

Abstract
Background
Government regulations require postmarket surveillance for cleared/approved medical devices. Trend analysis of newly marketed devices may help to confirm device-related safety or uncover other device or procedure-related problems.
Methods
Complaints related to the use of 3D-printed triangular titanium implants for sacroiliac joint (SIJ) fusion were compared with those of the prior machined version of the device manufactured with a titanium plasma spray (TPS) coating. Event rates were calculated either by dividing event counts by numbers of surgeries or, for late events, using Kaplan–Meier survival analysis.
Results
Three types of complaints with nontrial frequencies were identified. Issues in instruments occurred at a low and constant rate (1.3%). Using Kaplan–Meier analysis, pain-related complaints occurred at a low and similar rate in both groups (<0.5%). The 1-year cumulative probability of surgical revision was low in both the 3D and machined versions of the device (1.5% for machined and 1% for 3D printed, P=0.0408 for difference). No implant breakages or migrations were identified in either group, and overall rates were similar to a previously published report.
Conclusion
The 3D-printed version of triangular titanium implant was associated with complaint and adverse event rates similar to those for the prior machined version of the device.
Keywords: permanent implants, quality system, complaint analysis, adverse events, postmarket surveillance, sacroiliac joint fusion
Introduction
Following regulatory approvals, both medical device and pharmaceutical manufacturers must perform postmarket surveillance to ensure that use of their products in the commercial setting continues to be safe and effective. As per US1 and European regulations,2 medical device manufacturers must track and evaluate all complaints. US regulations define a complaint as “any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution”. A complaint can be solely a device-related issue without patient impact or an issue that causes harm to a patient. These records are commonly inspected by regulatory authorities. Such tracking aids manufacturers in confirming the safety use of commercialized devices and helps to detect potential trends in safety problems (eg, for hip implants).3 In the US, complaints resulting in patient harm are submitted to United States Food and Drug Administration and available for public review.4
The sacroiliac joint (SIJ) connects the lower lumbar spine with the pelvis. The SIJ is thought to be involved in 15–30% of all chronic lower back pain.5–9 Nonsurgical treatments for SIJ pain are commonly used, but evidence supporting long-term effectiveness is lacking and chronic persistent pain is relatively common. With the commercial availability of purpose-built devices, interest in surgical fusion of the SIJ is growing. Prospective trials of SIJ fusion using titanium plasma spray (TPS)-coated triangular titanium implants (iFuse; SI-BONE, Inc., Santa Clara, CA, USA) have shown the procedure to be safe and effective, providing long-term pain and disability relief for most patients with chronic SIJ dysfunction.10–13
Previously, we reported postmarket surveillance findings for both procedure-related safety events14 and surgical revisions after SIJ fusion using TPS-coated triangular titanium implants.15 The former study reported a relatively low rate of complaints resulting in patient harm. The reported rate was similar to the device and procedure-related adverse events subsequently observed in three prospective trials of the same device.10–12 The latter study reported an important but less common adverse event, namely pain requiring surgical revision.15 Surgical revision rates were low and decreased over time, possibly related to improved surgical techniques and/ or improved training.
Recently, a 3D-printed version of the iFuse implant (iFuse-3D) gained market clearance in the US and EU (Figure 1). The implant, made from the same underlying material (titanium, Ti-6Al-4V ELI, ASTM F136) but produced via additive manufacturing (3D printing) has an optimized porous surface as well as fenestrations designed to allow bone to grow onto and through the center of implant. Using methods derived from the prior published safety studies, we report a comparison of perioperative complaints as well as surgical revision rates of the TPS-coated and 3D-printed implants.
Figure 1.
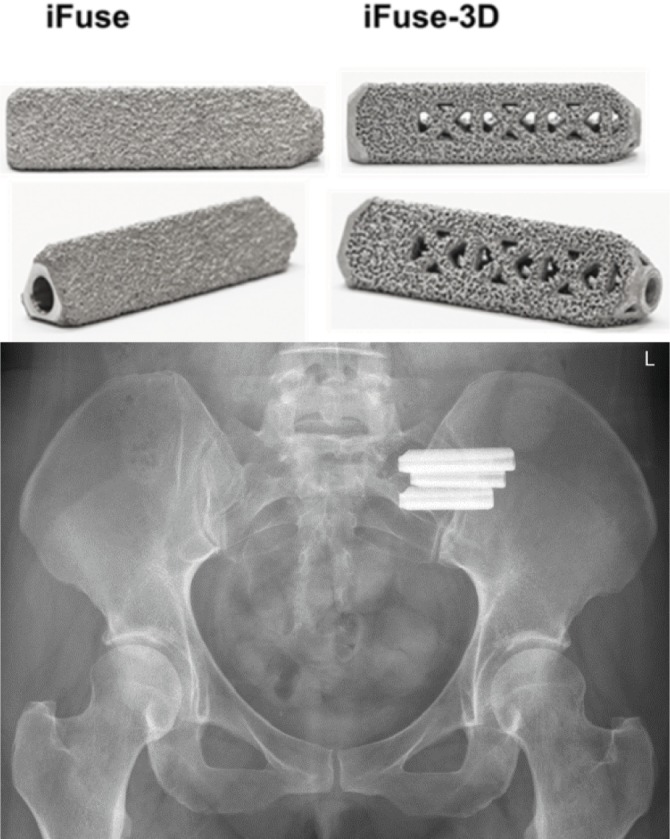
Top: triangular titanium implants for SIJ fusion. Left: machined iFuse implant (commercially available since 2009). Right: 3D-printed implant (iFuse-3D, commercially available since early 2017). Bottom: X-ray image of implants after placement across the left SIJ.
Abbreviation: SIJ, sacroiliac joint.
Methods
Complaint database
Since 2009, key data regarding product complaints reported to SI-BONE, Inc., have been actively maintained. Data captured include procedure date, surgeon name, implant catalog number/lot number, and complaint details. All complaints are investigated as to root cause, and appropriate actions are undertaken as required (eg, device redesign, fixing manufacturing issues, and surgeon retraining) in response to such root causes. When a complaint involves a potential adverse event, a staff surgeon (WCR) reviews the event and may contact the involved surgeon to gather further details. All complaints are evaluated for reportability as required by the US FDA’s Medical Device Reporting regulation1 as well as European2 and other relevant regulations. Institutional review board approval was not required for this study as it was based on an analysis of internal company data routinely collected during postmarket surveillance.
In this report, a surgical revision was defined as an additional surgical procedure on an SIJ treated with the company’s device (iFuse or iFuse-3D). Index cases representing unapproved uses or uses to address the failure of another device were excluded. In addition, analysis focused solely on complaints occurring in the USA and Canada, as these are likely reported with the greatest fidelity. The current analysis included complaints reported to the manufacturer between January 1, 2015, and June 30, 2018.
Statistical analysis
For acute intraoperative events (eg, instrument breakage) or immediate postoperative events (bleeding), rates are reported as the number of events per quarter divided by the number of surgical cases per quarter. Where relevant, acute events were compared across device types with a Fisher’s test.
For events that are more likely to be delayed (eg, surgical revision and pain), Kaplan–Meier survival analysis was additionally used. For patients with revision surgery, “days to event” was defined as days from index surgery date to revision surgery; for patients with pain complaints, days to event was defined as index surgery date to complaint date. Patients without such complaints were censored according to days from index surgery to a fixed date (July 1, 2018). Kaplan–Meier analyses were restricted to surgeries (and their associated complaints) taking place from January 1, 2015, to present. Given the relatively young mean age of patients undergoing this procedure (55.815), expected overall survival is high and lack of vital status information, which could cause early censoring, is not expected to meaningfully affect calculated rates. Survival rates were compared using the log-rank test.
An unknown but small (estimated 10%) proportion of patients undergo staged bilateral surgery, meaning index surgery on one side, followed weeks to months later by index surgery on the other side. The reported number of sides therefore slightly overestimates the number of treated patients.
Results
From January 1, 2015, to June 30, 2018, 14,210 SIJ fusions were performed using either iFuse implants (11,070 cases) or iFuse-3D implants (3,140 cases). iFuse cases occurred at a steady rate until the second quarter of 2017, when iFuse-3D was introduced. By the second quarter of 2018, iFuse-3D was used in >80% of all index surgeries.
During this period, 837 complaints (USA) were reported to SI-BONE, Inc. (Table 1). Most events occurred at a very low rate and were therefore not compared across implant types due to the lack of statistical power. Analysis below focuses on events occurring with more than very low frequencies, namely instrument problems, pain complaints, and revision surgeries. Table 2 shows days elapsed from index surgery date to complaint report date and includes events that typically occur soon after surgery (eg, cardiovascular events and infection) and events that can occur at later times (eg, pain recurrence and revision surgery). We note that the two cases of metal sensitivity were not confirmed via MELISA16,17 (metal-linked immunosorbent assay, see http://www.melisa. org) or LTT (lymphocyte transformation test,18,19 https://www.orthopedicanalysis.com/).
Table 1.
Complaints (USA) reported to manufacturer between January 1, 2015, and June 30, 2018
| Complaint type | 2015 | 2016 | 2017 | 2018 |
|---|---|---|---|---|
| Revision surgery | 140 | 101 | 138 | 56 |
| Pain complaints (general) | 69 | 56 | 33 | 15 |
| Instrument issue | 59 | 47 | 47 | 30 |
| Hematoma/seroma/bleeding | 4 | 2 | 3 | 2 |
| Other medical procedures | 4 | 0 | 2 | 0 |
| Embolism/aneurysm/DVT | 3 | 0 | 0 | 0 |
| iFuse use problem | 2 | 0 | 0 | 0 |
| Allergy (metal) | 1 | 0 | 1 | 0 |
| Bone fracture | 1 | 1 | 0 | 0 |
| iFuse implant product problem | 1 | 1 | 1 | 2 |
| Intraoperative issues | 1 | 0 | 1 | 0 |
| Off-label use | 1 | 2 | 3 | 0 |
| Others | 1 | 0 | 1 | 0 |
| Cardiac incident | 0 | 3 | 0 | 0 |
| Infection | 0 | 1 | 0 | 1 |
| Instrument use problem | 0 | 0 | 0 | 1 |
Abbreviation: DVT, deep venous thrombosis.
Table 2.
Days from index surgery to complaint by complaint type and device type. In some cases, surgery or complaint dates were not available.
| Complaint type | Device type
|
|||||||
|---|---|---|---|---|---|---|---|---|
| iFuse, days from index surgery to complaint
|
iFuse-3D, days from index surgery to complaint
|
|||||||
| N | Mean | SD | Range | N | Mean | SD | Range | |
| Allergy (metal) | 2 | 151.5 | 204.4 | 7–296 | 0 | – | – | – |
| Bone fracture | 2 | 270.5 | 301.9 | 57–484 | 0 | – | – | – |
| Cardiac incident | 3 | 20.7 | 35.8 | 0–62 | 0 | – | – | – |
| Embolism/aneurysm/DVT | 3 | 24 | 16.1 | 11–42 | 0 | – | – | – |
| Hematoma/seroma/bleeding | 8 | 12 | 25.6 | 0–73 | 1 | 15 | – | – |
| iFuse implant product problem | 2 | 4.5 | 6.4 | 0–9 | 0 | – | – | – |
| iFuse use problem | 1 | 0 | – | – | 0 | – | – | – |
| Infection | 1 | 8 | – | – | 1 | 13 | – | – |
| Instrument issue | 31 | 126.5 | 362 | 0–1,529 | 0 | – | – | – |
| Instrument use problem | 0 | – | – | – | 0 | – | – | – |
| Intraoperative issues | 3 | 0.3 | 0.6 | 0–1 | 1 | 0 | – | – |
| Off-label use | 0 | – | – | – | 0 | – | – | – |
| Others | 1 | 965 | – | – | 0 | – | – | – |
| Other medical procedures | 5 | 609 | 177.8 | 413–819 | 0 | – | – | – |
| Pain complaints (general) | 151 | 520.3 | 376.8 | 3–1,651 | 3 | 41 | 52 | 2–100 |
| Revision surgery | 405 | 542.8 | 533.6 | 0–2,626 | 26 | 72.1 | 100.9 | 2–408 |
| All | 618 | 497.4 | 495.1 | 0–2,626 | 32 | 63.3 | 93.7 | 0–408 |
Note: Endashes indicate no value or statistic not calculable. In 4 cases, revision or index surgery dates were not known.
Abbreviation: DVT, deep venous thrombosis.
Instrument problem
For iFuse implants, the rate of damaged instruments occurred at a mean rate of 1.3% with no obvious changes over time (Table 3). As the same instruments are used to place iFuse and iFuse-3D, no analysis across implant type was performed.
Table 3.
Instrument issue rate (USA) by quarter between January 1, 2015, and June 30, 2018
| Year/quarter | N complaints | N cases | Rate (%) |
|---|---|---|---|
| 2015/1 | 16 | 936 | 1.7 |
| 2015/2 | 22 | 957 | 2.3 |
| 2015/3 | 10 | 909 | 1.1 |
| 2015/4 | 11 | 921 | 1.2 |
| 2016/1 | 8 | 865 | 0.9 |
| 2016/2 | 9 | 944 | 1.0 |
| 2016/3 | 14 | 917 | 1.5 |
| 2016/4 | 16 | 1,067 | 1.5 |
| 2017/1 | 14 | 1,008 | 1.4 |
| 2017/2 | 8 | 1,018 | 0.8 |
| 2017/3 | 11 | 1,067 | 1.0 |
| 2017/4 | 14 | 1,226 | 1.1 |
| 2018/1 | 12 | 1,133 | 1.1 |
| 2018/2 | 19 | 1,242 | 1.5 |
Note: The number of iFuse and iFuse-3D cases is lumped since the same instrument sets are used independent of implant type.
Pain complaints
A total of 173 pain complaints (170 with iFuse and three with iFuse-3D) were reported to the manufacturer between January 2015 and June 2018. For surgeries performed after January 1, 2015, the probability of a pain complaint event, evaluated by Kaplan–Meier methodology, was very low (1-year rate <0.5%) and showed no difference across device type (log rank P=0.138). These complaints represented a variety of issues, including transient pain after surgery, wound infection, persistent pain, and pain recurrence. None of these complaints resulted in a surgical revision of the treated side.
Revision surgery
The company was notified of a total of 435 revision surgeries between January 2015 and June 2018: 409 in iFuse cases and 26 in iFuse-3D cases. For surgeries performed after January 1, 2015, the 1-year product-limit estimate of the cumulative rate of revision surgery was 1.5% for iFuse and 1.0% for iFuse-3D (P=0.0408, Figure 2). For these revisions, implant malposition resulting in symptomatic nerve impingement was the most common reason (n=151, 54% of all revisions, Table 4), occurring at a median of 29 days after index surgery. There were no differences in time to revision for symptomatic implant malposition across device types (29 days for iFuse vs 41 days for iFuse-3D, P=0.9). Revisions for other causes occurred at a median of 367–478 days after surgery. These late revisions include procedures performed to treat symptomatic pseudoarthrosis, lack of improvement in malpositioned implants (not placed fully across the SIJ), and a small number procedures where implants were removed because the patients obtained no pain relief from the index procedure, possibly due to misdiagnosis.
Figure 2.
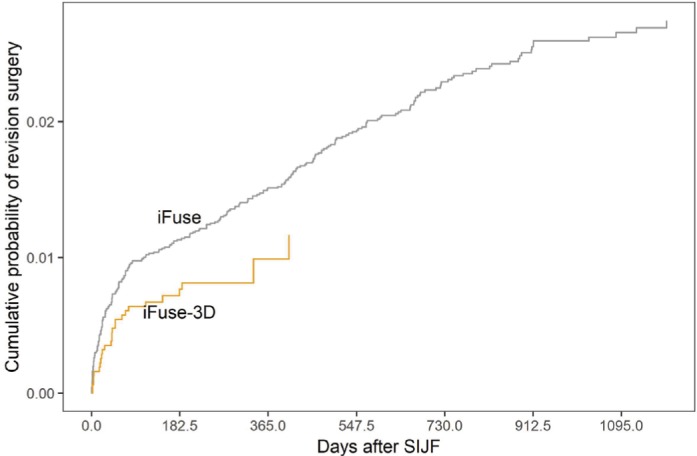
Cumulative probability of implant revision surgery after iFuse or iFuse-3D.
Abbreviation: SIJF, sacroiliac joint fusion.
Table 4.
Suspected cause for surgical revisions (USA only) by device type for surgeries performed between January 1, 2015, and June 30, 2018
| Suspected cause | iFuse
|
iFuse-3D
|
Total
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Percentage | Median | N | Percentage | Median | N | Percentage | Median | |
| Insufficient fixation | 51 | 20.2 | 408 | 1 | 3.8 | 63 | 52 | 18.7 | 400.5 |
| Lucency/halos | 26 | 10.3 | 477.5 | 0 | 0 | – | 26 | 9.4 | 477.5 |
| Malpositioned: nerve impingement | 127 | 50.4 | 29 | 24 | 92.3 | 41 | 151 | 54.3 | 29 |
| Malpositioned: not nerve-related | 19 | 7.5 | 402 | 1 | 3.8 | 182 | 20 | 7.2 | 367.5 |
| No pain relief | 15 | 6 | 456 | 0 | 0 | – | 15 | 5.4 | 456 |
| Others | 14 | 5.6 | 414 | 0 | 0 | – | 14 | 5 | 414 |
| Total | 252 | 100 | 189.5 | 26 | 100 | 42 | 278 | 100 | 146.5 |
Note: Median number of days from index surgery to revision surgery.
Discussion
The major findings of our postmarket surveillance analysis are as follows: 1) complaints related to use of triangular titanium implants for SIJ fusion occur at a low rate and 2) the complaint rate for iFuse-3D (newer version of device) appears to be qualitatively similar to the older, machined version of the device. Notably, we did not observe any instances of device breakage or migration. No new types of complaints related to iFuse-3D were identified.
The complaint rate for instrument issues (primarily damaged instruments) has been consistent at ~1.3% of all US cases. There were no reported adverse events related to damaged instruments. Instrument damage due to user error is the most common occurrence (eg, bending or cutting of guide pins due to noncollinearity during drilling, damaged chisel edges when the removal guide is not correctly lined up with the iFuse Implant and guide pin clamps that are not used correctly). Other instrument damages such as breaking of soft tissue protector heads, through which other instruments used for device placement are passed, may have been due to heat-related damage during re-sterilization.
The rate of pain complaints to the manufacturer was very low, and reported incidents were highly variable. None required revision surgery.
Surgical revisions after any surgical procedure are an important clinical outcome. Previously, we reported that surgical revision rates with triangular titanium implants observed in the commercial and clinical trial settings were both low and of similar magnitude.15 One-year surgical revision rates after SIJ fusion surgery (~1.5%) appear to be substantially lower than those reported for lumbar stenosis surgery (4.1% at 1 year)20 and lumbar arthrodesis (4–5%).21
Surgical revisions after SIJ fusion with iFuse implants fall into two general categories: early and late revisions. Early revisions are typically for symptomatic implant malposition, which can result in the impingement of the L5 or S1 nerve roots that causes immediate postoperative radiating leg pain. Pain typically responds favorably to repositioning of the offending implant. Late revisions are performed to treat symptomatic pseudoarthrosis (recurrent pain, sometimes associated with implant loosening) or to remove implants in cases where a patient never had pain relief (most likely as a result of misdiagnosis). Failure of pain to improve after SIJ fusion can be due to poor implant position with insufficient stabilization or initial misdiagnosis. Our data provide insight into the relative occurrence rates of these events. Based on reported complaints, approximately half of surgical revisions, typically performed soon after the index procedure, are due to implant malposition resulting in nerve impingement (Table 4). Of revisions performed after the perioperative period, causes are primarily insufficient fixation (~40%), radiolucency (23%), malposition (14%), and failure to relieve pain (13%). The number of late revisions with iFuse-3D was too small to make any rate comparisons across device type. Our data do not allow any conclusions regarding the rate of misdiagnosis (ie, patient had a condition other than SIJ pain).
Previously, we reported a reduction in the rate of surgical revisions from initial product launch (2009) to 2015, which we attributed to increased physician experience and perhaps improved training.15 The 2-year revision rate for 2012–2014 reported in the prior study (2.2%) was the same as that observed in the current study from 2015 to present (2.3%).
Our study has several strengths. The number of index surgeries performed is carefully recorded since implants, in almost all cases, are provided directly through company representatives. A company representative is almost always in attendance for both index procedures and surgical revisions. Revisions typically require special instrumentation manufactured by SI-BONE, Inc., and the removed implant is often replaced with another SI-BONE implant. Finally, company staff are instructed to report all complaints, especially revisions, to complaint-handling staff.
Limitations to the study are as follows. First, despite rigorous training, some complaints may not be reported and some physicians may choose not to report a complaint (eg, if a revision was done with another product) to the manufacturer; the extent of underreporting cannot be determined. Nonetheless, the revision rate calculated in the current study is similar to that reported in fully monitored prospective trials, suggesting that under-reporting may not be marked.22 Second, because iFuse-3D is newer, the follow-up period for patients undergoing treatment with the 3D-printed implant is shorter than with the machined implant. The relatively shorter follow-up period likely explains the higher proportion of revision cases in the 3D implant that are for acute causes (implant malposition resulting in nerve impingement). However, through the use of Kaplan–Meier methods to adjust for differences in follow-up times, our data provide relatively strong evidence that late revisions with the 3D products are not increased compared to the prior machined version of the device. iFuse-3D’s surface is designed to increase bone on-growth and allow through-growth, but whether these features result in lower revision rates will require further follow-up. Third, our analysis disregards the chance that patients could be censored due to death from other causes, which could result in the underestimation of revision rates.
Despite these limitations, our study provides evidence to support a postmarket surveillance safety profile for iFuse-3D that is similar to the original device.
Conclusion
Complaints and adverse events with the use of a 3D-printed triangular titanium implant for SIJ fusion occur at low rates similar to those of the prior machined version of the device.
Footnotes
Disclosure
All authors are employees of SI-BONE, Inc., which manufactures the implants described herein. The authors report no other conflicts of interest in this work.
References
- 1.FDA [webpage on the Internet] Medical Device Reporting. Code of Federal Regulations, Title 21, Part 803. [Accessed August 18, 2018]. Available from: http://www.access-data.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=803.
- 2.Guidelines on a medical devices vigilance system MEDDEV 2.12.1 rev 8. 2013. [Accessed September 10, 2018]. Available from: http://ec.europa.eu/DocsRoom/documents/15506.
- 3.Urban RM, Jacobs JJ, Tomlinson MJ, Gavrilovic J, Black J, Peoc’h M. Dissemination of wear particles to the liver, spleen, and abdominal lymph nodes of patients with hip or knee replacement. J Bone Joint Surg Am. 2000;82(4):457–477. doi: 10.2106/00004623-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 4.FDA [webpage on the Internet] MAUDE – Manufacturer and User Facility Device Experience. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm. Accessed July 11, 2018.
- 5.Bernard TN, Kirkaldy-Willis WH. Recognizing specific characteristics of nonspecific low back pain. Clin Orthop Relat Res. 1987;217:266–280. [PubMed] [Google Scholar]
- 6.Schwarzer AC, Aprill CN, Bogduk N. The sacroiliac joint in chronic low back pain. Spine. 1995;20(1):31–37. doi: 10.1097/00007632-199501000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Maigne JY, Aivaliklis A, Pfefer F. Results of sacroiliac joint double block and value of sacroiliac pain provocation tests in 54 patients with low back pain. Spine. 1996;21(16):1889–1892. doi: 10.1097/00007632-199608150-00012. [DOI] [PubMed] [Google Scholar]
- 8.Irwin RW, Watson T, Minick RP, Ambrosius WT. Age, body mass index, and gender differences in sacroiliac joint pathology. Am J Phys Med Rehabil. 2007;86(1):37–44. doi: 10.1097/phm.0b013e31802b8554. [DOI] [PubMed] [Google Scholar]
- 9.Sembrano JN, Polly DW. How often is low back pain not coming from the back? Spine. 2009;34(1):E27–E32. doi: 10.1097/BRS.0b013e31818b8882. [DOI] [PubMed] [Google Scholar]
- 10.Polly DW, Swofford J, Whang PG, et al. Two-Year Outcomes from a Randomized Controlled Trial of Minimally Invasive Sacroiliac Joint Fusion vs. Non-Surgical Management for Sacroiliac Joint Dysfunction. Int J Spine Surg. 2016;10 doi: 10.14444/3028. Article 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dengler JD, Kools D, Pflugmacher R, et al. 1-Year Results of a Randomized Controlled Trial of Conservative Management vs. Minimally Invasive Surgical Treatment for Sacroiliac Joint Pain. Pain Physician. 2017;20(6):537–550. [PubMed] [Google Scholar]
- 12.Duhon BS, Bitan F, Lockstadt H, Kovalsky D, Cher D, Hillen T, SIFI Study Group Triangular Titanium Implants for Minimally Invasive Sacroiliac Joint Fusion: 2-Year Follow-Up from a Prospective Multicenter Trial. Int J Spine Surg. 2016;10 doi: 10.14444/3013. Article 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanaclocha V, Herrera JM, Sáiz-Sapena N, Rivera-Paz M, Verdú-López F. Minimally Invasive Sacroiliac Joint Fusion, Radiofrequency Denervation, and Conservative Management for Sacroiliac Joint Pain: 6-Year Comparative Case Series. Neurosurgery. 2018;82(1):48–55. doi: 10.1093/neuros/nyx185. [DOI] [PubMed] [Google Scholar]
- 14.Miller LE, Reckling WC, Block JE. Analysis of postmarket complaints database for the iFuse SI Joint Fusion System®: a minimally invasive treatment for degenerative sacroiliitis and sacroiliac joint disruption. Med Devices. 2013;6:77–84. doi: 10.2147/MDER.S44690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cher D, Reckling WC, Capobianco R. Implant survivorship analysis after minimally invasive sacroiliac joint fusion using the iFuse Implant System®. Med Devices (Auckl) 2015;8:485–492. doi: 10.2147/MDER.S94885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stejskal VD, Cederbrant K, Lindvall A, Forsbeck M. MELISA-an in vitro tool for the study of metal allergy. Toxicol In Vitro. 1994;8(5):991–1000. doi: 10.1016/0887-2333(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 17.Valentine-Thon E, Schiwara HW. Validity of MELISA for metal sensitivity testing. Neuro Endocrinol Lett. 2003;24(1-2):57–64. [PubMed] [Google Scholar]
- 18.Hallab NJ. Lymphocyte transformation testing for quantifying metal-implant-related hypersensitivity responses. Dermatitis. 2004;15(2):82–90. doi: 10.2310/6620.2004.03054. [DOI] [PubMed] [Google Scholar]
- 19.Hallab NJ, Mikecz K, Vermes C, Skipor A, Jacobs JJ. Differential lymphocyte reactivity to serum-derived metal-protein complexes produced from cobalt-based and titanium-based implant alloy degradation. J Biomed Mater Res. 2001;56(3):427–436. doi: 10.1002/1097-4636(20010905)56:3<427::aid-jbm1112>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 20.Deyo RA, Martin BI, Kreuter W, Jarvik JG, Angier H, Mirza SK. Revision surgery following operations for lumbar stenosis. J Bone Joint Surg Am. 2011;93(21):1979–1986. doi: 10.2106/JBJS.J.01292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin BI, Mirza SK, Comstock BA, Gray DT, Kreuter W, Deyo RA. Reoperation rates following lumbar spine surgery and the influence of spinal fusion procedures. Spine. 2007;32(3):382–387. doi: 10.1097/01.brs.0000254104.55716.46. [DOI] [PubMed] [Google Scholar]
- 22.Dengler J, Duhon B, Whang P, et al. Predictors of Outcome in Conservative and Minimally Invasive Surgical Management of Pain Originating from the Sacroiliac Joint: A Pooled Analysis. Spine. 2017;42(21):1664–1673. doi: 10.1097/BRS.0000000000002169. [DOI] [PMC free article] [PubMed] [Google Scholar]