

Abstract
In the present work, we report the use of TiO2 nanotube (NT) layers with a regular intertube spacing that are decorated by Pt nanoparticles through the atomic layer deposition (ALD) of Pt. These Pt‐decorated spaced (SP) TiO2 NTs are subsequently explored for photocatalytic H2 evolution and are compared to classical close‐packed (CP) TiO2 NTs that are also decorated with various amounts of Pt by using ALD. On both tube types, by varying the number of ALD cycles, Pt nanoparticles of different sizes and areal densities are formed, uniformly decorating the inner and outer walls from tube top to tube bottom. The photocatalytic activity for H2 evolution strongly depends on the size and density of Pt nanoparticles, driven by the number of ALD cycles. We show that, for SP NTs, a much higher photocatalytic performance can be achieved with significantly smaller Pt nanoparticles (i.e. for fewer ALD cycles) compared to CP NTs.
Keywords: anodization, atomic layer deposition, photocatalysis, platinum, spaced TiO2 nanotubes
1. Introduction
Since the pioneering work of Fujishima and Honda, the production of H2 through the photocatalytic splitting of H2O on semiconductor materials has been intensively investigated.1 Among the various photocatalysts studied, titanium dioxide (TiO2) with a band gap energy E g≈3.0–3.2 eV has attracted wide attention, mostly owing to its suitable conduction and valence band‐edge position for photogenerated charge carriers to react with water as well as for its stability against photocorrosion.2, 3 The key to the photocatalytic activity of TiO2 in aqueous solutions arises from the fact that the conduction band edge of TiO2 lies higher than the redox potential of water (−0.45 V and 0 V vs. NHE); this enables it to reduce H2O to H2 with photoexcited electrons.
Throughout the past decades, nanotubular TiO2 geometries have received huge scientific attention in various fields; but, in particular in the photocatalytic field, as they combine a surface area with directionality to light and charge management.4, 5 Generally, anodic TiO2 nanotube arrays grow in a hexagonally close‐packed (CP) configuration, that is, they only have minor spacing between the tubes. Differently, spaced (SP) TiO2 nanotubular arrays that can be grown under specific anodization conditions offer a defined intertube spacing. Recently, it has been shown that spacing between NTs improves the accessibility of outer‐tube walls, and this results in better inter‐diffusion and the formation of interdigitated structures that often outperform CP tubes, even though as‐formed SP NTs have a less active surface area than conventional CP NTs.6, 7, 8
In photocatalysis, bare anatase TiO2 nanotubes exhibit a low efficiency for H2 production, owing to a kinetically slow electron transfer to reactants. To improve the photocatalytic activity, a common approach is to use a co‐catalyst that aids charge separation and transfer. As such, Pt generally leads to the highest activity towards hydrogen evolution, as Pt nanoparticles (NPs) provide a favorable solid‐state junction for TiO2 that can improve the electron‐transfer kinetics at the photocatalyst/environment interface.9 In addition, Pt can also catalyze the hydrogen atom recombination reaction, thus facilitating H2 gas formation (2H0→H2).10−12
However, the cost of Pt demands an optimized deposition technique that reduces the loading of Pt while yielding maximum photocatalytic activity. On 3D nanotubular arrays, it is hard to achieve uniform and well‐dispersed Pt particles with precisely controlled size and quantity through traditional deposition techniques (e.g. photo‐deposition,13 vapor deposition,14 electrochemical deposition,15 or sputter deposition16). In contrast, atomic layer deposition (ALD) can yield the uniform deposition of complex nanostructures and allows for a controlled loading amount by varying the number of ALD cycles.17, 18 Also, it provides precise thickness control conformability on high‐aspect‐ratio nanostructures.19, 20
In the present work, we evaluate the deposition of Pt by using ALD into SP and CP TiO2 NTs using different numbers of ALD cycles, which influences the size and density of Pt nanoparticles. The objective is to maximize the H2 evolution rate of platinized SP TiO2 NTs under UV and solar light, and compare the results with conventional CP TiO2 NTs.
2. Results and Discussion
The SP TiO2 NTs were produced by anodization in a DMSO‐based electrolyte (i.e. containing 0.3 wt % NH4F, 7 wt % H2O, and 4 wt % HF) by following the procedure described previously.21 Figure 1 a shows an SEM image of the SP NTs obtained at 30 V for 1 h (30 °C). Such nanotubular layers provide a distinct spacing of approximately 139±40 nm in between individual nanotubes from top to bottom. This in contrast to classical anodic nanotubes, used as a reference, that are fabricated in an EG‐based electrolyte and grow in a CP arrangement, as illustrated in Figure 1 b. The geometrical features of the as‐formed NTs are as follows: EG CP NTs have an inner diameter of 120–140 nm under an initiation layer (that stems from the two‐step anodization) and a wall thickness of 17 nm. SP NTs have an outer diameter of 127±9 nm, inner diameter of 112±8 nm, and wall thickness of 14±2 nm. Note that the SP NTs have a length of 2.5 μm, whereas the CP NTs are around 2 μm long.
Figure 1.
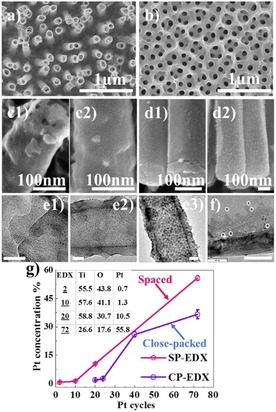
Top SEM images show bare a) spaced (SP) and b) close‐packed (CP) NTs. SEM side view images of SP NTs after c1) two cycles and c2) ten cycles, and CP NTs after d1) 20 cycles and d2) 26 cycles. High‐resolution TEM images of SP NTs after e1) two, e2) ten, and e3) 20 cycles. f) High‐resolution TEM image of CP NTs after 24 cycles. Scale bars are 30 nm if not stated otherwise. g) Variation of Pt atomic concentration of SP and CP NTs as a function of Pt ALD cycles (inset shows at % of Ti, O, and Pt of Pt deposited SP NTs), measured by using EDX.
The SP and CP NTs were decorated with Pt using ALD by applying different cycle numbers, as described in the Experimental Section. Figures 1 c –d and S1 display examples of SEM images of the SP and CP NTs with the Pt nanoparticle decoration after the ALD process. For both TiO2 NTs, Pt can be decorated uniformly on the inner and outer side of SP and CP NTs from top to bottom. The distribution and size of the Pt nanoparticles were evaluated from TEM images (examples are shown in Figures 1 e and 1 f). For the SP tubes, the Pt nanoparticles are apparent already for the lowest ALD cycle number used in this work (i.e. two cycles), and the amount and size increase with increasing number of ALD cycles. The total loading of Pt was evaluated by EDX, see Figure 1 g, where the atomic concentration of Pt increases from 0.7 to 55.8 at % with an increasing number of ALD cycles from 2 to 72. At low ALD cycle numbers, the increase in Pt loading is relatively slow, namely, it increases from 0.7 at % (after two cycles) to 1.3 at % (after ten cycles). The amount of Pt loading is determined as 0.003 mg cm−2 after two cycles and 0.42 mg cm−2 at ten cycles. In the case of CP NTs, only after 20 ALD cycles can particle growth be observed and an atomic concentration of 1.7 % is reached, which has a similar atomic concentration to the SP TiO2 NTs after ten cycles. At lower ALD cycle numbers (i.e. 8 or 16 cycles), Pt is not detectable on CP NTs through EDX analysis. On the other hand, at higher ALD cycle numbers (i.e. 72 cycles), Pt loading reaches 36 at % on CP NTs, whereas on SP NTs the deposited amount is approximately 56 at % Pt; see Figure 1 g. This finding indicates the first advantageous feature of SP NTs over CP NTs, that is, the spacing enables significant initiation of Pt particle growth on the NT walls.
In the literature, the importance of surface termination on the nucleation and growth of ALD is discussed in detail.22−24 To determine the influence of the chemical state of the surface, we performed XPS analysis of reference SP and CP NTs (annealed at 450 °C in air), see Ti 1s and fitted O 1s peaks in Figure S2. The findings show that both SP and CP morphologies have a similar surface composition/termination and contain −OH and C−O bonds on the surface. Therefore, the higher growth rate of Pt particles on SP NTs is attributed to the loose morphology of nanotubes, which improves the diffusion of precursor and is not attributed to surface termination of the different tubes.
Figure 2 and Figure S3 give further TEM images of SP NTs after 2, 10, and 20 ALD cycles. As can be seen from the high‐resolution TEM image, SP NTs after ten cycles provide a lattice spacing of 3.49 Å, corresponding to (101) anatase crystal (JPCD 00–021–1272), and 2.31 Å (111), corresponding to metallic Pt (JPCD 01–087–0644). The selected area diffraction (SAD) can be assigned to contributions of the anatase response from different planes, namely, (101), (103), (105), (213), and to Pt (200) planes (Figure 2 b).
Figure 2.
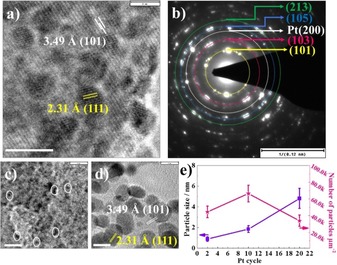
a) High‐resolution TEM image of SP NTs after ten cycles, showing the lattice spacing of anatase [3.49 Å, (101) plane] and metallic Pt [2.31 Å, (111)]. b) Selected area diffraction (SAD) pattern of SP NTs after ten cycles, demonstrating the anatase phase of TiO2 and metallic Pt. High‐resolution TEM images of SP NTs after c) two cycles and d) 20 cycles, illustrating the lattice spacing of anatase [3.49 Å, (101)] and metallic Pt [2.31 Å, (111)]. Scale bars of TEM images are 5 nm if not stated otherwise. e) The variations of particle size and number of particles per μm2 as a function of Pt ALD cycle number.
By using TEM images, statistical analyses of the Pt nanoparticles was carried out, which is summarized in Figure 2 e. For the SP tubes, the size and density of the particles vary as a function of ALD cycle number. The particle size is 0.9±0.2 nm after two cycles, 1.85±0.9 nm after ten cycles, and 4.9±1 nm after 20 cycles. Similarly, the number of particles (particle areal density) increases with ALD cycle number until coalescence occurs. This demonstrates the fact that particle size, density, and coalescence can be controlled by the number of ALD cycles. In addition, these results confirm that sub‐nanometer nanoparticles in TiO2 NTs can be deposited deep in a 3D structure in a homogenous manner by using ALD. It is important to note that, for CP tubes, an onset of deposition is observed only after ≥20 cycles for 7 μm CP NTs,25 demonstrating a strong difference to SP NTs, for which ALD creates Pt deposition already after only two deposition cycles. This finding is ascribed to the enhanced diffusion of ALD precursors toward SP tube walls in comparison with CP NTs. The formation of sparse particles can be explained by the surface diffusion of Pt atoms that occurs during the initial cycles or the early stage of ALD, leading to a distinct distribution of Pt particles on the surface rather than a continuous monolayer of atoms. The size of the sparse particles increases with the cycle number, eventually leading to coalescence into a continuous film.26, 27 In line with the literature, for higher ALD cycle numbers, Pt deposition loses its particulate nature and Pt fully covers the tube walls like a continuous film, in this case after 72 cycles (see Figure S4 in the Supporting Information). Clearly, at 72 cycles, Pt forms a continuous film on the outer as well as on the inner wall of nanotubes.
Figure 3 a shows the XRD patterns of the SP NTs with/without Pt decoration. The as‐formed TiO2 NTs are amorphous. After annealing, TiO2 NTs crystallize in the form of anatase, and the peak is recognized at 25.2° [(101), JCPDS 00–021–1272] and a rutile peak is identified at 27.4° [(110), JCPDS 00–021–1276]. In the case of Pt‐deposited NTs, the XRD pattern confirms the presence of metallic Pt by the main peak positioned at 38.7° [(111), JCPDS 087–0644]. The visibility of the Pt peak improves with an increasing number of ALD cycles. The size of the Pt particles is calculated by using the Scherrer equation [Eq. (1) in the Experimental Section] and determined as 10 nm after 20 cycles (note that particle size is calculated as 25.7 nm at 72 cycles; however, the continuous film‐like structure of Pt after 72 cycles makes it difficult to define the particle size by using SEM or TEM images). These TEM size results agree with the particle size calculated by the Scherrer equation. The minor difference between the particle size determined by using TEM and XRD is ascribed to the shape factor (i.e. the shape of Pt particle is not truly spherical) or strain broadening of the XRD peak.
Figure 3.
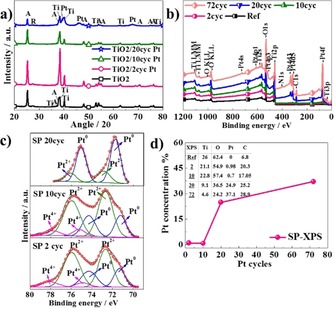
a) XRD pattern of bare and Pt‐deposited SP TiO2 NTs (2, 10, and 20 cycles). b) XPS survey spectra of bare and Pt‐deposited SP TiO2 NTs (2, 10, 20, and 72 cycles). c) Fitted high‐resolution XPS of Pt 4f spectra of Pt‐deposited SP TiO2 NTs (2, 10, and 20 cycles). d) Variation of Pt atomic concentration as a function of ALD cycle number for SP NTs (inset shows at % of Ti, O, Pt, C of bare and Pt deposited SP NTs), measured by using XPS.
The chemical compositions of the reference and Pt‐deposited tubular layers were analyzed by using XPS. Figure 3 b displays XPS survey spectra of reference/bare and Pt ALD‐deposited SP NTs after 2, 10, 20, and 72 cycles. According to the survey spectra, the NTs contain Ti, O, Pt, and C. Fitting of the high‐resolution spectrum in the Pt 4f region (see Figure 3 c) gives evidence of the presence of metallic Pt for Pt 4f7/2 at 71.3 and 74.3 eV, besides a high contribution of oxides is observed at low number of ALD cycles (two and ten cycles); PtII (PtO) is apparent at binding energies of 72.7 and 75.9 eV, and PtIV (PtO2) can be found at 74.9 and 78.1 eV. After 20 ALD cycles, essentially all Pt is presented as metallic Pt, and only a small amount of PtII species can be observed. The oxidation of Pt is frequently found for very small sized Pt, which is more susceptible to oxidation.28−31
The chemical composition of layers with different cycle numbers, which is extracted from XPS analysis, is outlined in Figure 3 d. Although the Pt concentration does not vary considerable after two (0.98 at %) or ten cycles (0.72 at %), it is significantly enhanced after 20 cycles, reaching 24.9 at %. Note that the variation between at % of Pt obtained by EDX and XPS analysis arises because of the difference in working principle of EDX and XPS. While the information depth of EDX is several micrometers, XPS gives information about the top most layer (i.e. only a few nanometers).
Figure 4 a summarizes the photocatalytic H2 evolution results as a function of the number of ALD cycles for SP and CP NTs, using either a UV laser as a light source or a solar light (AM 1.5, 100 mW cm−2). The bare SP TiO2 nanotubes show a photocatalytic activity of 1.05 and 0.72 μL h−1 under UV and solar light, respectively. For the Pt‐deposited NTs, the maximum H2 generation is observed for ten ALD cycles (171.6 μL h−1), followed by that for two cycles (150.75 μL h−1) under UV light. Likewise, under solar illumination, the highest activity towards H2 generation is measured after ten ALD cycles (10.53 μL h−1) followed by that for two cycles (9.41 μL h−1). The large difference between the evolved H2 under UV and solar light is attributed to the different light absorption characteristics of Pt‐decorated TiO2 NT layers at different wavelengths.25
Figure 4.
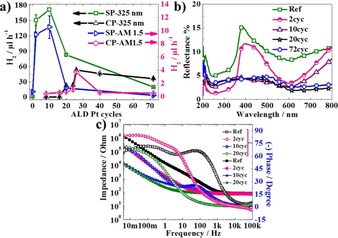
a) H2 evaluation rate of SP and CP TiO2 NTs as a function of ALD cycle number under UV (λ=325 nm) and solar light. b) Total reflectance spectra of bare and Pt‐deposited SP NTs after two, ten, 20, and 72 ALD cycles. c) Impedance and phase variation graph of bare and Pt deposited SP NTs after two, ten, and 20 cycles.
When the number of ALD cycles is increased to 20 or 72, the nanostructured electrodes exhibit a lower activity towards H2 generation, that is, 20 ALD cycles leads to 83 μL h−1 and 72 cycles yields 20 μL h−1 under UV light. Under solar illumination, SP NTs at a higher number of ALD cycles display poor photocatalytic hydrogen generation rate, for instance, 1.29 μL h−1 after 20 cycles and 0.16 μL h−1 after 72 cycles. Clearly, the photocatalytic H2 generation rate enhances initially with increasing Pt content, reaches a maximum, and then declines once the Pt content is beyond an optimized amount. The reduction in the photocatalytic activity is attributed to 1) an oxide shading effect by Pt, that is, a large amount of Pt deposition shades the photosensitive TiO2 surface or hinders the light absorption, thus decreasing the surface concentration of electrons and holes available for reaction;32 2) larger Pt particles or higher metal loadings may provide more recombination sites for photogenerated electrons and holes.33
Furthermore, we evaluated CP NTs deposited with Pt by using a similar approach. For the CP NTs, the highest photocatalytic H2 generation is measured to be 53 μL h−1 after 26 cycles under a 325 nm laser and 3.69 μL h−1 under solar illumination. Although, after 26 cycles, CP TiO2 NTs have a higher amount of Pt than SP TiO2 NTs after ten cycles, they show a photocatalytic H2 generation rate that is three times lower. For instance, CP NTs formed after 20 ALD cycles, which have a similar Pt loading tp SP NTs after ten cycles (representing an optimized amount), present a photocatalytic hydrogen generation rate that is seven times lower (22.5 μL h−1). Note that we also performed H2 evolution for CP NTs at lower ALD cycles; a hydrogen generation rate of 0.43 μL h−1 after eight cycles and 0.5 μL h−1 after 16 cycles were observed under solar illumination. The experimental findings clearly suggest that spacing between NTs leads to a higher photocatalytic activity at low ALD cycle numbers. This may be ascribed to two factors: 1) spacing improves the accessibility of external tube walls (diffusivity of Pt precursor) and leads to more Pt deposition at low cycles, and 2) spacing improves the light absorption behavior of TiO2 NTs and leads to better use of active surface area (diffusivity of reactants). Furthermore, we compared the photocatalytic hydrogen generation rate of ALD Pt‐decorated spaced NTs (with a loading similar to 2, 10, and 20 ALD cycles) to decoration by using a conventional sputtering approach; see Figure S6 in the Supporting Information. In this approach, SP NTs were sputter deposited with 0.66 at % (CA1), 1.68 at % (CA2), and 9.3 at % Pt (CA3). Clearly, the sputter‐deposited SP NTs display a poor photocatalytic activity; the amounts of generated H2 for the CA1, CA2, and CA3 samples were 5, 4.3, and 1.5 μl h−1 under solar illumination, respectively. The amount of generated H2 is two times lower than the amount of H2 produced from ALD Pt‐deposited SP NTs. The lower performance for the sputtered samples can be ascribed to the non‐uniform deposition (thicker Pt layer at the tube top and low or no Pt loading on the inner or outer tube walls) and the resulting shading effect of Pt.
We also tested the stability and reusability of SP NTs after ten Pt ALD cycles through a photocatalytic cycling test. As shown in Figure S7 a, the result indicates a minor variation in the photocatalytic activity after four cycles, demonstrating good stability of the photocatalyst. Even after a four‐day cycling test, the H2 evolves steadily over time (Figure S7 b). This illustrates the high stability of the Pt‐deposited SP nanotubular arrays.
The optical behavior of bare and Pt‐deposited SP NTs was evaluated by using reflectance measurements. Figure 4 b illustrates the total reflectance of the bare and Pt‐decorated SP nanotubular layers. Although bare NTs show low reflectance in the UV range, it increases in the visible range (as light penetrates the nanotube layer and is reflected back from the underlying metal34). Also, Pt‐decorated NTs after two ALD cycles demonstrate a similar reflectance behavior to the bare NTs. However, for higher ALD cycle numbers (i.e. ten cycles and more), Pt nanoparticles change the light absorbance behavior over the entire spectral range (i.e. corresponding to a coherent metallic behavior).
We additionally measured EIS to examine the influence of Pt deposition on the charge‐transfer behavior of the NTs. Figure 4 c illustrates the impedance spectra in a Bode plot for bare NTs and Pt‐deposited SP NTs after two, ten, and 20 ALD cycles. Clearly, the bare TiO2 NTs exhibit the highest charge‐transfer resistance. The resistance then decreases with an increasing number of ALD cycles. The largest decrease in the impedance occurs with increasing cycle number between two and ten cycles. This coincides with the occurrence of a Pt‐metallic response in the reflectance data.35 In other words, this seems to represent the percolation threshold from individual particles to a Pt‐metal connected network behavior.
3. Conclusions
Overall, the experimental results show that SP NTs already lead to a maximum photocatalytic H2 generation efficiency after a few ALD cycles; this in contrast to CP NTs that give an optimized activity only after several tens of ALD cycles (≥20 cycles). Although an optimized loading is reached with a similar amount of Pt, a much higher efficiency is obtained for SP NTs. This finding can be ascribed to the smaller particle size and to a much higher particle density on SP NTs, and to better diffusion/access of the reactants. Additionally, we find that, for the SP NTs, a percolation network along the tube walls, in view of electronic behavior, occurs between two and ten ALD cycles. This finding illustrates the advantageous feature of a distinct spacing that improves the accessibility of tube walls and enhances the photocatalytic activity of NTs. We believe that this concept of loading spaced tubular arrays with a synergistic secondary material is not only beneficial for an improved photocatalytic performance of TiO2 nanotubular structures, but can also find wider applications in further functional hierarchical structures and different loading species.
Experimental Section
TiO2 NTs were formed on 0.1 mm thick Ti foils (99.6 % pure temper annealed, ADVENT) in a two‐electrode configuration with Pt as the cathode and the Ti substrate as the anode, using an O‐ring cell with a 1 cm2 surface area (IMP‐Series Jaissle Potentiostat). Prior to anodization, the Ti foils were degreased by sonicating in acetone, ethanol, and distilled water, respectively, and dried under a nitrogen (N2) stream.
Anodization consists of two steps. The first anodization or pretreatment step was performed at 80 V for 10 min in ethylene glycol (EG, 99.5 %, Sigma–Aldrich)‐based electrolyte containing 0.5 wt % ammonium fluoride (NH4F, 98 %, Sigma–Aldrich) and 2 m H2O. Subsequently, the tubular layer was removed in deionized water (DIW) by sonication and the degreasing/cleaning step was repeated. In the second step, to fabricate spaced nanotubes (SP NTs), anodization was conducted in dimethyl sulfoxide (DMSO, VWR Chemicals) electrolyte with additions of 4 wt % HF (40 %, Sigma Aldrich), 7 wt % H2O (water content of HF is also considered in the calculations), and 0.3 wt % NH4F (98 %, Sigma Aldrich) at 30 V (30 °C) for 1 h. After anodization, the samples were immersed in ethanol for 2 h and dried in a N2 stream.
The closed‐packed (CP) reference NTs were produced in EG‐based electrolyte containing 1.5 m lactic acid (LA, 90 %, Sigma–Aldrich), 0.1 m NH4F, and 5 wt % H2O. The anodization experiments were carried out at 120 V for an anodization time of 10 min. After the first anodization step, samples were sonicated to remove the nanotubular layer and a dimpled structure was left on the Ti substrate. The second anodization was carried out under the same conditions (electrolyte, voltage) for an anodization time of 2 min.
One of the main differences between CP and SP NTs is the wall morphology, that is, a double‐walled (consisting of carbon‐rich inner shell) versus single‐walled structure. Although DMSO‐based SP TiO2 NTs have a single‐walled morphology, EG‐based CP TiO2 NTs have a double‐walled structure. To obtain a single‐walled structure, as‐grown CP TiO2 NTs were treated by using the following the procedure. The as‐grown tubes were annealed at 150 °C for 1 h in air, with a heating and cooling rate of 30 °C min−1 by using a rapid thermal annealer (Jipelec JetFirst100). Afterwards, the annealed nanotubes were etched with piranha solution (1:3 vol% of H2O2:H2SO4) at 70 °C for 6 min.36 Before ALD, the SP and CP nanotubular layers were annealed at 450 °C for 1 h in air.
To obtain a conformal Pt deposition, an ALD tool (thermal ALD, TFS 200, Beneq) was utilized. The cycle number was varied (from 2 to 72 cycles) to see the influence on density of Pt particles and particle size. Within the ALD procedure, (trimethyl)‐methyl‐cyclopentadienyl‐platinum (IV) (Strem elec. Grade, 99 % heated up to 80 °C to obtain a proper vapor pressure) and oxygen (Messer, 99.95 %) were utilized as Pt precursor and oxidizing agent, respectively. One ALD deposition cycle was defined by the following sequence: Pt pulse (1 s)–N2 purge (5 s)–O2 pulse (1.5 s)–N2 purge (5 s). The deposition temperature was 300 °C, using N2 (99.9999 %) as the carrier and purging gas at a flow rate of 400 standard cubic centimeters per minute (sccm) for both purposes.
For comparison, deposition of a thin Pt film (1, 10 and 50 nm‐thick) on SP TiO2 NTs was carried out by using a sputter coater with an Ar‐plasma sputtering system (EM SCD500, Leica). As the Pt source, a Pt target (99.99 %, Hauner MetallischeWerkstoffe) was used. The pressure of the sputtering chamber was reduced to 10−4 mbar, and then maintained at 10−2 mbar of Ar during the sputtering. The applied current was 16 mA. The amount of sputtered Pt was determined by using an automated quartz crystal film‐thickness sensor.
The morphology was characterized by field‐emission SEM (S4800 Hitachi) coupled with an energy‐dispersive X‐ray detector (EDX, Genesis 4000). X‐ray diffraction (XRD, X‘pert Philips MPD with a Panalytical X‘celerator detector) was performed by using graphite monochromatized Cu Kα radiation (wavelength 1.54056 Å). The approximate crystallite size was determined from the peak broadening of XRD reflections by the Scherrer formula37 given in Equation (1):
| (1) |
where D is the crystallite size, k is the Scherrer constant (0.9 in this study), k is the wavelength of the X‐ray radiation (0.15418 nm for Cu Kα), and β is the full width at half‐maximum (FWHM) in X‐ray diffraction reflections of the anatase (101) and rutile (110) peaks of TiO2.
The chemical composition was characterized with X‐ray photoelectron spectroscopy (XPS, PHI 5600, USA) using Al Kα monochromatized radiation (spectra were shifted to Ti 2p a 458.5 eV) and the peaks were fitted with Multipak software. Further morphological and structural characterizations were carried out with transmission electron microscopy (TEM, CM 30 TEM/STEM, Philips). The size as well as the number of particles per μm2 were measured from different TEM images and average values were considered.
The photocatalytic H2 evolution experiments were carried out in a 20 vol % ethanol–high purity water mixture inside the sealed quartz tube. The ethanol–water mixture amounted to 7 mL, and the cell headspace was 8 mL that was purged with N2 for 15 min before photocatalytic experiments. The experiments were conducted by using two different light sources: 1) a CW‐laser emitting UV light (I 0=60 mW cm−2, λ=325 nm) and 2) an AM 1.5 solar simulator (300 W Xe, I 0=155 mW cm−2, light spot size ca. 20 cm2, Solarlight) calibrated to 100 mW cm−2. To assess the amount of generated H2, the gas that evolved under irradiation was accumulated within the headspace of the quartz reactor and was then analyzed by gas chromatography (using a GCMS‐QO2010SE chromatograph, Shimadzu), withdrawing 200 μL samples with a gas tight syringe.
The total reflectance was measured by using an Avantes spectroscopy system with AvaLight‐DH‐S‐BAL balanced power source and AvaSpec‐2048L Starline versatile fiber‐optic spectrometer detector (PTFE that was utilized as a reference standard).
To determine the charge‐transfer behavior, electrochemical impedance spectroscopy (EIS) was measured by using an electrochemical workstation “IM6eX”, a potentiostat “XPot”, and the corresponding “Thales”‐Software (Zahner‐Elektrik)) in 0.1 m Na2SO4 (in DIW) at open‐circuit conditions (OCP) and a frequency range of 4 mHz to 100 kHz using an amplitude of 10 mV.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge the ERC (projects no. 340511, 638857), the DFG, the DFG Engineering of Advanced Materials’ cluster of excellence, DFG “funCOS” and Ministry of Youth, Education and Sports of the Czech Republic (projects no. LM2015082, LQ1601) for financial support. The authors also thank to Helga Hildebrand for X‐ray photoelectron spectroscopy and Ulrike Marten‐Jahns for X‐ray diffraction analyses.
S. Ozkan, J. Yoo, N. T. Nguyen, S. Mohajernia, R. Zazpe, J. Prikryl, J. M. Macak, P. Schmuki, ChemistryOpen 2018, 7, 797.
References
- 1. Fujishima A., Honda K., Nature 1972, 283, 37–38. [DOI] [PubMed] [Google Scholar]
- 2. Fujishima A., Zhang X., Tryk D. A., Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar]
- 3. Chen X., Mao S. S., Chem. Rev. 2007, 107, 2891–2959. [DOI] [PubMed] [Google Scholar]
- 4. Lee K., Mazare A., Schmuki P., Chem. Rev. 2014, 114, 9385–9454. [DOI] [PubMed] [Google Scholar]
- 5. Macak J. M., Zlamal M., Krysa J., Schmuki P., Small 2007, 3, 300–304. [DOI] [PubMed] [Google Scholar]
- 6. Ozkan S., Nguyen N. T., Hwang I., Mazare A., Schmuki P., Small 2017, 13, 1603821. [DOI] [PubMed] [Google Scholar]
- 7. Nguyen N. T., Ozkan S., Hwang I., Mazare A., Schmuki P., Nanoscale 2016, 8, 16868–16873. [DOI] [PubMed] [Google Scholar]
- 8. Ozkan S., Cha G., Mazare A., Schmuki P., Nanotechnology 2018, 29, 195402. [DOI] [PubMed] [Google Scholar]
- 9. Linsebigler A. L., Lu G., Yates J. T., Chem. Rev. 1995, 95, 735–758. [Google Scholar]
- 10. Anpo M., Takeuchi M., J. Catal. 2003, 216, 505–516. [Google Scholar]
- 11. Das C., Kot M., Rouissi Z., Kędzierski K., Henkel K., Schmeißer D., ACS Omega 2017, 2, 1360–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kemppainen E., Bodin A., Sebok B., Pedersen T., Seger B., Mei B., Bae D., Vesborg P. C. K., Halme J., Hansen O., Lund P. D., Chorkendorff I., Energy Environ. Sci. 2015, 8, 2991–2999. [Google Scholar]
- 13. Ishitani O., Inoue C., Suzuki Y., Ibusuki T., J. Photochem. Photobiol. A 1993, 72, 269–271. [Google Scholar]
- 14. Honciuc A., Laurin M., Albu S., Sobota M., Schmuki P., Libuda J., Langmuir 2010, 26, 14014–14023. [DOI] [PubMed] [Google Scholar]
- 15. Almeida L. C., Zanoni M. V. B., J. Braz. Chem. Soc. 2014, 25, 579–588. [Google Scholar]
- 16. Paramasivam I., Macak J. M., Schmuki P., Electrochem. commun. 2008, 10, 71–75. [Google Scholar]
- 17. Johnson R. W., Hultqvist A., Bent S. F., Mater. Today 2014, 17, 236–246. [Google Scholar]
- 18. Zazpe R., Knaut M., Sopha H., Hromadko L., Albert M., Prikryl J., Gärtnerová V., Bartha J. W., Macak J. M., Langmuir 2016, 32, 10551–10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. George S. M., Chem. Rev. 2010, 110, 111–131. [DOI] [PubMed] [Google Scholar]
- 20. Ng S., Kuberský P., Krbal M., Prikryl J., Gärtnerová V., Moravcová D., Sopha H., Zazpe R., Yam F. K., Jäger A., Hromádko L., Beneš L., Hamáček A., Macak J. M., Adv. Eng. Mater. 2018, 20, 1700589. [Google Scholar]
- 21. Ozkan S., Nguyen N. T., Mazare A., Hahn R., Cerri I., Schmuki P., Electrochem. commun. 2017, 77, 98–102. [Google Scholar]
- 22. Vervuurt R. H. J., Kessels W. M. M. E., Bol A. A., Adv. Mater. Interfaces 2017, 4, 1700232. [Google Scholar]
- 23. Lee H.-B.-R., Bent S. F., Chem. Mater. 2012, 24, 279–286. [Google Scholar]
- 24. Ande C. K., Knoops H. C. M., de Peuter K., van Drunen M., Elliott S. D., Kessels W. M. M., J. Phys. Chem. Lett. 2015, 6, 3610–3614. [DOI] [PubMed] [Google Scholar]
- 25. Yoo J., Zazpe R., Cha G., Prikryl J., Hwang I., Macak J. M., Schmuki P., Electrochem. commun. 2018, 86, 6–12. [Google Scholar]
- 26. Dasgupta N. P., Liu C., Andrews S., Prinz F. B., Yang P., J. Am. Chem. Soc. 2013, 135, 12932–12935. [DOI] [PubMed] [Google Scholar]
- 27. Anitha V. C., Zazpe R., Krbal M., Yoo J., Sopha H., Prikryl J., Cha G., Slang S., Schmuki P., Macak J. M., J. Catal. 2018, 365, 86–93. [Google Scholar]
- 28. Mackus A. J. M., Verheijen M. A., Leick N., Bol A. A., Kessels W. M. M., Chem. Mater. 2013, 25, 1905–1911. [Google Scholar]
- 29. Zhang J., Chen C., Chen S., Hu Q., Gao Z., Li Y., Qin Y., Catal. Sci. Technol. 2017, 7, 322–329. [Google Scholar]
- 30. Mayrhofer K. J. J., Blizanac B. B., Arenz M., Stamenkovic V. R., Ross P. N., Markovic N. M., J. Phys. Chem. B 2005, 109, 14433–14440. [DOI] [PubMed] [Google Scholar]
- 31. Baker L., Cavanagh A. S., Seghete D., George S. M., Mackus A. J. M., Kessels W. M. M., Liu Z. Y., Wagner F. T., J. Appl. Phys. 2011, 109, 84333. [Google Scholar]
- 32. Kiwi J., Graetzel M., J Phys Chem. 1984, 88, 1302–1307. [Google Scholar]
- 33. Bamwenda G. R., Tsubota S., Nakamura T., Haruta M., J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar]
- 34. So S., Kriesch A., Peschel U., Schmuki P., J. Mater. Chem. A 2015, 3, 12603–12608. [Google Scholar]
- 35. Bao X.-Q., Liu L. F., Mater. Chem. Phys. 2015, 149–150, 309–316. [Google Scholar]
- 36. Hwang I., So S., Mokhtar M., Alshehri A., Al-Thabaiti S. A., Mazare A., Schmuki P., Chem. Eur. J. 2015, 21, 9204–9208. [DOI] [PubMed] [Google Scholar]
- 37. Patterson A. L., Phys. Rev. 1939, 56, 978–982. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary