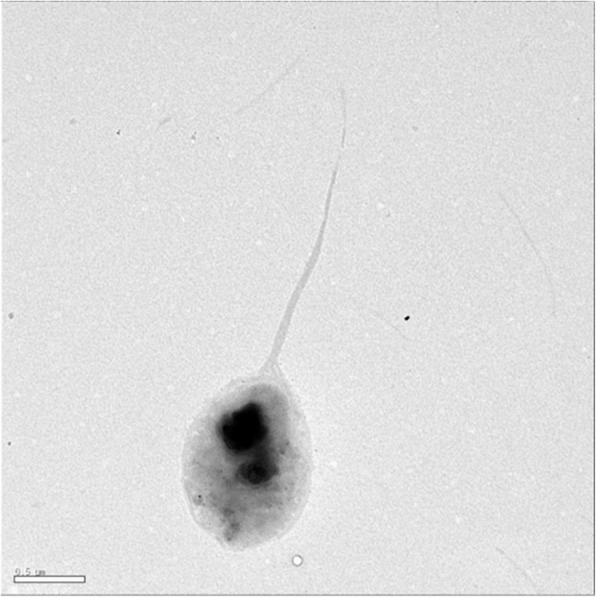
Transmission electron micrograph (TEM) of K. konosiri X49T. The TEM (JEM-1010; JEOL) image was obtained from a previous study [1]
Chemotaxonomic data
The predominant cellular fatty acids (> 10% of the total) in K. konosiri X49T were C16:0, C18:1 ω7c, Summed feature 3 (C16:1 ω7c and/or C16:1 ω6c), and C12:0 3OH. The respiratory quinone was ubiquinone Q9, and minor quinones were ubiquinone Q8 and Q10. The polar lipids contained diphosphatidylglycerol, phosphatidylglycerol, phosphatidylethanolamine, phosphatidylserine, two unidentified aminophospholipids, two unidentified phospholipids, and two unidentified lipids.
Genome sequencing information
Genome project history
K. konosiri X49T was selected for genome sequencing based on its environmental potential and this genome sequencing was part of the Agricultural Microbiome R&D Program (grant number: 914006–4) at the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry (IPET) funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA). The genome sequence was deposited in DDBJ/EMBL/GenBank under accession number CP021323, and the genome project was deposited in the GOLD [11] under Gp0223024. The sequencing and annotation were performed by Macrogen (Seoul, Korea). The details of the project information and the associations with MIGS [12] are shown in Table 2.
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | 20 kb SMRTbell library |
| MIGS-28.2 | Number of reads | 160,304 sequencing reads |
| MIGS-29 | Sequencing platforms | PacBio RSII platform |
| MIGS-31.2 | Fold coverage | × 224 |
| MIGS-30 | Assemblers | HGAP v.3 |
| MIGS-32 | Gene calling method | IMG annotation pipeline v.4.15.1. |
| Locus Tag | B9G99 | |
| GenBank ID | CP021323 | |
| GenBank Date of Release | 2017-05-24 | |
| GOLD ID | Gp0223024 | |
| BIOPROJECT | PRJNA383456 | |
| MIGS-13 | Source material identifier | KACC 14623T, JCM 16805T |
| Project relevance | Environmental |
Growth conditions and genomic DNA preparation
K. konosiri X49T (lab stored, = KACC 14623T = JCM 16805T) was cultured aerobically in LB broth (BD, USA) containing NaCl (4% w/v) at 30 ± 1 °C for 3 days. The genomic DNA of K. konosiri X49T was extracted using a MG™ Genomic DNA Purification kit (Macrogen, Korea) according to the manufacturer’s instructions.
Genome sequencing and assembly
For library preparation, gDNA was sheared with g-TUBE (Covaris Inc., USA) and then used for library preparation by ligating SMRTbell adaptors (20 kb SMRTbell library). The sequences of the generated library were sequenced using the PacBio RSII system, SMRT sequencing with DNA Sequencing Reagent Kit P6 and SMRT Cells 8Pac V3 (Pacific Biosciences). The sequencing generated 1,199,776,790 bp with 160,304 reads. After filtering of sequences that were shorter than 50 bp, 1,199,771,927 bp sequences with 160,189 subreads remained. Assembly was performed using software RS HGAP v3, which consists of pre-assembly, de novo assembly with Celera® Assembler, and assembly polishing with Quiver. Assembly resulted in one scaffold with the complete genome in circular form and 244-fold coverage.
Genome annotation
Annotation of the assembled genome was performed using the DOE-JGI Microbial Genome Annotation Pipeline v.4.15.1 [13]. The gene prediction was carried out using the IMG-ER platform. Comparisons of the predicted ORFs using the KEGG [14], NCBI COG [15], Pfam [16], TIGRfam [17], and InterPro [18] databases were conducted during gene annotation. Additional gene prediction analyses and functional assignment were carried out using the NCBI PGAP [19] and the RAST with the gene caller classicRAST [20] based on the SEED [21]. CRISPR system analysis was carried out using the web-based interface CRISPRFinder (http://crispr.i2bc.paris-saclay.fr/). The chromosome map of K. konosiri X49T was obtained from the output of the IMG pipeline (Fig. 3).
Fig. 3.

Circular map of the complete K. konosiri X49T genome. Marked characteristics are shown from the outside to the center: the number of bases, COG on forward strand, COG on reverse strand, RNA genes (tRNAs, green bars; rRNAs, red bars; other RNAs, black bars), GC content, and GC skew. Individual genes are colored according to COG categories
Genome properties
The genome of K. konosiri X49T comprised a single circular chromosome with a length of 3,584,631 bp and a G + C content of 59.1% (Fig. 3 and Table 3). Of the 3347 predicted genes, 3261 were protein-coding. According to tRNAscan-SE and RNAmmer 1.2, 66 tRNA, 12 rRNA (four 5S rRNA, four 16S rRNA, and four 23S rRNA genes), and 8 miscRNA genes were found in the genome. The genome contained one CRISPR structure with 25 spacers of 28 bp and two putative CRISPRs. The number of CDSs including signal peptides was 239 (7.14%). The majority of the protein-coding genes (2815 genes; 84.1%) were assigned to functional categories, while the remainder were annotated as hypothetical proteins (446 genes). The properties and statistics of the genome are summarized in Table 3, and the distributions of genes among the functional categories of COG are shown in Table 4.
Table 3.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,584,631 | 100% |
| DNA coding (bp) | 3,211,842 | 89.60% |
| DNA G + C (bp) | 2,118,687 | 59.10% |
| DNA scaffolds | 1 | 100% |
| Total genes | 3347 | 100% |
| Protein coding genes | 3261 | 97.43% |
| RNA genes | 86 | 2.57% |
| Pseudo genes | 73 | 2.18% |
| Genes in internal clusters | 611 | 18.26% |
| Genes with function prediction | 2815 | 84.11% |
| Genes assigned to COGs | 2568 | 76.73 |
| Genes with Pfam domains | 2913 | 87.03% |
| Genes with signal peptides | 239 | 7.14% |
| Genes with transmembrane helices | 823 | 24.59% |
| CRISPR repeats | 1 | 0 |
Table 4.
Number of genes associated with general COGs functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 222 | 6.81% | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03% | RNA processing and modification |
| K | 179 | 5.49% | Transcription |
| L | 112 | 3.43% | Replication, recombination and repair |
| B | 1 | 0.03% | Chromatin structure and dynamics |
| D | 32 | 0.98% | Cell cycle control, cell division, chromosome partitioning |
| V | 54 | 1.66% | Defense mechanisms |
| T | 134 | 4.11% | Signal transduction mechanisms |
| M | 195 | 5.98% | Cell wall/membrane/envelope biogenesis |
| N | 80 | 2.45% | Cell motility |
| U | 35 | 1.07% | Intracellular trafficking, secretion, and vesicular transport |
| O | 108 | 3.31% | Posttranslational modification, protein turnover, chaperones |
| C | 197 | 6.04% | Energy production and conversion |
| G | 216 | 6.62% | Carbohydrate transport and metabolism |
| E | 264 | 8.10% | Amino acid transport and metabolism |
| F | 84 | 2.58% | Nucleotide transport and metabolism |
| H | 179 | 5.49% | Coenzyme transport and metabolism |
| I | 108 | 3.31% | Lipid transport and metabolism |
| P | 174 | 5.34% | Inorganic ion transport and metabolism |
| Q | 73 | 2.24% | Secondary metabolites biosynthesis, transport and catabolism |
| R | 268 | 8.22% | General function prediction only |
| S | 138 | 4.23% | Function unknown |
| – | 779 | 23.89% | Not in COGs |
The total is based on the total number of protein coding genes (3261) in the genome
Insights from the genome sequence
Comparative genomics
To determine the genomic relatedness between K. konosiri X49T and closest relative strain K. marisflavi SW32T, the ANI value was calculated using an online calculator (https://www.ezbiocloud.net/tools/ani). The ANI value between two whole genome sequences was 89.32%. This value was well below the threshold of 95%; this suggested that two strains represent genotypically distinct species.
Hypersaline adaptation
Kushneria members possess hypersaline resistance and K. konosiri X49T can grow optimally at 11–19% (w/v) NaCl and survive in the presence of 26% (w/v) NaCl [1]. To balance the osmotic pressure between the inside and outside of the cell in the hypersaline habitat, theses halophile microorganisms increase the internal osmolarity of the cytoplasm using inorganic ions (mostly potassium ions) or organic compounds (mainly ectoine, choline, glycine betaine, and proline betaine, etc.), which are known as compatible solutes for the exclusion of salt ions. Genome analysis through the IMG pipeline revealed that K. konosiri X49T can adapt its salt tolerance using several functional genes (Fig. 4). In response to changes in the external osmolality, potassium is accumulated in the cytoplasm by transport via the Trk-type transport systems, KUP system, or Kdp two-component system [22–24]. With increasing potassium levels, osmoprotective compounds are accumulated by synthesis and/or uptake in the cytoplasm as a bi-phasic response [22]. Also, expression of membrane proteins such as OMPs and of MDOs is affected by environmental osmolarity [25, 26].
Fig. 4.

Genomic prediction of the osmoregulation in K. konosiri X49T. Trk, K+ transport; KUP, K+ uptake permease; Kdp, K+-dependent ATPase; BCCT, betaine-carnitine-choline transport; Osm, osmotically inducible protein; MDO, membrane-derived oligosaccharides; OOP, OmpA-OmpF Porin; EctA, L-2,4-diaminobutyric acid acetyltransferase; EctC, L-ectoine synthase; EctD, ectoine hydrolase; BetA, choline dehydrogenase; BetB, betaine aldehyde dehydrogenase; BetC, choline sulfatase protein
To regulate the osmolarity between cytoplasm and environment, the genome of K. konosiri X49T encodes potassium uptake-related loci: the Trk system (low-affinity potassium transport; two TrkA and two TrkH genes), KUP system (two potassium uptake permeases), and Kdp two-component system (one sensor histidine kinase KdpD and one operon response regulator KdpE). The X49T genome also encodes a variety of organic compound regulation systems including ectoine biosynthesis-related genes (one EctA, L-2,4-diaminobutyric acid acetyltransferase; one EctC, L-ectoine synthase; and two EctD, ectoine hydrolases), betaine uptake-related genes (ten members of the betaine-carnitine-choline transport family; two BetPT, proline-betaine transporters; and six OsmC, F, V, and Y, glycine-betaine transporters), betaine biosynthesis-related genes (one BetA, choline dehydrogenase; two BetB, betaine aldehyde dehydrogenases; and one BetC, choline sulfatase), and one glycerol uptake facilitator protein. The osmoregulated membrane protein-related genes (three OmpA-OmpF Porin Family and osmoregulated periplasmic glucan biosynthesis) and osmoregulated periplasmic glucan-related genes (two MdoC, glucan biosynthesis proteins; one MdoB, phosphoglycerol transferase MdoB-like AlkP superfamily enzyme) were also found in the genome (Fig. 4).
Carotenoid biosynthesis
Carotenoids are naturally occurring pigments that not only act as antioxidants but also enhance salt stress tolerance [27]. The first step of carotenoid biosynthesis starts by formation of one phytoene from two molecules of GGPP, and phytoene is converted to α- or β-carotene along the biosynthesis pathway. α- or β-carotene is transformed into xanthophyll by obtaining an oxygen atom (Fig. 5). The colonies of K. konosiri X49T were orange. The genome of K. konosiri X49T encodes genes related to carotenoid biosynthesis: two GGPP synthases, one phytoene synthase, two phytoene desaturases, one lycopene β-cyclase, one β-carotene hydroxylase, and one enhancing lycopene biosynthesis protein 2. However, there is no xanthophyll cycle-related gene, ZEP, which is key for conversion of zeaxanthin to violaxanthin, suggesting that the orange color of the colony of K. konosiri is derived from β-carotene or β-cryptoxanthin. These carotenoids may act also as the potential osmoprotectants (Fig. 5).
Fig. 5.
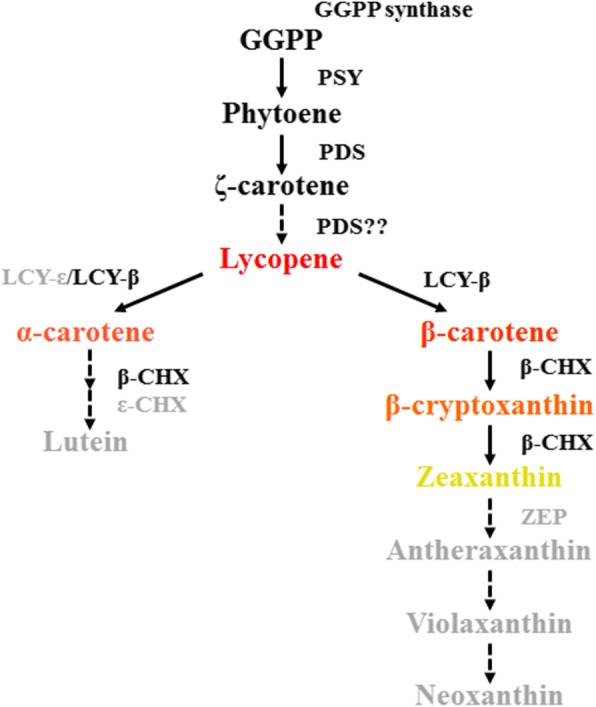
Carotenoid biosynthesis pathway in K. konosiri X49T. GGPP synthase, geranylgeranyl diphosphate synthase; PSY, phytoene synthase; PDS, phytoene desaturase; LCY-β, lycopene beta-cyclase; LCY-ε, lycopene ε-cyclase; β-CHX, beta-carotene 3-hydroxylase; ε-CHX, ε-carotene 3-hydroxylase. The font in gray color represents missing enzyme-coding genes and unproduced pigment in K. konosiri X49T
Conclusions
The orange pigmented K. konosiri X49T was isolated from a salt-fermented food, Daemi-jeot, and was resistant to a hypersaline environment. Whole genome sequence analysis and physiological observations leads us to conclude that K. konosiri X49T is a orange-coloured halophile and its capabilities of a cellular response are enabled by a variety of genes determining the ‘carotenoid biosynthesis’ and ‘inorganic or organic transport and metabolism’. The presence of ectoine and betaine biosynthesis genes or transport system related genes demonstrates the possibility of a cellular response to high osmolarity through biosynthesis of ectoine and betaine to protect the cell from stress. As ectoine or betaine can play as a protectant under stress condition, the genome sequence of K. konosiri X49T may provide the molecular basis for its hypersaline tolerance and may lead to new development in its diverse biotechnological applications comprising environmental, medical and biofuel industries.
Acknowledgments
Funding
This study was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry (IPET) through the Agricultural Microbiome R&D Program, funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (grant number: 914006–4).
Abbreviations
- ANI
Average-nucleotide identity
- COG
Clusters of orthologous groups
- CRISPR
Clustered regularly interspaced short palindromic repeat
- DOE
Department of energy
- GGPP
Geranylgeranyl diphosphate
- GOLD
Genomes on line database
- JGI
Joint genome Institute
- KEGG
Kyoto encyclopedia of genes and genomes
- LB
Luria–bertani
- MIGS
Minimum information about a genome sequence
- MOD
Membrane-derived oligosaccharides
- OMP
Outer membrane proteins
- PGAP
Prokaryotic genome annotation pipeline
- RAST
Rapid annotation using subsystem technology
- SMRT
Single-molecule real-time
- ZEP
Zeaxanthin epoxidase
Authors’ contributions
JHY performed the experiments, analyzed the data, prepared and displayed elements, and wrote the main manuscript text. HJS and HSK performed the experiments. EJT, WRK, JYL, DWH, and PSK prepared elements. JWB is the guarantor of this work, has full access to all the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analyses. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yun JH, Park SK, Lee JY, Jung MJ, Bae JW. Kushneria konosiri sp. nov., isolated from the Korean salt-fermented seafood Daemi-jeot. Int J Syst Evol Microbiol. 2017;67:3576–3582. doi: 10.1099/ijsem.0.002170. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Porro C, de la Haba RR, Soto-Ramirez N, Marquez MC, Montalvo-Rodriguez R, Ventosa A. Description of Kushneria aurantia gen. nov., sp. nov., a novel member of the family Halomonadaceae, and a proposal for reclassification of Halomonas marisflavi as Kushneria marisflavi comb. nov., of Halomonas indalinina as Kushneria indalinina comb. nov. and of Halomonas avicenniae as Kushneria avicenniae comb. nov. Int J Syst Evol Microbiol. 2009;59:397–405. doi: 10.1099/ijs.0.001461-0. [DOI] [PubMed] [Google Scholar]
- 3.Soto-Ramirez N, Sanchez-Porro C, Rosas S, Gonzalez W, Quinones M, Ventosa A, Montalvo-Rodriguez R. Halomonas avicenniae sp. nov., isolated from the salty leaves of the black mangrove Avicennia germinans in Puerto Rico. Int J Syst Evol Microbiol. 2007;57:900–905. doi: 10.1099/ijs.0.64818-0. [DOI] [PubMed] [Google Scholar]
- 4.Cabrera A, Aguilera M, Fuentes S, Incerti C, Russell NJ, Ramos-Cormenzana A, Monteoliva-Sanchez M. Halomonas indalinina sp. nov., a moderately halophilic bacterium isolated from a solar saltern in Cabo de Gata, Almeria, southern Spain. Int J Syst Evol Microbiol. 2007;57:376–380. doi: 10.1099/ijs.0.64702-0. [DOI] [PubMed] [Google Scholar]
- 5.Yoon JH, Choi SH, Lee KC, Kho YH, Kang KH, Park YH. Halomonas marisflavae sp. nov., a halophilic bacterium isolated from the Yellow Sea in Korea. Int J Syst Evol Microbiol. 2001;51:1171–1177. doi: 10.1099/00207713-51-3-1171. [DOI] [PubMed] [Google Scholar]
- 6.Bangash A, Ahmed I, Abbas S, Kudo T, Shahzad A, Fujiwara T, Ohkuma M. Kushneria pakistanensis sp. nov., a novel moderately halophilic bacterium isolated from rhizosphere of a plant (Saccharum spontaneum) growing in salt mines of the Karak area in Pakistan. Antonie Van Leeuwenhoek. 2015;107:991–1000. doi: 10.1007/s10482-015-0391-9. [DOI] [PubMed] [Google Scholar]
- 7.Zou Z, Wang G. Kushneria sinocarnis sp. nov., a moderately halophilic bacterium isolated from a Chinese traditional cured meat. Int J Syst Evol Microbiol. 2010;60:1881–1886. doi: 10.1099/ijs.0.013797-0. [DOI] [PubMed] [Google Scholar]
- 8.Yun Ji-Hyun, Bae Jin-Woo. Complete genome sequence of the halophile bacterium Kushneria marisflavi KCCM 80003 T , isolated from seawater in Korea. Marine Genomics. 2018;37:35–38. doi: 10.1016/j.margen.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The genomes on line database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010;38:D346–D354. doi: 10.1093/nar/gkp848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 14.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–D269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41:D387–D395. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ly A., Henderson J., Lu A., Culham D. E., Wood J. M. Osmoregulatory Systems of Escherichia coli: Identification of Betaine-Carnitine-Choline Transporter Family Member BetU and Distributions of betU and trkG among Pathogenic and Nonpathogenic Isolates. Journal of Bacteriology. 2003;186(2):296–306. doi: 10.1128/JB.186.2.296-306.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Ting, You Yibo, Hong De, Sun Haipeng, Sun Baolin. The Staphylococcus aureus KdpDE Two-Component System Couples Extracellular K+Sensing and Agr Signaling to Infection Programming. Infection and Immunity. 2011;79(6):2154–2167. doi: 10.1128/IAI.01180-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dominguez-Ferreras A., Munoz S., Olivares J., Soto M. J., Sanjuan J. Role of Potassium Uptake Systems in Sinorhizobium meliloti Osmoadaptation and Symbiotic Performance. Journal of Bacteriology. 2009;191(7):2133–2143. doi: 10.1128/JB.01567-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Csonka LN, Hanson AD. Prokaryotic osmoregulation: genetics and physiology. Annu Rev Microbiol. 1991;45:569–606. doi: 10.1146/annurev.mi.45.100191.003033. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy E. P. Osmotic regulation and the biosynthesis of membrane-derived oligosaccharides in Escherichia coli. Proceedings of the National Academy of Sciences. 1982;79(4):1092–1095. doi: 10.1073/pnas.79.4.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang L, Ji CY, Kim SH, Ke Q, Park SC, Kim HS, Lee HU, Lee JS, Park WS, Ahn MJ, et al. Suppression of the beta-carotene hydroxylase gene increases beta-carotene content and tolerance to abiotic stress in transgenic sweetpotato plants. Plant Physiol Biochem. 2017;117:24–33. doi: 10.1016/j.plaphy.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrity GM, Bell JA, Lilburn T, Phylum XIV. Proteobacteria. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria 2nd ed. New York: Springer; 2005. p. 1. [Google Scholar]
- 30.Garrity GM, Bell JA, Lilburn T, Class III. Gammaproteobacteria. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria, 2 edn. New York: Springer; 2005. p. 1. [Google Scholar]
- 31.Garrity GM, Bell JA, Lilburn T, Order VIII. Oceanospirillales. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria 2nd ed. New York: Springer; 2005. pp. 270–323. [Google Scholar]
- 32.Garrity GM, Bell JA, Lilburn T, Family IV. Halomonadaceae. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria 2nd ed. New York: Springer; 2005. pp. 300–323. [Google Scholar]