

Abstract
Background
Hybridization and introgression are common phenomena among oak species. These processes can be beneficial by introducing favorable genetic variants across species (adaptive introgression). Given that drought is an important stress, impacting physiological and morphological variation and limiting distributions, our goal was to identify drought-related genes that might exhibit patterns of introgression influenced by natural selection. Using RNAseq, we sequenced whole transcriptomes of 24 individuals from three oaks in southern California: (Quercus engelmannii, Quercus berberidifolia, Quercus cornelius-mulleri) and identified genetic variants to estimate admixture rates of all variants and those in drought genes.
Results
We found 398,042 variants across all loci and 4352 variants in 139 drought candidate genes. STRUCTURE analysis of all variants revealed the majority of our samples were assignable to a single species, but with several highly admixed individuals. When using drought-associated variants, the same individuals exhibited less admixture and their allele frequencies were more polarized between Engelmann and scrub oaks than when using the total gene set. These findings are consistent with the hypothesis that selection may act differently on functional genes, such as drought-associated genes, and point to candidate genes that are suggestive of divergent selection among species maintaining adaptive differences. For example, the drought genes that showed the strongest bias against engelmannii-fixed oak variants in scrub oaks were related to sugar transporter, coumarate-coA ligases, glutathione S-conjugation, and stress response.
Conclusion
This pilot study illustrates that whole transcriptomes of individuals will provide useful data for identifying functional genes that contribute to adaptive divergence among hybridizing species.
Electronic supplementary material
The online version of this article (10.1186/s12863-018-0677-9) contains supplementary material, which is available to authorized users.
Keywords: Adaptation, Candidate genes, Drought stress, Hybridization, Introgression, RNAseq, Transcriptome, Quercus spp.
Background
The availability of genetic variants for evolutionary response of local populations to environmental change will depend on existing genetic variation, new mutations, gene flow among populations, or in some cases, interspecific gene flow. In many plant taxa, hybridization and backcrossing can lead to introgression of alleles from one species into the gene pool of another [1, 2]. As a result, it is possible for adaptive genetic variation to cross species boundaries [3–5] This interspecific gene flow can lead to adaptive evolution to novel ecological contexts [3–6]. In contrast, divergent selection can act to favor species-specific adaptive alleles and select against introgression at ecologically important loci [7–9], after initial F1 hybrid formation or across generations of backcrosses. Nonetheless, a first step in understanding whether introgression could enhance evolutionary response is to look for alleles shared across species.
Oaks (Quercus spp.) represent an excellent system to investigate selection favoring or acting against introgressed alleles [10] because of their well-known ability to hybridize but maintain ecological distinctiveness [11–14]. Many studies have documented the responses of Quercus species to drought [15–18]. Because so many Quercus species are adapted to drought-prone environments by ability either to avoid or tolerate water stress or both [19], introgression of genetic variants underlying these traits may provide the genetic material for species to adapt to climate change.
The overall goal of this paper is to examine a set of previously identified drought-associated genes that might be useful for future studies of climate-adaptive introgression and divergence. The study system is comprised of three sympatric species of white oaks (Section Quercus) found in southern California: one tree oak Quercus engelmannii and two drought-tolerant scrub oaks, Quercus berberidifolia and Quercus cornelius-mulleri. Engelmann oak is distantly related to the two recently diverged California endemic scrub white oak species [20–23]. These three species have overlapping distributions, but with different habitat preferences within southern California, a geographic region with a history of high temperatures and low rainfall [24]. We have evidence of contemporary hybridization among all three species [25] as well as ancient introgression between Q. engelmannii and Q. berberidifolia [26]. Using a sample of 24 study specimens representing the three species and putative hybrids sampled throughout the southern California region, we identified sequence variants across all expressed genes based on RNA-Seq data [27, 28] that were mapped to a previously published reference transcriptome for a related oak, Quercus lobata [29]. First, we described the overall admixture to assign species identity across the 24 individuals. Second, using variants derived from drought-associated genes that were identified through other studies [30–32], we examined the admixture to compare with the admixture based on the entire set of variants. Third, we looked for evidence of selection by testing whether alleles fixed in Q. engelmannii that are shared by a subset of the scrub oak species are less frequent in the two drought-tolerant scrub oaks when the variants are associated with drought genes compared to variants across all genes. Finally, we described the functions of drought associated genes that were outliers in the previous analysis.
Methods
Study specimen and sampling
The study species are three oak species of southern California that are sometimes sympatric with each other in many parts of their ranges [26, 33, 34]: Q. engelmannii, Q. berberidifolia and Q. cornelius-mulleri. Engelmann oak is native to southern California, USA and northern Baja California, Mexico), with the core of the species’ distribution range in San Diego County (USA), where the species is generally distributed in scattered patches often consisting of a few individuals [33, 35]. Engelmann oak is associated with more mesic local habitats than the southern California scrub oaks [24] and is more closely related to a Mexican/Arizona group of white oaks then to the scrub oaks [20]. The scrub oaks, Q. berberidifolia and Q. cornelius-mulleri, are both endemic to California and northern Baja California [20], but they belong to different genetic clusters within the white scrub oak complex [23, 26, 36]. Q. berberidifolia is widely distributed from northern California to Baja California and overlaps extensively but not completely the entire geographical range occupied by Engelmann oak [24, 33]. In contrast, Q. cornelius-mulleri, has a distribution restricted to southern California and northern Baja California usually in desert ecosystems, located more inland than Q. berberidifolia [24, 33]. They are known to hybridize with each other and hybrids show intermediate leaf traits and tend to be more shrub-like than tree-like [24, 33, 37].
This study includes 24 individuals sampled from 9 different localities of Southern California (Table 1, Fig. 1). In some sites, only one species was present so it was intended that the individuals would represent putatively “pure” species, while in other sites more than one species was present creating the opportunity for contemporary hybridization. Nixon [20] includes a hybrid species between Engelmann and Muller oaks, Q. ×acutidens, but we did not attempt to determine whether the samples collected for this study could be considered as part of that nothospecies. Preliminary species and hybrid identifications of field samples were made by VLS and confirmed based on genetic information from the transcriptomes, which provided the bases of final assignments to species or hybrid. Voucher specimens are available through VLS.
Table 1.
Localities of 24 individuals sampled from three southern California oak species: Q. berberidifolia, Q. cornelius-mulleri, and Q. engelmannii
| Location | Sample IDs | Latitude | Longitude | Altitude (m) |
|---|---|---|---|---|
| Pala Reserve | 8, 20, 21, 22, 23, 24 | 33° 22.53’ | − 117° 02.71’ | 267 |
| Oak Knoll | 19 | 33° 17.92’ | − 116° 55.29’ | 713 |
| Lake Henshaw | 18 | 33° 16.56’ | −116° 51.24’ | 711 |
| Lake Wohlford | 7, 15, 16 | 33° 09.87’ | −117° 00.23’ | 256 |
| Santa Ysabel | 1, 6 | 33° 06.55’ | −116° 40.16’ | 999 |
| Julian | 14 | 33° 04.90’ | −116° 34.42’ | 1683 |
| Laguna Mountain | 9, 13, 17 | 32° 50.97’ | −116° 29.14’ | 1516 |
| Alpine | 2 | 32° 48.99’ | −116° 45.80’ | 595 |
| McCain Valley | 3, 4, 5, 10, 11, 12 | 32° 41.95’ | −116° 15.50’ | 1114 |
Localities are presented from northern to southern latitudes
Fig. 1.

Map of region with location of sets of individuals from three species of oaks—Q. berberidifolia, Q. cornelius-mulleri, Q. engelmannii—and their hybrids found within southern California. Size of circle indicates number of individuals sampled. (See Table 1 for details of localities)
RNA extraction and sequencing
Fresh young leaf samples were frozen on dry ice in the field and stored at − 80 °C until total RNA extraction. Preliminary RNA precipitation [29, 30] as performed prior to total RNA extraction with the Qiagen RNeasy Plant Mini Kit protocol with DNase treatment (Qiagen, Hilden, Germany). RNA-Seq libraries with insert length 100–380 bp (mode = 170 bp) were prepared from 4 μg of RNA using an Illumina TruSeq RNA Sample Prep Kit. Each library was uniquely tagged using 12 TruSeq indexed adapters (numbers 1–12) to enable 12-plexing of samples in each Illumina HiSeq 2000 lane.
Variant calling
We aligned a minimum of 13.8 million 50 bp reads per sample to our Q. lobata reference transcriptome [29] using BWA MEM [38]. Prior to mapping, the reference transcriptome was concatenated into 90 contigs of approximately 1 Mb in length. The original contigs were ordered by length and separated by runs of 200 Ns. This concatenation facilitates the analysis because GATK [39] and PLINK [40] were not designed for use with large numbers of reference contigs. GATK 3.2–2 [39] was used for variant discovery and filtering with the following tools, HaplotypeCaller, VariantFiltration and SelectVariants. To determine appropriate parameters for hard filtering with VariantFiltration, we inspected variant calls along with the read alignments using IGV [41]. The following cutoffs were applied: FS > 30.0, QD < 2.0 and QUAL < 30. Variants from the smallest contigs (737 bases and below) were discarded, leaving 858,564 variants on the first 58 of the 1 Mb concatenated contigs. The ratio of Ts/Tv for these variants was 1.76. The vcftools 0.1.15 package [42] was used to remove sites with mean depth coverage of less than 5 (−-min_meanDP 5) and with more than 5% of samples with missing data (−-max-missing 0.95) leaving 398,042 variants (Ts/Tv = 1.87). For STRUCTURE analysis, LD pruning was applied for r2 correlations above 0.1 within 50 bp windows, sliding by 5 bp using PLINK v1.90b3.36 [40] (indep-pairwise, based on correlations between genotype allele counts), leaving 30,809 variants (Ts/Tv = 1.93). For our allele frequency analysis, variants were limited to those for which a single allele was found across the four Q. engelmannii samples, leaving 219,407 variants (Ts/Tv = 1.85). The bedtools [43] package was used to facilitate allele frequency analyses.
Drought gene selection
We selected 139 drought candidate genes for oaks that were identified in the literature [30, 32, 44] and located on the Q. lobata reference transcriptome [29].
Structure analysis
Based on morphology of the samples that were assigned to the three species, we expected three genetic clusters. To test this hypothesis, we ran structure with K values from 1 to 9. The results were imported to Structure Harvester [45] to choose the “optimal” number of clusters following the methods of Evanno et al. [46]. STRUCTURE [47] was run on the filtered, LD pruned variants. We ran 10,000 burn-in and 20,000 experimental repetitions. Population label information was not used in the inference. The degree of admixture parameter, α, was inferred from the data. The allele frequency prior was estimated with preliminary runs (inferalpha = 0) on the full set of variants and set at 0.66 for all further runs, and correlated allele frequencies were assumed (freqscorr = 1). All other parameters were default. Membership coefficients for each sample were plotted using Distruct 1.1 [48] (Fig. 2).
Fig. 2.

Assignment of species ancestry of 24 individuals collected from three species —Q. berberidifolia, Q. cornelius-mulleri, Q. engelmannii— across southern California, using STRUCTURE (K = 3). (a) Ancestry assignments are based on 219,407 LD-pruned variants across all sequences (b). Ancestry assignments are based on 30,809 LD-pruned variants associated with drought-related genes
As a complement to STRUCTURE, we also examined the relationships among species using principal component analysis, which illustrates relationships among samples without assumptions about the assignment to species. For the PCA we used the R package SNPRelate [49], limiting the analysis to the 378,489 biallelic variants for all genes (Fig. 3a) and 4138 biallelic variants for the drought genes (Fig. 3b).
Fig. 3.

Principal component analysis of 24 individuals from three species using same data as Fig. 2. (a) Variants associated with all genes. (b) Variants associated with drought-related genes. Hybrids are identified based on STRUCTURE results
Allele frequency analysis
We examined evidence of species-specific selection against drought gene introgression by selecting variants that are “fixed” in Q. engelmannii samples and variable in the scrub oak samples. We used quality-filtered, biallelic variant loci for which a single allele was found across the four Q. engelmannii samples. Four samples are not sufficient to determine fixation in the species, but for simplicity we refer to these loci as “engelmannii-fixed”. For each engelmannii-fixed variant locus, we measured the fraction of the engelmannii-fixed variant in the 20 scrub oak samples, i.e., the minimum of zero Q. engelmannii alleles = 0, and the maximum of 40 Q. engelmannii alleles = 1. We repeated this measure for the subset of engelmannii-fixed loci found in the 139 drought genes. We tested the significance of the increase in low Q. engelmannii allele frequencies by comparing the proportion of low Q. engelmannii loci in our drought genes with the same from 1000 randomly drawn sets of 139 genes. These random sets were drawn from the 13,159 transcriptome genes with the highest confidence annotations (i.e., those including 5′- and 3′-UTR, start and stop codons and no predicted introns).
Functional descriptions of outlier genes
From the set of engelmannii-fixed variants, we selected those with less than 10% of the Q. engelmannii allele across the scrub oaks as outliers and identified overlapping drought genes to be functionally described. Additional information about putative functions were identified using BLASTX via the Gramene Server [50] to identify Arabidopsis homologs of the Q. lobata transcriptome contigs. Links for the top matching Arabidopsis gene were followed to the UniProtKB/Swiss-Prot server [51], from which protein names and protein function descriptions were taken. Most of the Q. lobata genes matched the Arabidopsis genes at greater than 80% amino acid identity and close to full length. All were greater than 58% identical (Table 2).
Table 2.
Drought genes that contain variants that are “fixed” in Q. engelmannii and at low frequency and under-represented across 20 scrub oak samples
| Gene (transcriptome contig) | Biallelic variants | engelmannii-fixed variants | Scrub oak variants with less than 10% engelmannii | % Identity to Arabidopsis protein | Uniprot Protein Name | Gene | Uniprot Function Description |
|---|---|---|---|---|---|---|---|
| m01oak10430CC | 163 | 138 | 8 | 80% | CHY-type/CTCHY-type/RING-type Zinc finger protein | None | none |
| m01oak03512CC | 71 | 55 | 7 | 71% | Monosaccharide-sensing protein 3 | MSSP3 | Sugar transport |
| m01oak02926cC | 109 | 42 | 7 | 87% | Phosphoenolpyruvate carboxylase 1 | PPC1 | Through the carboxylation of phosphoenolpyruvate (PEP) it forms oxaloacetate, a four-carbon dicarboxylic acid source for the tricarboxylic acid cycle. Contributes probably to the adaptation to inorganic phosophate (Pi) deprivation |
| m01oak01922jc | 71 | 49 | 5 | 92% | Cellulose synthase A catalytic subunit 2 | CESA2 | Catalytic subunit of cellulose synthase terminal complexes (‘rosettes’), required for beta-1,4-glucan microfibril crystallization, a major mechanism of the cell wall formation. Involved in the primary cell wall formation. |
| m01oak00521CC | 61 | 53 | 3 | 87% | Heat shock 70 kDa protein 10, mitochondrial;Mitochondrial HSO70 2 isoform 2 | HSP70 | In cooperation with other chaperones, Hsp70s stabilize preexistent proteins against aggregation and mediate the folding of newly translated polypeptides in the cytosol as well as within organelles. These chaperones participate in all these processes through their ability to recognize nonnative conformations of other proteins. They bind extended peptide segments with a net hydrophobic character exposed by polypeptides during translation and membrane translocation, or following stress-induced damage (By similarity). |
| m01oak09381CC | 22 | 21 | 2 | 81% | Amino acid permease 3 | AAP3 | Amino acid-proton symporter. Stereospecific transporter with a broad specificity for GABA, tryptophan and both neutral and basic amino acids. High affinity transport of cationic amino acids. |
| m01oak03200CC | 66 | 46 | 2 | 79% | Polyol transporter 5 | PLT5 | Plasma membrane broad-spectrum sugar-proton symporter. Mediates the uptake of linear polyols such as sorbitol, xylitol, erythritol or glycerol. Can transport the cyclic polyol myo-inositol and different hexoses, pentoses (including ribose), tetroses and sugar alcohols. |
| m01oak01473cC | 42 | 33 | 1 | 77% | Aspartic proteinase A1 | APA1 | Involved in the breakdown of propeptides of storage proteins in protein-storage vacuoles (By similarity). Possesses aspartic protease activity in vitro |
| m01oak03575jC | 32 | 29 | 1 | 88% | Cryptochrome-1 | CRY1 | Photoreceptor that mediates primarily blue light inhibition of hypocotyl elongation and photoperiodic control of floral initiation, and regulates other light responses, including circadian rhythms, tropic growth, stomata opening, guard cell development, root development, bacterial and viral pathogen responses, abiotic stress responses, cell cycles, programmed cell death, apical dominance, fruit and ovule development, seed dormancy, and magnetoreception |
| m01oak00924cC | 53 | 34 | 1 | 84% | Auxin-responsive protein IAA8 | IAA8 | Aux/IAA proteins are short-lived transcriptional factors that function as repressors of early auxin response genes at low auxin concentrations. Repression is thought to result from the interaction with auxin response factors (ARFs), proteins that bind to the auxin-responsive promoter element (AuxRE). Formation of heterodimers with ARF proteins may alter their ability to modulate early auxin response genes expression |
| m01oak01539cC | 15 | 10 | 4 | 84% | Translation initiation factor SUI1 family protein (TAIR database) | AT5G11900 | none |
Results and discussion
Using the reference transcriptome for a related oak species, Q. lobata [29], we aligned RNA-seq data for 24 individuals and identified 398,042 variants including 12,068 indels and 358,974 SNPs. After LD pruning, 1061 indels and 29,748 SNPs remained. We examined 139 drought-associated genes for introgression, which are listed in Additional file 1: Table S1. The transcriptome contigs for these genes had 4352 variants, with 330 remaining after LD pruning.
Based on the LD pruned data for all contigs/genes, we found support for three genetic clusters (Additional file 2: Figure S1; Table S2). None of our samples appear to have ancestry from only one species (Fig. 2a), although most trees could be assigned to one primary species that corresponded to our field identifications. We cannot explain why all samples included at least some genetic variation that was assigned to Q. engelmannii (Fig. 2a). It might be that shared genetic variation to all the species is assigned statistically to Q. engelmannii when it may be the signature of common ancestor or ancient introgression. The findings also indicate that two of the scrub oak samples (PR-22 and PR-24) are hybrids of Q. engelmannii and Q. berberidifolia, one sample (MV-12) is almost 50% from each scrub oak species and a fourth sample (MV-10) also shows marked admixture from both species. Several other Q. cornelius-mulleri samples included more than 10% of their genetic ancestry from Q. berberidifolia (MV-3, MV-5, MV-11). These patterns of assignments are likely indicative of introgression among the scrub oaks, but we cannot rule out common ancestry. Phylogenetic analysis indicates that these somewhat sympatric species are in different clades, but only recently diverged [26, 36].
The STRUCTURE analysis of assigned ancestry of the drought genes alone indicates similar patterns of introgression to those of the whole variant data set. One notable difference is that the five hybrids of Q. cornelius-mulleri and Q. berberidifolia, which show admixture of approximately 20--40% based on all variants (Fig. 2a), seem to be almost pure Q. cornelius-mulleri when looking at the drought genes alone (Fig. 2b). One must interpret these findings cautiously because the admixture analysis when based on drought genes has a reduced sample size of 330 variants compared to the 30,000+ variants across all sequences. Within the drought genes all the samples showed less admixture, which raises the question of whether selection is acting against variants not enhancing the fitness of a given species occurring within its own niche. The outcome is to make each species appear to have less introgression than indicated by the full set of variants. Because an alternative explanation for the ancestry patterns based on drought genes is sampling error, larger sample sizes would allow the permutation tests needed to test whether the pattern we observe is due to chance.
The PCA plots indicate differences in the relationships among individuals based on the total gene and drought gene sets (Fig. 3). The analyses for both samples of variants identifies the same two likely F1-hybrids between Q. engelmannii and Q. berberidifolia, but the scrub oak hybrids suggested by the STRUCTURE analysis cluster closely with Q. berberidifolia samples, suggesting that the introgression we observe is more likely to be due to admixture between Q. engelmannii and Q. berberidifolia, even though when looking at the distribution of variants on a one-by-one basis, it is not easy to say from which scrub oak they are shared. These findings are consistent with a concurrent study that demonstrated evidence of ancient introgression between the scrub white oaks and Q. engelmannii was largely due to shared alleles between Q. engelmannii and Q. berberidifolia and not with Q. cornelius-mulleri [26].
To find evidence that selection is acting on drought-associated genetic variants, we looked for a decrease of allele number from the more mesic Q. engelmannii found within genes associated with drought compared to all genes. To do this, we identified variants that are “fixed” in Q. engelmannii, i.e., having the same allele for all eight Q. engelmannii haplotypes (across four samples), and looked at the frequencies of these alleles across the scrub oaks. We discovered 219,407 engelmannii-fixed loci of which 2422 were located within the 139 drought genes. As one would expect since most variants have low minor allele frequencies, the vast majority of the “engelmannii-fixed” alleles were shared among greater than 90% of the scrub oaks. However, “engelmannii-fixed” alleles that are low frequency (defined as less than 50%) in the scrub oaks (2121 SNPs in 726 genes) are underrepresented in the scrub oaks compared to low-frequency alleles in the total gene data set (Fig. 4). This result is apparent in the left skew on the histogram of drought gene allele frequencies relative to other gene allele frequencies. The difference is seen across all tested bins of low allele frequencies (Fig. 4b) and is mostly reversed when looking at the high frequency bins. The observation that Q. engelmannii drought gene alleles are rarer in scrub oak than expected based on allele frequencies from other genes might suggest selection against Q. engelmannii alleles in the scrub oaks and thus adaptive divergence among species.
Fig. 4.

(a) Frequencies of Q. engelmannii-fixed alleles in the 20 scrub oaks: The frequencies of engelmannii-fixed alleles for each variant site is calculated as the number of Q. engelmannii-fixed alleles in the scrub oaks divided by the total number of alleles across all scrub oaks at that locus, usually 40 depending on missing data. The proportion of variants falling into each allele frequency bin is reported as a percentage of the total number of variants, 219,407 for all genes and 2421 for the drought genes. These variant sets are non-LD-pruned engelmannii-fixed biallelic loci with no more than 5% missing data. (b) Allele frequencies less than 0.5. Overall 8746 (4.0%) sites have a Q. engelmannii-fixed allele frequency in scrub oaks of 0.5 or less, and 137 (5.7%) drought gene variant sites have a Q. engelmannii-fixed allele frequency in scrub oaks of 0.5 or less. The difference between all tested bins of low allele frequencies mostly reversed at high frequency bins
In order to test the significance of this difference, we compared the results to 1000 random sets of 139 genes (equivalent to the number of drought genes) (Fig. 5). We then examined the results with the proportion of low-frequency engelmannii-fixed alleles for all genes (shown in blue; Fig. 5) with the proportion for drought genes (shown in red) and the proportion for the 1000 sets of random genes (shown in white). We see a fairly normal distribution with the drought genes skewed to having a higher proportion of low frequency Q. engelmannii alleles. Only 105 out of 1000 random gene sets have a higher proportion of low frequency Q. engelmannii alleles, yielding p = 0.1. Thus, the analysis suggests that the scrub oaks are not significantly less likely to carry Q. engelmannii drought gene alleles than expected from patterns of overall allele sharing across the three species. The initial findings reported here are consistent with the notion that Q. engelmannii variants within the drought genes are rarer in the scrub oaks than expected and might suggest divergent selection among species at these ecological relevant loci.
Fig. 5.
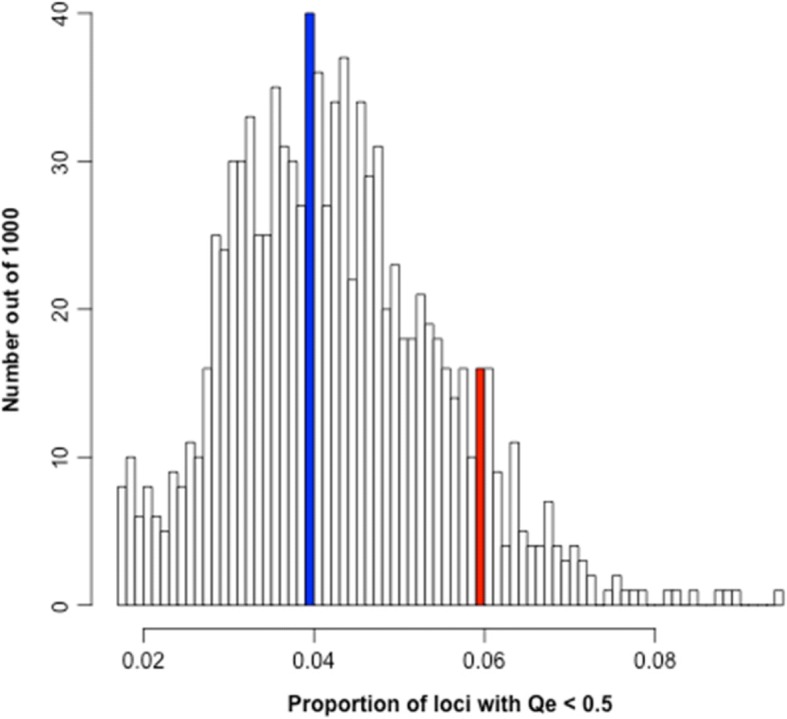
The proportion of variant sites with Q. engelmannii allele frequencies below 50% in the scrub oaks is shown in blue for all genes, red for the 139 drought genes and white for 1000 random sets of 139 genes
Finally, we use these findings to narrow down our set of candidate genes potentially involved in drought resistance to a set of genes that vary across species and warrant further study. Drought-associated genes that are fixed across oaks and or that vary but with no species-specific signature will not allow analysis of adaptive introgression. However, drought genes that are species-species can be informative. Looking only at variants with less than 10% of the Q. engelmannii alleles found across the scrub oaks, we find 41 variants fall within 11 of the 139 candidate drought genes (Table 2). Two of our hybrid samples from one of the localities offered the opportunity to look for additional loci where the engelmannii-fixed alleles might show evidence that selection has acted against them: PR-22 and PR-24. For each of the two hybrid samples, we identified engelmannii-fixed loci where the hybrid is homozygous for the alternate allele, i.e., it has zero of the Q. engelmannii allele. Out of 219,407 engelmannii-fixed loci in our dataset, PR-22 had 1993 loci with zero of the Q. engelmannii allele, of which 33 were within 4 drought genes (Table 3), 2 of which were also identified in Table 2. PR-24 had 1863 loci with zero of the Q. engelmannii allele, of which 14 were within 5 drought genes (Table 4).
Table 3.
Drought genes in hybrid PR-22 lacking variants found in Q. engelmannii allele, as candidates for genes involved in local adaptation to warm or dry habitats
| Gene (transcriptome contig) | Biallelic variants | engelmannii-fixed variants | Variants with no Q. engelmannii allele in hybrid PR-22 | % Identity to Arabidopsis | Uniprot Protein Name | Gene | Uniprot Function Description |
|---|---|---|---|---|---|---|---|
| m01oak02926cC (also in Table 2) | 109 | 42 | 18 | 87% | Phosphoenolpyruvate carboxylase 1 | PPC1 | Through the carboxylation of phosphoenolpyruvate (PEP) it forms oxaloacetate, a four-carbon dicarboxylic acid source for the tricarboxylic acid cycle. Contributes probably to the adaptation to inorganic phosophate (Pi) deprivation |
| m01oak03200CC (also in Table 2) |
66 | 46 | 11 | 79% | Polyol transporter 5 | PLT5 | Plasma membrane broad-spectrum sugar-proton symporter. Mediates the uptake of linear polyols such as sorbitol, xylitol, erythritol or glycerol. Can transport the cyclic polyol myo-inositol and different hexoses, pentoses (including ribose), tetroses and sugar alcohols. |
| m01oak01188Ct | 7 | 5 | 1 | 58% | Yellow leaf specific gene 9 | YLS9 | Up-regulated in leaves during natural senescence |
| m01oak04613CC | 43 | 12 | 3 | 68% | ACT domain-containing protein | ACR8 | May bind amino acids; induced by abscisic acid (ABA), and cold and salt stresses |
Table 4.
Drought genes in hybrid PR-24 lacking variants found in Q. engelmannii, as candidates for genes involved in local adaptation to warm or dry habitats
| Gene (transcriptome contig) | Biallelic variants | engelmannii-fixed variants | Variants with no engelmannii allele in hybrid PR-24 | % Identity to Arabidopsis protein | Uniprot Protein Description | Gene | Uniprot Function Description |
|---|---|---|---|---|---|---|---|
| m01oak09012cC | 61 | 47 | 6 | 80% | 4-coumarate--CoA ligase-like 7 | 4CLL7 | Contributes to jasmonic acid biosynthesis by initiating the beta-oxidative chain shortening of its precursors. |
| m01oak03358CC | 56 | 24 | 5 | 81% | 4-coumarate--CoA ligase 1 | 4CL1 | Produces CoA thioesters of a variety of hydroxy- and methoxy-substituted cinnamic acids, which are used to synthesize several phenylpropanoid-derived compounds, including anthocyanins, flavonoids, isoflavonoids, coumarins, lignin, suberin and wall-bound phenolics |
| m01oak00399CT | 12 | 9 | 1 | 63% | Glutathione S-transferase F7 | GSTF7 | May be involved in the conjugation of reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles and have a detoxification role against certain herbicides (also possible response to salt stress) |
There were many interesting potential functions amongst this narrowed down set, which include several sugar transporters, two separate homologs to coumarate-coA ligases, two genes involved in glutathione S-conjugation, and several stress response genes. Other studies have found other genes such as FBA1 [52], BURP [53, 54], USP [55], GH16_XET [56], CYP707 gene family [57], HSF [58], AAI_LTSS [59] (Additional file 1: Table S1) to be involved in drought resistance that we would have expected to be important in this study but did not show species-specific signatures in this study. The genes we report these here so that future studies of local adaptation might use them for comparison.
Conclusion
This exploratory study found evidence of modest admixture among two scrub white oak species and one tree oak based on a transcriptome-wide set of loci. When we examined a subset of drought-associated genes, variants that were most associated with the more mesic Engelmann oak were under-represented across the scrub oaks, which is consistent with the hypothesis that these variants are selected against by the local environment. Specifically, we identified eleven genes that may be examples of species-specific adaptation associated with drought. Of course, one limitation of this study is that some genes may be so extensively introgressed that we would not be able to distinguish them from shared common ancestry. Future work would be able to do so through larger samples sizes that can more sensitively identify more species-specific variants, and complementary analyses using genome-wide DNA sequence data that can test alternative demographic models that test for introgression while accounting for incomplete lineage sorting. Nonetheless, this study illustrates that the transcriptomes of individuals across hybridizing species have the potential to provide useful data to study adaptive introgression because they can identify functional genes associated with specific environmental factors.
Additional files
Candidate gene list associated with drought stress. Table S1. Candidate loci were taken from evoltree database depending on search tool for drought and hypoxia relates stress (target trait) and matched the sequences to the Quercus lobata reference transcriptome [29]. The information of gene names, protein locus and functions was taken from NCBI database based on given information in evoltree database [32]. (XLS 48 kb)
Calculations of Structure Harvester and Evanno method results. Figure S1. Structure Harvester Results. Structure was run with settings as described in the methods section for all K values between 1 and 9. The results were imported to the online version of Structure Harvester [45], yielding support for K = 3 being the best approximation. Table S2. Based on Evanno et al. [46] method results. This table lists Delta K values for K values from 1 to 9 associated with samples that were assigned to the three species of oaks based on morphology. (DOCX 93 kb)
Acknowledgements
We thank Krista Beckley for assistance with project and members of the Sorklab for feedback on the study.
Funding
S. O-B was supported by The Scientific and Technological Research Council of Turkey International Postdoctoral Research Scholar Program during this study at University of California, Los Angeles in USA. Seed funding to VLS from University of California, Los Angeles supported the RNAseq Analyses, S. F-G, and PFG. JMC was supported by a fellowship from the Chinese Academy of Sciences. The valley oak genome assembly v.0.5 used for this study was supported by National Science Foundation Plant Genome Research Program to VLS and PFG (IOS-1444611). None of the agencies providing support for individuals or the project were involved in collecting, analyzing, or interpreting the data or in preparation of the manuscript.
Availability of data and materials
Illumina RNA sequence reads are available through NCBI BioProject accession [PRJNA489462].
Abbreviations
- AL
Alpine
- BWA
Burrows-Wheeler Aligner
- FS
FisherStrand
- J
Julian
- LD
Linkage disequilibrium
- LH
Lake Henshaw
- LM
Laguna Mountain
- LW
Lake Wohlford
- MV
McCain Valley
- OK
Oak Knoll
- PCA
Principal component analysis
- PR
Pala Reserve
- QD
QualByDepth
- SNPs
Single nucleotide polymorphisms
- SY
Santa Ysabel
- Ts/Tv
Transitions to tranversions
Author’s contributions
SO-B, SF-G, and VLS equally contributed to the preparation of the manuscript. JMC designed and collected samples and conducted preliminary analyses with VLS. JMC and PFG performed RNA-Seq analysis and helped revised the manuscript. SF-G and SO-B conducted the bioinformatics and STRUCTURE statistical analyses. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable: This study does not directly involve humans or animals. Plant collection permits were not required because leaf samples were collected along public right-of-way road corridors and no species are endangered or threatened.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Signem Oney-Birol, Email: sobirol@mehmetakif.edu.tr.
Sorel Fitz-Gibbon, Email: sorel@ucla.edu.
Jin-Ming Chen, Email: jmchen@wbgcas.cn.
Paul F. Gugger, Email: pgugger@umces.edu
Victoria L. Sork, Email: vlsork@ucla.edu
References
- 1.Anderson E, Stebbins GL. Hybridization as an evolutionary stimulus. Evolution. 1954;8(4):378–388. doi: 10.1111/j.1558-5646.1954.tb01504.x. [DOI] [Google Scholar]
- 2.Rieseberg LH, Wendel JF. Introgression and its consequences in plants. In: Harrison RG, editor. Hybrid zones and the evolutionary process. Oxford: Oxford University Press; 1993. pp. 70–109. [Google Scholar]
- 3.Arnold ML. Transfer and origin of adaptations through natural hybridization: were Anderson and Stebbins right? Plant Cell. 2004;16:562–570. doi: 10.1105/tpc.160370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold ML, Martin NH. Adaptation by introgression. J Biol. 2009;8:1–3. doi: 10.1186/jbiol176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Carvalho D, Ingvarsson PK, Joseph J, Suter L, Sedivy C, Macaya-Sanz D, Cottrell J, Heinze B, Schanzer I, Lexer C. Admixture facilitates adaptation from standing variation in the European aspen (Populus tremula L.), a widespread forest tree. Mol Ecol. 2010;19:1638–1650. doi: 10.1111/j.1365-294X.2010.04595.x. [DOI] [PubMed] [Google Scholar]
- 6.Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato K, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C. Major ecological transitions in annual sunflowers facilitated by hybridization. Science. 2003;301:1211–1216. doi: 10.1126/science.1086949. [DOI] [PubMed] [Google Scholar]
- 7.Wu CI. The genic view of the process of speciation. J Evol Biol. 2001;14:851–865. doi: 10.1046/j.1420-9101.2001.00335.x. [DOI] [Google Scholar]
- 8.Minder AM, Widmer A. A population genomic analysis of species boundaries: neutral processes, adaptive divergence and introgression between two hybridizing plant species. Mol Ecol. 2008;17:1552–1563. doi: 10.1111/j.1365-294X.2008.03709.x. [DOI] [PubMed] [Google Scholar]
- 9.Rieseberg LH, Blackman BK. Speciation genes in plants. Ann Bot. 2010;106:439–455. doi: 10.1093/aob/mcq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lexer C, Heinze B, Alia R, Rieseberg LH. Candidate gene polymorphisms associated with salt tolerance in wild sunflowers: implications for the origin of Helianthus paradoxus, a diploid hybrid species. New Phytol. 2004;161:225–233. doi: 10.1046/j.1469-8137.2003.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller C. Ecological control of hybridization in Quercus: a factor in the mechanism of evolution. Evolution. 1952;6:147–161. [Google Scholar]
- 12.Whittemore AT, Schaal BA. Interspecific gene flow in sympatric oaks. Proc Natl Acad Sci U S A. 1991;88:2540–2544. doi: 10.1073/pnas.88.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petit RJ, Bodénès C, Ducousso A, Roussel G, Kremer A. Hybridization as a mechanism of invasion in oaks. New Phytol. 2004;161:151–164. doi: 10.1046/j.1469-8137.2003.00944.x. [DOI] [Google Scholar]
- 14.Valen Leigh Van. Ecological Species, Multispecies, and Oaks. Taxon. 1976;25(2/3):233. doi: 10.2307/1219444. [DOI] [Google Scholar]
- 15.Tyree MT, Cochard H. Summer and winter embolism in oak: impact on water relations. Ann For Sci. 1996;53:173–180. doi: 10.1051/forest:19960201. [DOI] [Google Scholar]
- 16.Cavender-Bares J, Bazzaz FA. Changes in drought response strategies with ontogeny in Quercus rubra: implications for scaling from seedlings to mature trees. Oecol. 2000;124(1):8–18. doi: 10.1007/PL00008865. [DOI] [PubMed] [Google Scholar]
- 17.Spieß N, Oufir M, Matušíková I, Stierschneider M, Kopecky D, Homolka A, Burg K, Fluch S, Hausman JF, Wilhelm E. (2012) Ecophysiological and transcriptomic responses of oak (Quercus robur) to long-term drought exposure and rewatering. Environ Exp Bot. 2012;77:117–126. doi: 10.1016/j.envexpbot.2011.11.010. [DOI] [Google Scholar]
- 18.Gugger PF, Peñaloza-Ramírez JM, Wright JW, Sork VL. Whole-transcriptome response to water stress in a California endemic oak. Quercus lobata Tree Physiol. 2017;37:632–44. [DOI] [PubMed]
- 19.Abrams MD. Adaptations and responses to drought in Quercus species of North America. Tree Physiol. 1990;7:227–238. doi: 10.1093/treephys/7.1-2-3-4.227. [DOI] [PubMed] [Google Scholar]
- 20.Nixon K. The oak (Quercus) biodiversity of California and adjacent regions. In: Standiford DMRB, Purcell KL, editors. Proceedings of the fifth symposium on oak woodlands: oaks in California’s changing landscape. San Diego: USDA Forest Service Gen Tech Rep; 2002. pp. 3–20. [Google Scholar]
- 21.Pearse IS, Hipp AL. Phylogenetic and trait similarity to a native species predict herbivory on non-native oaks. Proc Natl Acad Sci U S A. 2009;106:18097–18102. doi: 10.1073/pnas.0904867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortego J, Noguerales V, Gugger PF, Sork VL. (2015) evolutionary and demographic history of the Californian scrub white oak species complex: an integrative approach. Mol Ecol. 2015;24:6188–6208. doi: 10.1111/mec.13457. [DOI] [PubMed] [Google Scholar]
- 23.Sork VL, Ortego J, Riordan E, Gugger PF, Fitz-Gibbon S, Wei X. Phylogeny and introgression of California scrub white oaks (Quercus sect. Quercus). Int. Oak J. 2016;27:61–74. [Google Scholar]
- 24.Riordan EC, Gugger PF, Ortego J, Smith C, Gaddis K, Thompson P, Sork VL. Association of genetic and phenotypic variability with geography and climate in three southern California oaks. Am J Bot. 2016;103:73–85. doi: 10.3732/ajb.1500135. [DOI] [PubMed] [Google Scholar]
- 25.Ortego Joaquín, Gugger Paul F., Riordan Erin C., Sork Victoria L. Influence of climatic niche suitability and geographical overlap on hybridization patterns among southern Californian oaks. Journal of Biogeography. 2014;41(10):1895–1908. doi: 10.1111/jbi.12334. [DOI] [Google Scholar]
- 26.Kim B, Wei X, Fitz-Gibbon S, Lohmueller K, Ortego J, Gugger PF, Sork VL. RADseq data reveal ancient, but not pervasive, introgression within the Californian scrub white oak species complex (Quercus: Fagaceae). Mol Ecol. 2018; 10.1111/MEC.14869. [DOI] [PubMed]
- 27.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive Transcriptome analysis. Curr Protoc Mol Biol. 2010;89:1–4. doi: 10.1002/0471142727.mb0411s89. [DOI] [PubMed] [Google Scholar]
- 29.Cokus SJ, Gugger PF, Sork VL. Evolutionary insights from de novo transcriptome assembly and SNP 2 discovery in California white oaks. BMC Genomics. 2015;6:552. doi: 10.1186/s12864-015-1761-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gugger PF, Cokus SJ, Pellegrini M, Sork VL. Association of transcriptome-wide sequence variation with climate gradients in valley oak (Quercus lobata) Tree Genet Genomes. 2016;12(2):15. doi: 10.1007/s11295-016-0975-1. [DOI] [Google Scholar]
- 31.Sork VL, Squire K, Gugger PF, Steele S, Levy ED, Eckert AJ. Landscape genomic analysis of candidate genes for climate adaptation in a California endemic oak, Quercus lobata Née (Fagaceae) Am J Bot. 2016;103:33–46. doi: 10.3732/ajb.1500162. [DOI] [PubMed] [Google Scholar]
- 32.Evolution of trees as drivers of terrestrial biodiversity: http://www.evoltree.eu (2014). Accessed 15 Nov. 2014.
- 33.Roberts FM. Illustrated guide to the oaks of the Southern California Floristic Province. California: F.M. Roberts Publications; 1995. [Google Scholar]
- 34.Nixon KC, sect MCHQL. Quercus white oaks. In: Flora of North America north of Mexico, editor. Flora of North America Editorial Committee. New York: Oxford University Press; 1997. pp. 436–506. [Google Scholar]
- 35.Scott TA. Conserving California’s rarest oak, Quercus engelmannii. Fremontia. 1990;18:26–29. [Google Scholar]
- 36.Fitz-Gibbon S, Hipp A, Pham K, Manos P, Sork VL. Phylogenomic inferences from reference-mapped and de novo assembled short-read sequence data using RADseq sequencing of California white oaks (Quercus sect. Quercus) Genome. 2017;60:743–755. doi: 10.1139/gen-2016-0202. [DOI] [PubMed] [Google Scholar]
- 37.Ortego J, Riordan EC, Gugger PF, Sork VL. Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol. 2012;21:3210–3223. doi: 10.1111/j.1365-294X.2012.05591.x. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang CC, Christopher C, Chow L, Cam T, Shashaank V, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4(1):7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature Biotech. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker M, Lunter G, Marth G, Sherry ST, et al. (2011) the variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sork VL, Fitz-Gibbon ST, Puiu D, Crepeau M, Gugger PF, Sherman R, Stevens K, Langley CH, Pellegrini M, Salzberg SL. First draft assembly and annotation of the genome of a California endemic oak. Quercus lobata Née (Fagaceae). G3. 2016c;11:3485–95. [DOI] [PMC free article] [PubMed]
- 45.Earl DA, von Holdt BM. Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 46.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 47.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 49.Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28(24):3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tello-Ruiz MK, Stein J, Wei S, Preece J, Olson A, Naithani S, Amarasinghe V, Dharmawardhana P, Jiao Y, Mulvaney J, et al. Gramene 2016: comparative plant genomics and pathway resources. Nucleic Acids Res. 2016;44(D1):133–140. doi: 10.1093/nar/gkv1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon Seok-Jun, Shin Dong-Jin, Kim Beom-Gi, Byun Myung-Ok. Putative fructose-1,6-bisphosphate aldolase 1 (AtFBA1) affects stress tolerance in yeast and Arabidopsis. Journal of Plant Biotechnology. 2012;39(2):106–113. doi: 10.5010/JPB.2012.39.2.106. [DOI] [Google Scholar]
- 53.Xu H, Li Y, Yan Y, Wang K, Gao Y, Hu Y. Genome-scale identification of soybean BURP domain-containing genes and their expression under stress treatments. BMC Plant Biol. 2010;10:197. doi: 10.1186/1471-2229-10-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harshavardhan VT, Seiler C, Junker A, Weigelt-Fischer K, Klukas C, Altmann T, et al. AtRD22 and AtUSPL1 members of the plant-specific BURP domain family involved in Arabidopsis thaliana drought tolerance. PLoS One. 2014;9:e110065. doi: 10.1371/journal.pone.0110065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isokpehi RD, Simmons SS, Cohly HHP, Ekunwe SIN, Begonia GB, Ayensu WK. Identification of drought-responsive universal stress proteins in Viridiplantae. Bioinform Biol Insights. 2011;5:41–58. doi: 10.4137/BBI.S6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson RS, Stewart CN, Gou J, et al. Development and use of a switchgrass (Panicum virgatum L.) transformation pipeline by the BioEnergy science center to evaluate plants for reduced cell wall recalcitrance. Biotechnol Biofuels. 2017;10:309. doi: 10.1186/s13068-017-0991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kushiro T, Okamoto M, Nakabayashi K, et al. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: key enzymes in ABA catabolism. EMBO J. 2004;23(7):1647–1656. doi: 10.1038/sj.emboj.7600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dossa K, Diouf D, Cissé N. Genome-wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front Plant Sci. 2016;7:1522. doi: 10.3389/fpls.2016.01522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seo SG, Jeon SB, Kim JS, et al. Characterization and expression pattern of IbPRP1 and IbPRP2 stress-related genes from sweet potato. Genes Genom. 2010;32:487. doi: 10.1007/s13258-010-0072-z. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Candidate gene list associated with drought stress. Table S1. Candidate loci were taken from evoltree database depending on search tool for drought and hypoxia relates stress (target trait) and matched the sequences to the Quercus lobata reference transcriptome [29]. The information of gene names, protein locus and functions was taken from NCBI database based on given information in evoltree database [32]. (XLS 48 kb)
Calculations of Structure Harvester and Evanno method results. Figure S1. Structure Harvester Results. Structure was run with settings as described in the methods section for all K values between 1 and 9. The results were imported to the online version of Structure Harvester [45], yielding support for K = 3 being the best approximation. Table S2. Based on Evanno et al. [46] method results. This table lists Delta K values for K values from 1 to 9 associated with samples that were assigned to the three species of oaks based on morphology. (DOCX 93 kb)
Data Availability Statement
Illumina RNA sequence reads are available through NCBI BioProject accession [PRJNA489462].