

Hutchins and Bronner show that a transient pulse of the secreted molecule Draxin regulates the timing and progression of cranial neural crest EMT along the anterior to posterior embryonic body axis by modulation of canonical Wnt signaling.
Abstract
Neural crest cells undergo a spatiotemporally regulated epithelial-to-mesenchymal transition (EMT) that proceeds head to tailward to exit from the neural tube. In this study, we show that the secreted molecule Draxin is expressed in a transient rostrocaudal wave that mirrors this emigration pattern, initiating after neural crest specification and being down-regulated just before delamination. Functional experiments reveal that Draxin regulates the timing of cranial neural crest EMT by transiently inhibiting canonical Wnt signaling. Ectopic maintenance of Draxin in the cranial neural tube blocks full EMT; while cells delaminate, they fail to become mesenchymal and migratory. Loss of Draxin results in premature delamination but also in failure to mesenchymalize. These results suggest that a pulse of intermediate Wnt signaling triggers EMT and is necessary for its completion. Taken together, these data show that transient secreted Draxin mediates proper levels of canonical Wnt signaling required to regulate the precise timing of initiation and completion of cranial neural crest EMT.
Introduction
The neural crest is a stem cell population that originates within the forming central nervous system but subsequently delaminates from the neuroepithelium by undergoing a spatiotemporally regulated epithelial-to-mesenchymal transition (EMT) with a sequence of initiation that progresses from rostral to caudal. In amniotes, the process of neural crest emigration starts at the level of the caudal forebrain/rostral midbrain and then propagates progressively caudally. After delamination, mesenchymal neural crest cells migrate sometimes long distances to populate numerous sites in the periphery, where they differentiate into many different cell types including peripheral ganglia, melanocytes of the skin, and bone and cartilage of the face (Green et al., 2015; Martik and Bronner, 2017; Gandhi and Bronner, 2018).
The mechanisms of neural crest EMT appear to differ at different levels of the body axis, with cranial neural crest cells undergoing a more collective emigration process whereas trunk neural crest cells delaminate as individuals (Théveneau et al., 2007). Consistent with this, cranial neural crest cells possess different gene regulatory subcircuits than trunk neural crest cells (Simoes-Costa and Bronner, 2016); for example, the premigratory cranial but not trunk neural crest expresses transcription factors like the Wnt effector Axud1 (Simões-Costa et al., 2015) and Ets-1, proposed to confer collective EMT (Théveneau et al., 2007).
Several secreted factors (e.g., FGFs, BMPs, and Wnts) have been proposed to regulate various aspects of neural crest specification, delamination/EMT, and migration. For example, cranial neural crest specification is thought to require intermediate levels of BMP signaling together with canonical Wnt signaling (Tribulo et al., 2003; Steventon et al., 2009; Steventon and Mayor, 2012). The latter is mediated by the transcription factor Axud1, which then directly interacts with neural plate border genes Pax7 and Msx1 to activate neural crest specifier genes (Simões-Costa et al., 2015). Wnt signaling is also critical for controlling proliferation of neural crest precursors in the dorsal neural tube (Dickinson et al., 1995). Later, noncanonical Wnt signaling is essential for neural crest migration (Carmona-Fontaine et al., 2008a; Matthews et al., 2008), whereas canonical Wnt signaling is involved in lineage specification, with high Wnt biasing neural crest cells toward neuronal differentiation and low Wnt toward mesenchymal derivatives (Hari et al., 2012). However, it has been difficult to separate the effects of these factors on these different processes given that they occur sequentially, particularly because blocking specification can indirectly influence later events. An important unanswered question is how Wnt signaling controls so many different aspects of neural crest development.
In this study, we focus on how neural crest precursor cells transition from specification to EMT by examining the mechanisms underlying initiation of neural crest EMT along the neural axis. We show that the secreted molecule Draxin is expressed in a transient rostrocaudal wave within the premigratory neural crest and that its down-regulation mirrors initiation of cranial neural crest emigration. Intriguingly, ectopic maintenance of Draxin in the cranial neural tube inhibits emigration. In probing the molecular mechanism, we find that modulating Draxin expression alters canonical Wnt reporter activity in vivo and that Draxin interacts with the canonical Wnt coreceptor LRP5. Maintenance of Draxin blocks the emigration process, whereas its loss results in premature delamination, but both gain and loss block mesenchymalization. Collectively, these data suggest that a transient pulse of intermediate Wnt signaling mediated by Draxin is required for proper neural crest EMT. Thus, Draxin acts as a rheostat that transiently inhibits canonical Wnt signaling to correctly time cranial neural crest delamination and completion of EMT.
Results
Draxin expression tightly correlates with cranial neural crest EMT
Draxin was identified in a screen for genes enriched in premigratory cranial neural crest cells but intriguingly was absent from migrating cranial neural crest cells (Simões-Costa et al., 2014). To elucidate its spatiotemporal expression pattern, we performed whole-mount in situ hybridization and first detected Draxin at Hamburger–Hamilton stage 9− (HH9−) in the dorsal neural tube within the region that will give rise to the cranial neural crest (Fig. 1 A). The Draxin expression domain expanded more caudally by HH9, following the rostrocaudal progression of neural crest development, while remaining restricted to the dorsal neural folds (Fig. 1 B). By HH9+, Draxin was down-regulated within the cranial neural crest, though its expression persisted further caudally (Fig. 1 C).
Figure 1.
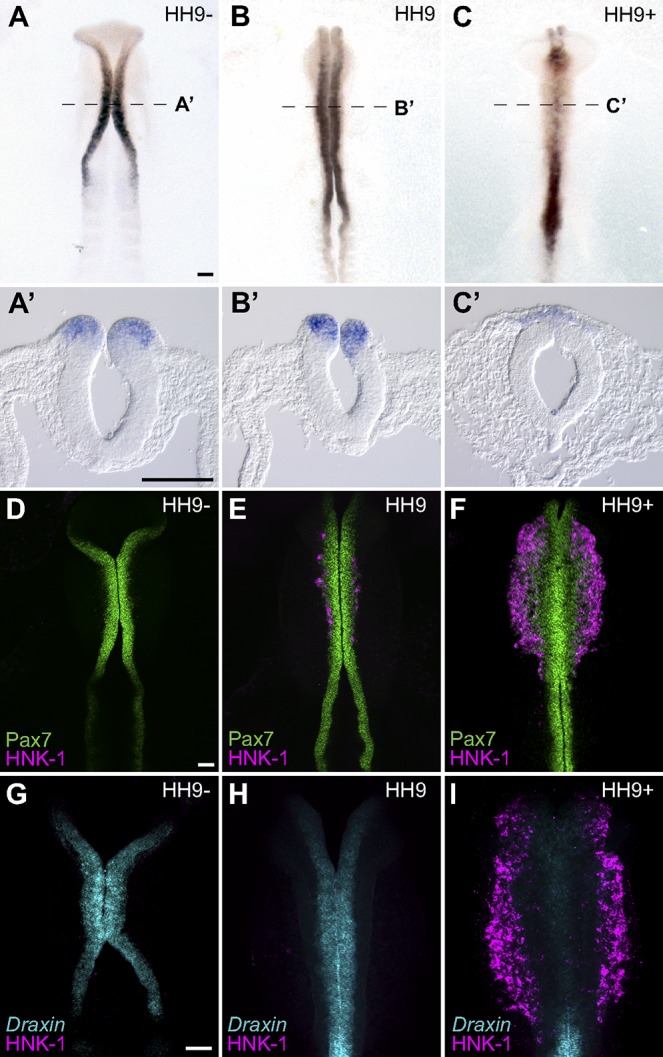
Draxin is expressed in premigratory cranial neural crest and down-regulated at the onset of EMT. (A–C) Representative images of chromogenic whole-mount in situ hybridization for Draxin in stage HH9− (A and A’), HH9 (B and B’), and HH9+ (C and C’) chick embryos. Dotted lines in A, B, and C indicate cross sections shown in A’, B’, and C’, respectively. (D–F) Immunostaining for Pax7 (green) and HNK1 (magenta) in stage-matched embryos. (G–I) Fluorescent in situ hybridization for Draxin (cyan) with immunostaining for HNK1 (magenta) in stage-matched embryos. Bars, 100 µm.
To better define its expression relative to the progression of cranial neural crest development, we compared expression patterns for the neural crest markers Pax7 and HNK-1 with Draxin. Pax7 is an early marker for the neural plate border and premigratory neural crest, and it persists in early migrating neural crest cells (Basch et al., 2006; Betancur et al., 2010a), whereas HNK-1 marks migratory neural crest cells that have exited the neural tube and completed EMT (Bronner-Fraser, 1986). Immunohistochemical analysis revealed that Pax7 expression at HH9−/9 preceded and subsequently overlapped with Draxin expression within the neural folds (Fig. 1, D and E; compare with Fig. 1, G and H). By HH9+, a wave of migratory neural crest was observed, restricted to the cranial level (mesencephalon through the second rhombomere) of neural tube as indicated by HNK-1 labeling (Fig. 1 F). Notably, robust staining for HNK-1 at HH9+ coincided with a loss of endogenous Draxin from the cranial neural folds (Fig. 1 I). Thus, Draxin is expressed in a transient fashion within the neural folds, initiating after neural crest specification (Simões-Costa et al., 2015), and then it is rapidly down-regulated before EMT.
Draxin knockdown induces premature delamination of cranial neural crest
Because of the mutually exclusive expression of HNK-1 and Draxin and because Draxin is specifically expressed within premigratory cranial neural crest cells, we hypothesized that Draxin must be down-regulated for cranial neural crest EMT to proceed. To test its function within premigratory neural crest, we knocked down Draxin using an antisense morpholino (MO)-mediated knockdown approach. To this end, we performed unilateral HH4 electroporations with either a control MO (Fig. 2 A) or a translation-blocking Draxin MO (Fig. 2 B). Loss of Draxin caused defects in cranial neural crest emigration at stage HH9+ compared with control, reducing the distance neural crest traveled away from the midline (Fig. 2 C; 60.2 ± 3.1% vs. 86.5 ± 1.9% of the relative maximum migration distance of the unelectroporated side, respectively). The penetrance of this phenotype was similar to CRISPR/Cas9-mediated Draxin knockdown (Fig. S1, A–C). Draxin loss of function had no effect on the levels of cell death or proliferation (Fig. S1, D–I).
Figure 2.

Draxin knockdown caused cranial neural crest to prematurely delaminate and accumulate at the dorsal midline. (A and B) Representative images for Pax7 immunostaining (green) in embryos unilaterally electroporated with FITC-labeled (magenta) control MO (A) or a translation-blocking Draxin MO (B). Bars, 100 µm. Arrowhead highlights defects in neural crest emigration resulting from perturbation. (C) Quantification of relative maximum migration distance indicated Draxin knockdown (B) significantly reduced the emigration of Pax7+ neural crest cells from the neural tube compared with control (A). Each circle represents the average of six measurements per embryo taken over a 300-µm anterior-to-posterior region of cranial neural crest (n = 11 and n = 17 control MO and Draxin MO embryos, respectively). *, P < 0.0001; two-tailed t test. Black bars indicate mean ± SEM. (D and E) Representative cross sections of embryos electroporated with control MO (D) or Draxin MO (E) immunostained for Pax7 (green) at HH9+. To visually compare the number of Pax7+ neural crest cells undergoing delamination, the extreme dorsal aspect of the neural tube epithelium is indicated by a dotted line. Bars, 20 µm. (F) Quantification of Pax7+ cells on electroporated versus unelectroporated sides of embryo (three sections/embryo; n = 3 embryos/condition) yielded no significant difference (P = 0.36, two-tailed t test) in total Pax7+ cells between control MO and Draxin MO–electroporated embryos, although a significant increase in Pax7+ cells that delaminated and left the dorsal neural tube (*, P = 0.04; two-tailed t test), and a significant decrease in Pax7+ cells that were retained within the dorsal neural tube (*, P = 0.003; two-tailed t test) were observed with Draxin knockdown. Black bars indicate mean ± SEM.
To parse the mechanistic events that caused this emigration phenotype, we examined cross sections of the dorsal neural tube of MO-electroporated embryos at stage HH9+. Pax7 immunostaining revealed increased cranial neural crest delamination (i.e., departure from the neuroepithelium) with Draxin knockdown (133.9 ± 10.7% of the control) as well as reduced neural crest retention within the dorsal neural tube (65.5 ± 4.2% of the control), although the total number of Pax7+ cells was unchanged (Fig. 2, D and F). To identify potential downstream effects on neural crest specification, we also examined the early neural crest specifier genes Snail2 and Sox9, both of which are expressed in premigratory neural crest following Pax7 (Betancur et al., 2010a). In contrast with Pax7, Draxin knockdown resulted in a decrease in the total numbers of Snail2+ and Sox9+ cells (78.6 ± 3.5% and 68.0 ± 8.1% of the control, respectively; Fig. 3, A–F). Further, whereas both exhibited reduced neural crest retention within the dorsal neural tube (48.2 ± 5.1% and 63.4 ± 10.9% of the control, respectively), the number of Sox9+ cells that had delaminated was also decreased (Fig. 3, D and F; 67.7 ± 7.6% of the control). Interestingly, the number of Snail2+ cells that delaminated was not significantly different from the control (Fig. 3, B and E); moreover, quantification of the relative fluorescence intensity of the delaminated Snail2+ cells revealed a significant increase in the level of Snail2 after Draxin knockdown (Fig. 3, B and B′; 167.1 ± 2.0% of the control; P = 0.0004, two-tailed t test).
Figure 3.

Draxin knockdown altered expression of EMT and neural crest markers. (A–F) Representative cross sections of embryos electroporated with control MO (A and C) or Draxin MO (B and D) immunostained for Snail2 (A and B) and Sox9 (C and D) at HH9/9+. To visually compare the number of Snail2+ and Sox9+ neural crest cells undergoing delamination, the extreme dorsal aspect of the neural tube epithelium is indicated by a dotted line. (E and F) Quantification of Snail2+ (E) and Sox9+ (F) cells on electroporated versus unelectroporated sides of embryo (three sections/embryo; n = 3 embryos/condition) yielded significant reductions in both the total and neural tube (NT) number of Snail2+ cells (*, P = 0.03 and P = 0.007, respectively; two-tailed t test) and Sox9+ cells (*, P = 0.02 and P = 0.03, respectively; two-tailed t test) between control MO and Draxin MO–electroporated embryos. Although a significant decrease was also observed for Sox9+ cells that delaminated and left the dorsal neural tube (P = 0.2; two-tailed t test), no significant difference was observed for delaminated Snail2+ cells (P = 0.6; two-tailed t test) with Draxin knockdown. (G–L) Representative cross sections of embryos electroporated with control MO (G and J) or Draxin MO (H and K) immunostained for Cad6B (G and H) and HNK-1 (J and K) at HH9/9+. Quantitation of the relative fluorescence intensity for both Cad6B (I) and HNK-1 (L) found significant reduction in fluorescence intensity between control MO and Draxin MO–electroporated embryos (*, P = 0.0005 and P = 0.007, respectively; two-tailed t test; three sections/embryo; n = 3 embryos/condition). Boxes in G–K indicate zoomed areas in G’–K’. Arrowheads indicate ROIs for comparison. Bars, 20 µm. Dotted arrows in H′ and K′ illustrate changes in fluorescence intensity compared with control. Data are presented as mean ± SEM.
Given that Snail2 has long been established as a regulator of EMT (Nieto et al., 1994), we next examined the effects of Draxin knockdown on the expression of Cadherin 6B (Cad6B), which is a direct target of Snail2 (Taneyhill et al., 2007; Strobl-Mazzulla and Bronner, 2012) and whose expression in premigratory neural crest is down-regulated during EMT, paralleling Draxin expression (Nakagawa and Takeichi, 1995). Quantitation of Cad6B immunostaining showed a significant decrease in the level of Cad6B protein in the dorsal neural tube at premigratory stages after Draxin knockdown (Fig. 3, G–I; 41.0 ± 2.1% of the control), which may account for the increased delamination observed in Draxin MO–electroporated embryos. Taken together, these data suggest that Draxin is necessary to retain premigratory neural crest within the dorsal neural tube and maintain the appropriate level of expression of neural crest specifier genes.
Despite the increased detachment from the neural tube after loss of Draxin, we hypothesized that the delaminated neural crest cells failed to complete EMT to become mesenchymal and migratory. To test this, we examined expression of the migratory neural crest marker, HNK-1, which is expressed on migratory neural crest cells after the delamination and completion of EMT (Bronner-Fraser, 1986; Giovannone et al., 2015). Indeed, we observed robust expression of HNK-1 on control-treated neural crest cells that had mesenchymalized and begun their emigration away from the neural tube (Fig. 3 I). In contrast, Draxin MO–electroporated embryos displayed a significant loss of the HNK-1 epitope (Fig. 3, K and L; 30.5 ± 7.6% of the control). Thus, these data suggest that Draxin is required within premigratory neural crest to control the precise timing of EMT onset and completion.
Ectopic maintenance of Draxin inhibits cranial neural crest EMT but not specification
Given that Draxin expression appears to be tightly regulated during neural crest development, we asked whether prolonging the expression of Draxin such that it persists during EMT would negatively affect cranial neural crest migration. To this end, we subcloned full-length chick Draxin with a C-terminal FLAG tag into an expression vector under control of a chick β-actin (CAG) promoter upstream of an internal ribosome entry site (IRES)-driven H2B-RFP (pCI-H2B-RFP; Betancur et al., 2010b); when this construct (Draxin-FLAG) was electroporated into gastrula stage HH4 chick embryos, it expressed and secreted full-length, FLAG-tagged Draxin after its endogenous expression was turned off (Fig. S2, A–D). No changes were noted in cell death or proliferation (Fig. S2, E–J).
To assess the effects of ectopic Draxin on neural crest migration, we individually electroporated Draxin-FLAG or pCI-H2B-RFP (as a control) into the right side of HH4 embryos. After electroporation, embryos were allowed to develop to stage HH9+ following the onset of cranial neural crest EMT, and they screened for successful electroporation by H2B-RFP expression. Pax7 immunostaining was used to assay effects on cranial neural crest migration. We measured the relative distances Pax7+ cells had migrated away from the midline compared with the unelectroporated, contralateral control side of the same embryo. pCI-H2B-RFP electroporation had little or no effect on neural crest EMT as Pax7+ neural crest cells on the electroporated side migrated 95.6 ± 2.8% (n = 12) of the distance of the unelectroporated side (Fig. 4, A, E, and F). In contrast, ectopic maintenance of Draxin significantly reduced cranial neural crest migration in Draxin-FLAG–electroporated embryos to 71.3 ± 2.8% (n = 17) of the distance of the unelectroporated side (Fig. 4, B–F; P < 0.00001, two-tailed t test). Similar effects were noted at caudal levels of cranial neural crest (rostral hindbrain through rhombomere 5; Fig. 4, G and G′).
Figure 4.
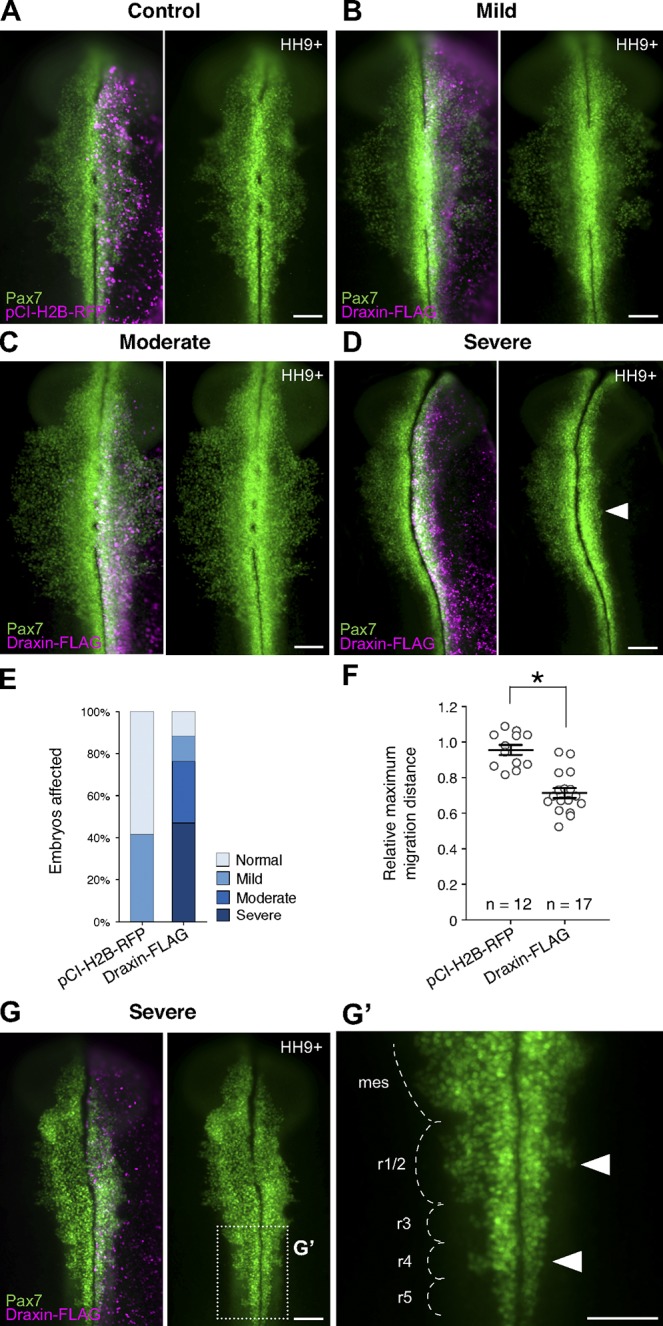
Draxin overexpression reduced cranial neural crest emigration from the neural tube. (A–D) Immunostaining against Pax7 (green) in pCI-H2B-RFP (A; control) or Draxin-FLAG–electroporated (B–D) embryos. H2B-RFP fluorescence (magenta) indicated successful electroporation for both constructs. (B–D) Representative images for mild (B), moderate (C), and severe (D) phenotypes of Pax7-immunostained (green) embryos unilaterally electroporated with Draxin-FLAG. Arrowhead in D highlights defects in neural crest emigration resulting from perturbation. (E) Quantification of phenotype penetrance for embryos unilaterally electroporated for pCI-H2B-RFP (control) or Draxin-FLAG. Data are presented as fraction of the total number of pCI-H2B-RFP– and Draxin-FLAG–electroporated embryos (n = 12 and n = 17, respectively; pooled from more than three independent experiments). Relative maximum migration distance data are represented in this figure as “normal” (within SEM of pCI-H2B-RFP average), “mild” (between minimum limit of SEM of pCI-H2B-RFP average and maximum limit of SEM of Draxin-FLAG average), “moderate” (within SEM of Draxin-FLAG average), and “severe” (less than the minimum limit of SEM of Draxin-FLAG average). (F) Quantification of relative maximum migration distance indicated that Draxin-FLAG overexpression significantly reduced the emigration of Pax7+ neural crest cells from the neural tube compared with pCI-H2B-RFP control (A). Each circle represents the average of six measurements per embryo taken over a 300-µm anterior-to-posterior region of cranial neural crest. *, P < 0.0001, two-tailed t test. Black bars, mean ± SEM. (G) Closer examination of the hindbrain region of a Draxin-FLAG overexpression embryo with a severe phenotype revealed a similar inhibition of neural crest emigration at rhombomeres 1/2 and 4 (arrowheads). Box in G indicates zoomed area in G’. Mes, mesencephalon; r, rhombomere. Bars, 100 µm.
Because defects in neural crest migration could result from errors in earlier events of neural crest development, such as induction or specification, we next examined pCI-H2B-RFP and Draxin-FLAG–electroporated embryos in cross section at HH9+ to more clearly delineate how Draxin overexpression affects neural crest EMT. In pCI-H2B-RFP–electroporated embryos, migratory cranial neural crest delaminated from the dorsal neural tube and underwent mesenchymalization (i.e., detached from each other and migrated away from the neural tube) to complete EMT (Fig. 5, A–C). However, in Draxin-FLAG–electroporated embryos, neural crest appeared to have delaminated to a lesser extent and failed to mesenchymalize, remaining adjacent to the neural tube and frequently tightly clumped together (Fig. 5, D–F). To determine whether the effects of Draxin overexpression were limited to EMT, we performed Pax7 and Sox9 cell counts and found that neither pCI-H2B-RFP nor Draxin-FLAG altered the total number of Pax7+ or Sox9+ neural crest cells on the electroporated versus unelectroporated side, although clear defects in the number of neural crests cells that delaminated from the neuroepithelium were observed (Fig. 5, G and I). Taken together, these data suggest that Draxin antagonizes the delamination step of neural crest EMT, but the perdurance of its expression has no effect on induction or specification.
Figure 5.

Draxin overexpression altered expression of markers of neural crest EMT. (A–F) Cross sections of pCI-H2B-RFP (A–C; control) or Draxin-FLAG–electroporated (D–F) embryos (magenta) immunostained for Pax7 (A and D), Snail2 (B and E), and Sox9 (C and F). To visually compare the number of Pax7+, Snail2+, and Sox9+ neural crest cells undergoing delamination, the extreme dorsal aspect of the neural tube epithelium is indicated by a dotted line. (G–I) Quantification of Pax7+ (G), Snail2+ (H), and Sox9+ (I) cells on electroporated versus unelectroporated sides of embryo (three sections/embryo; n = 3 embryos/condition). Draxin-FLAG overexpression yielded no significant difference in the total numbers of Pax7+ and Sox9+ cells (P = 0.13 and P = 0.6, respectively; two-tailed t test) between pCI-H2B-RFP (A and C) or Draxin-FLAG–electroporated (D and F) embryos, although a significant decrease in Pax7+ and Sox9+ cells that delaminated and left the dorsal neural tube (*, P = 0.009 and P = 0.03, respectively; two-tailed t test) and a significant increase in Pax7+ cells that were retained within the dorsal neural tube (NT; *, P = 0.03 and P = 0.04, respectively; two-tailed t test) were observed with Draxin-FLAG overexpression. Draxin-FLAG overexpression yielded significant decreases in both the total and delaminated numbers of Snail2+ cells (*, P = 0.008 and P = 0.002, respectively; two-tailed t test) and a significant increase in the number of NT Snail2+ cells (*, P = 0.03; two-tailed t test). (J–O) Representative cross sections of embryos electroporated with pCI-H2B-RFP (J and M) or Draxin-FLAG (K and N) immunostained for Cad6B (J and K) and HNK-1 (M and N) at HH9/9+. Boxes in J–N indicate zoomed areas in J’–N’. Arrowheads indicate ROIs for comparison. Quantitation of the relative fluorescence intensity for Cad6B (L) and HNK-1 (O) found significant increase in fluorescence intensity for Cad6B with Draxin-FLAG overexpression (*, P = 0.04; two-tailed t test) and a significant reduction in fluorescence intensity for HNK-1 with Draxin-FLAG overexpression (*, P = 0.02; two-tailed t test). Bars, 20 µm. Dotted arrows in K′ and N′ illustrate changes in fluorescence intensity compared with control. Data are presented as mean ± SEM.
Given Draxin’s effects on EMT and its potential role as a regulator of Snail2, we also examined the expression of Snail2 with Draxin-FLAG overexpression. Although we observed defects in the number of delaminated versus neural tube Snail2+ cells similar to those for Pax7 and Sox9 (Fig. 5, G–I), we also found a significant reduction in the number of Snail2+ neural crest cells in total (Fig. 5 H). As observed with Draxin knockdown (Fig. 3), we detected changes in expression for both Cad6B and HNK-1 after Draxin overexpression. Consistent with a loss of its repressor, Snail2 (Fig. 5 H), Cad6B intensity was significantly up-regulated (Fig. 5, J–L; 129.5 ± 9.2% compared with control); interestingly, we observed aberrant maintenance of Cad6B expression in delaminated neural crest cells (Fig. 5 K′). In addition, Draxin-FLAG overexpression significantly reduced HNK-1 levels (Fig. 5, M–O; 63.6 ± 8.0% compared with control). Thus, these data suggest that Draxin expression alters the levels of Snail2 and, consequently, Cad6B, to regulate the delamination of cranial neural crest. Disruption of the timing of delamination has negative consequences for the ability of neural crest to induce HNK-1 expression, an indicator of their mesenchymalization, and ultimately impedes the migratory capacity of neural crest.
Cysteine-rich domain (CRD) of Draxin is necessary and sufficient for inhibition of cranial neural crest EMT
To parse the molecular mechanism by which Draxin inhibits cranial neural crest EMT, we performed deletion analysis to identify the functional domains of Draxin mediating its action. Full-length Draxin protein contains an N-terminal signal peptide for secretion and a highly conserved C-terminal 60–amino acid CRD that has homology with the C-terminal cysteine-rich colipase fold domain of Dickkopf-related protein-1 (Dkk-1), the canonical Wnt antagonist (Fig. S3, A and B; Niehrs, 2006; Miyake et al., 2009). To determine whether Draxin and Dkk-1 might be in competition for binding partners, we performed chromogenic in situ hybridization for chick Dkk-1 (cDkk-1) at stages HH9− to HH9+. cDkk-1, although robustly expressed in the somites at these stages, was completely absent from the neural tube and, subsequently, migrating neural crest (Fig. S3, C–E). Thus, it is unlikely that Draxin is in competition with Dkk-1 during cranial neural crest EMT.
To test the role of Draxin’s CRD on neural crest EMT, we generated three mutated forms of Draxin: one coding for Draxin-FLAG with a deletion of the C-terminal 76 amino acids (Draxin-ΔCRD), one coding for only the CRD with the signal peptide and FLAG tag (CRD-FLAG), and one coding for only the CRD and FLAG tag but absent the signal peptide (CRD-ΔSP; Fig. 6 A). Electroporation of Draxin-ΔCRD failed to inhibit cranial neural crest EMT (Fig. 4 B); the relative neural crest migration distance for Draxin-ΔCRD (Fig. 4 E; 92.1 ± 4.3%, n = 6) was not significantly different from pCI-H2B-RFP control (P = 0.81, one-way ANOVA with Tukey’s post hoc; compared with Fig. 4). In contrast, electroporation of CRD-FLAG was sufficient to reduce relative neural crest migration (Fig. 4, C and E; 75.0 ± 3.1%, n = 7) similarly to full-length Draxin-FLAG (P = 0.91, one-way ANOVA with Tukey’s post hoc; compared with Fig. 4). Importantly, electroporation of CRD-ΔSP also failed to inhibit neural crest migration (Fig. 4, D and E; 90.5 ± 4.3%, n = 6), comparable with the pCI-H2B-RFP control (P = 0.32, one-way ANOVA with Tukey’s post hoc; compared with Fig. 4). Thus, these data suggest that the CRD of Draxin mediates its inhibition of cranial neural crest EMT via secretion.
Figure 6.

The CRD of Draxin was both necessary and sufficient for its inhibition of cranial neural crest EMT. (A) Schematic highlighting important features in full-length and mutated FLAG-tagged Draxin constructs. Draxin contains an N-terminal signal peptide (SP; green) and a C-terminal CRD (yellow). For all constructs, a FLAG tag (blue) was placed at the C terminus. (B–D) Immunostaining against Pax7 (green) in Draxin-ΔCRD (B; n = 6), CRD-FLAG (C; n = 7), or CRD-ΔSP–electroporated (D; n = 6) embryos. Bars, 100 µm. Arrowhead in C′ highlights defects in neural crest emigration resulting from perturbation. (E) Quantification of relative maximum migration distance indicated expression of Draxin-ΔCRD (B) did not affect emigration of Pax7+ neural crest cells from the neural tube (P = 0.81 vs. pCI-H2B-RFP control), whereas expression of CRD-FLAG (C) significantly reduced the emigration of Pax7+ neural crest cells (P = 0.001 vs. pCI-H2B-RFP control) to levels comparable with full-length Draxin-FLAG (P = 0.91). Expression of CRD-ΔSP (D) had no effect on emigration (P = 0.75 vs. pCI-H2B-RFP control). For ease of visual comparison, the data for relative maximum migration distance for pCI-H2B-RFP and Draxin-FLAG from Fig. 4 are presented in this figure again as gray circles. Each circle represents the average of six measurements per embryo taken over a 300-µm anterior-to-posterior region of cranial neural crest. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. *, P ≤ 0.01. Black bars, mean ± SEM.
Draxin antagonizes canonical Wnt signaling in vivo
The zebrafish homologue of Draxin was previously suggested to be a neural-specific gene involved in stabilization of β-catenin during canonical Wnt signaling (Miyake et al., 2009, 2012). As Wnt signaling is well known to play a role in neural crest development and EMT (García-Castro et al., 2002; Theveneau and Mayor, 2012; Simões-Costa et al., 2015) and Draxin’s CRD has homology to a known canonical Wnt antagonist, we tested whether Draxin MO and Draxin-FLAG affected Wnt signaling during cranial neural crest EMT. To measure Draxin’s actions on canonical Wnt signaling, we coelectroporated a TCF/Lef:H2B-GFP Wnt reporter (Ferrer-Vaquer et al., 2010) with either control MO or Draxin MO (Fig. 7 A) or pCI-H2B-RFP or Draxin-FLAG (Fig. 7 B) on different sides of the same embryo. Quantitative Western blot analyses revealed a significant increase in GFP reporter output with Draxin knockdown (Fig. 7 A; P < 0.0001, two-tailed t test) as well as a drastic reduction in GFP reporter output with Draxin-FLAG electroporation compared with control (Fig. 7 B; P = 0.015, two-tailed t test). To confirm that Draxin’s inhibition of canonical Wnt signaling has implications for neural crest EMT in vivo, next we examined cross sections of bilaterally electroporated embryos for reporter activity in cranial neural crest that were in the process of undergoing EMT. Neural crest cells that had successfully delaminated from the neural tube, yet were still adjacent to the tube and in the process of mesenchymalization (Fig. 7 C, circled region, left), exhibited qualitatively higher levels of GFP reporter output compared with surrounding cells with coelectroporation of pCI-H2B-RFP control. However, GFP reporter expression was markedly reduced in the same population of neural crest cells after coelectroporation of Draxin-FLAG (Fig. 7, C and D; 45.3 ± 9.0% compared with control). These data suggest that Draxin inhibits cranial neural crest EMT via modulation of canonical Wnt signaling.
Figure 7.
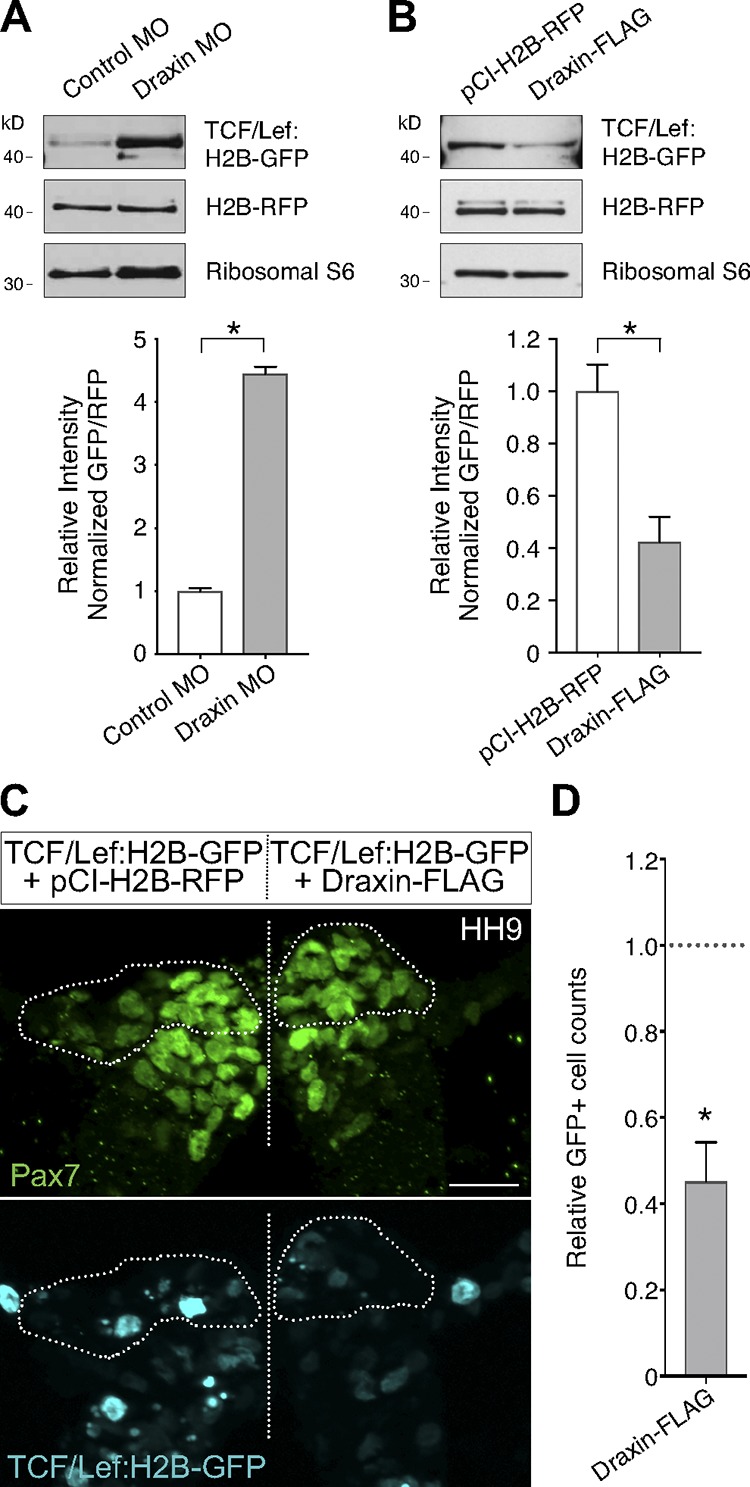
Draxin perturbation altered canonical Wnt reporter expression in vivo. (A) Quantitative Western blots compared normalized levels of GFP relative to RFP between control MO and Draxin MO–electroporated sides of the same embryos, each coelectroporated with a TCF/Lef:H2B-GFP canonical Wnt reporter construct and grown to HH9. Quantification revealed a significant increase in GFP expression when Draxin was knocked down (*, P < 0.0001; two-tailed t test; n = 12 pooled half embryos). (B) Quantitative Western blots compared normalized levels of GFP relative to RFP between pCI-H2B-RFP and Draxin-FLAG–electroporated sides of the same embryos, each coelectroporated with a TCF/Lef:H2B-GFP canonical Wnt reporter construct and grown to HH9. Quantification revealed a significant reduction in GFP expression when Draxin-FLAG was overexpressed (*, P = 0.015; two-tailed t test; n = 15 pooled half embryos). (C) Representative cross section of embryo coelectroporated with TCF/Lef:H2B-GFP (cyan) and pCI-H2B-RFP (left) or Draxin-FLAG (right) immunostained for Pax7 (green). To visually compare TCF/Lef:H2B-GFP reporter expression levels in neural crest cells undergoing EMT, Pax7+ cells that have delaminated from the neural tube are circled. Straight dotted line indicates the midline. Bar, 20 µm. (D) Quantitation of GFP+ cells on pCI-H2B-RFP (left) versus Draxin-FLAG (right) sides of embryo (three sections/embryo; n = 3 embryos/condition) yielded a significant decrease (*, P = 0.02; two-tailed t test) in total GFP+ cells on the Draxin-FLAG–electroporated side compared with the control side. Data are presented as mean ± SEM.
Draxin genetically interacts with canonical Wnt signaling components to modulate EMT
In the canonical Wnt pathway, a secreted Wnt ligand binds Frizzled and LRP5/6 coreceptors to form a ternary complex that is required to stabilize β-catenin for its subsequent transcriptional activities. Dkk family proteins interrupt this pathway by sequestering LRP5/6, thereby inhibiting β-catenin nuclear translocation and transcriptional activation (Niehrs, 2006). Because Draxin’s inhibition of neural crest EMT is mediated by its Dkk-homologous CRD and LRP5 has been shown to be necessary for zebrafish cranial neural crest migration (Willems et al., 2015), we hypothesized that Draxin antagonizes Wnt via sequestration of LRP5. To test this, we performed an epistasis experiment with full-length human LRP5 (hLRP5; MacDonald et al., 2011), which shares 88% amino acid identity with chick LRP5; if Draxin binds LRP5 to inhibit neural crest migration, overexpression of LRP5 would be predicted to act as a molecular sponge to titrate out the excess exogenous Draxin, thereby rescuing EMT. Consistent with this, when hLRP5 is coelectroporated with Draxin-FLAG, relative neural crest migration is rescued to levels similar to pCI-H2B-RFP control (Fig. 8, A and C; 89.4 ± 3.0%, n = 6). These data place Draxin upstream of LRP5 in the canonical Wnt pathway.
Figure 8.

Draxin-mediated defects on cranial neural crest EMT were rescued by canonical Wnt pathway components. (A) Immunostaining against Pax7 (green) and LRP5 (cyan) in hLRP5 and Draxin-FLAG–coelectroporated (magenta) embryos. (B) Immunostaining against Pax7 (green) and GFP (cyan) in embryos that were coelectroporated with Draxin-FLAG (magenta) and a β-catenin gain-of-function mutant construct, which lacks the first 90 amino acids of the mouse protein, is fused to a C-terminal EGFP and is driven by the FoxD3 NC1 enhancer (NC1-Δ90βcat). Bars, 100 µm. (C) Quantification of relative maximum migration distance indicated coelectroporation of either hLRP5 (n = 6) or NC1-Δ90βcat (n = 8) with Draxin-FLAG significantly rescued emigration of Pax7+ neural crest cells from Draxin-FLAG overexpression. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. *, P = 0.02, P ≥ 0.50. For ease of visual comparison, the means of relative maximum migration distance for pCI-H2B-RFP and Draxin-FLAG from Fig. 4 are presented in this figure again as gray circles. Each circle represents the average of six measurements per embryo taken over a 300-µm anterior-to-posterior region of cranial neural crest. Black bars, mean ± SEM. (D) Schematic of proposed model for Draxin function. When canonical Wnt signaling is active, Wnt forms a ternary complex with the Frizzled (Fz) and LRP receptors, causing stabilization of β-catenin (B-CAT) and its subsequent localization to the nucleus, where it functions as a transcriptional activator. When Draxin (D) is present, it attenuates canonical Wnt signaling by interacting with LRP extracellularly, occluding Wnt/Fz association and resulting in β-catenin degradation, which can then no longer activate transcription.
To solidify Draxin’s role in the canonical Wnt pathway during neural crest EMT, we sought to rescue Draxin’s inhibition of neural crest migration with the canonical pathway effector, β-catenin. To this end, we generated a gain-of-function mutant construct (NC1-Δ90βcat) in which the FoxD3 NC1 enhancer (Simões-Costa et al., 2012) drives a β-catenin variant that lacks the first 90 amino acids of the mouse protein and is fused to a C-terminal EGFP (Wrobel et al., 2007). This construct restricts expression of the gain-of-function mutant, which significantly up-regulates Wnt reporter output (Fig. S4), to the neural crest forming region following specification, in order to bypass deleterious effects of Wnt pathway modulation on induction and specification events. When coelectroporated with Draxin-FLAG (Fig. 8 B), as with hLRP5, neural crest migration was no longer adversely affected, and relative migration distance was restored to 92.5 ± 6.5% (n = 8) of the unelectroporated side (Fig. 8 C). Together, these data suggest that Draxin acts via LRP5 to modulate canonical Wnt signaling to control cranial neural crest EMT progression (Fig. 8 D).
Discussion
Canonical Wnt signaling has reiterative functions throughout different stages of neural crest development, from induction and specification to EMT and later differentiation (Wu et al., 2003; Yanfeng et al., 2003; Simões-Costa and Bronner, 2015; Simões-Costa et al., 2015). How it elicits different responses during these various events has been unknown. Our data provide the first mechanistic evidence that modulation of Wnt signaling output is necessary to control the timing and progression of cranial neural crest delamination/EMT and that the secreted molecule Draxin is a critical modulator of these signaling events.
Our results suggest the following model for the role of Draxin in EMT. The process of neural crest EMT can be divided into two steps: delamination from the neural tube and mesenchymalization (Rogers et al., 2013). Onset of Draxin expression within premigratory neural crest attenuates Wnt signaling to prevent early delamination. We show that loss of this attenuation can trigger the EMT program prematurely, with subsequent deleterious effects on cranial neural crest migration. After Draxin loss of function, the increase in Snail2 combined with decreased Cad6B causes premature delamination of neural crest from the dorsal neural tube (Fig. 9 A). This premature exit from the neural tube negatively impacts the maintenance of neural crest specifier gene expression and prevents up-regulation of the mesenchymal marker, HNK-1. Given that Sox9 and Snail2 cooperate to promote completion of EMT (Cheung et al., 2005; Liu et al., 2013), this likely explains why neural crest cells delaminate but fail to mesenchymalize and migrate away. In contrast, Draxin overexpression causes a decrease in Snail2 levels with concomitant increase in Cad6B, impeding separation from the neural tube (Fig. 9 B). The interference of delamination observed after Draxin overexpression also disrupts up-regulation of HNK-1 and, subsequently, completion of EMT.
Figure 9.
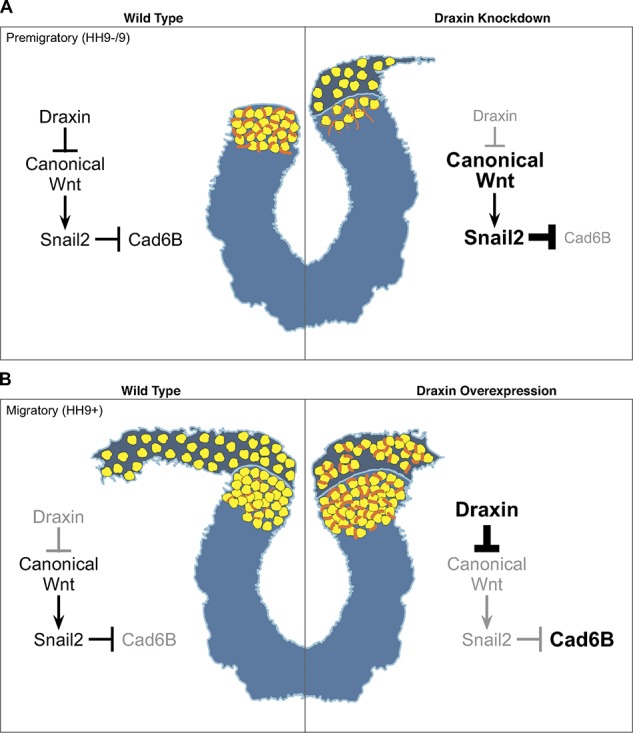
A model for Draxin’s control of cranial neural crest EMT. (A) Under WT conditions, Draxin is expressed in premigratory cranial neural crest between stages HH9− and HH9. While Draxin is present (left), tight control of canonical Wnt activation and inhibition maintains a balance of Snail2 and Cad6B expression, and neural crest cells are retained within the neural tube. When Draxin is knocked down (right) before EMT, there is an increase in canonical Wnt signaling, which causes an increase in Snail2 and decrease in Cad6b; this induces premature delamination of neural crest from the neural tube with a subsequent reduction in neural crest cell numbers and migration. (B) Under WT conditions, Draxin is down-regulated when cranial neural crest becomes migratory at stage HH9+. When Draxin is absent at this stage (left), the loss of canonical Wnt inhibition induces Cad6B repression via Snail2, and neural crest cells delaminate from the neural tube and mesenchymalize to migrate away from the midline. When Draxin is overexpressed at HH9+ (right), increased inhibition of canonical Wnt up-regulates levels of Cad6B both within the neural tube, reducing neural crest delamination, and in the migratory neural crest; however, maintenance of Cad6B expression in migratory neural crest reduces their mesenchymalization and migratory ability. Yellow circles indicate neural crest cells; orange lines indicate Cad6B.
The effects on Snail2 expression with Draxin perturbation strongly correlate with Draxin’s control of Wnt signaling output, given that Snail2 (Slug) has been shown to be a target of the canonical Wnt pathway in neural crest (LaBonne and Bronner-Fraser, 1998; Monsoro-Burq et al., 2005; Wu et al., 2005). This suggests that the levels of canonical Wnt signaling are tightly regulated and timed for correct progression of the EMT program. We hypothesize that premigratory neural crest cells need to remain within the neuroepithelium long enough to potentiate their transition to a mesenchymal cell type and that Draxin is a critical rheostat for regulating Wnt signaling during this process. Interfering with this potentiation by either inhibiting or maintaining Draxin results in failure to mesenchymalize and correctly complete the EMT program, thus impeding neural crest migration.
Our finding that a pulse of Draxin expression is required to transiently inhibit canonical Wnt within premigratory cranial neural crest for completion of EMT is consistent with recent work connecting premigratory signaling events with contact inhibition of locomotion (CIL; Scarpa et al., 2015). CIL, the process by which neural crest cells migrate, is controlled by noncanonical (β-catenin independent) Wnt/planar cell polarity (PCP) signaling (De Calisto et al., 2005; Carmona-Fontaine et al., 2008b). We speculate that perturbation of the degree and timing of canonical Wnt inhibition may impede the transition from canonical to noncanonical Wnt signaling and/or the switch from an epithelial to mesenchymal cell type, which may contribute to the negative consequences on neural crest migration resulting from altered Draxin expression. Further, our work corroborates a recent hypothesis that canonical Wnt signaling has differing effects on neural crest delamination and migration (Maj et al., 2016) and provides a mechanism for how canonical Wnt is tightly regulated in this context.
Recent work in the trunk neural crest has shown that transient intracellular inhibition of β-catenin–dependent Wnt signaling by the scaffold proteins Dact1/2 is necessary for trunk neural crest migration, though not for neural crest motility (Rabadán et al., 2016). Our results showing that modulation of Wnt signaling is important for chick cranial neural crest EMT agrees with the conclusions of Rabadán et al. (2016), who suggest that Wnt signaling needs to be transiently and reversibly inhibited for progression of EMT in the chick trunk region. However, the mechanisms underlying Wnt modulation appear to differ from those observed in our study on cranial neural crest EMT, likely because of well-known differences in the EMT mechanisms between the cranial and trunk neural crest in the chick (del Barrio and Nieto, 2002; Théveneau et al., 2007). Interestingly, Dact1/2 expression in the cranial region appears to be mutually exclusive from that of Draxin (Fig. 1); Dact1/2 expression is absent from premigratory neural crest cells that are retained within the dorsal neural tube and express Draxin but is up-regulated in neural crest cells that have delaminated from the neural tube (Alvares et al., 2009) when Draxin is down-regulated. This is consistent with our findings, where we hypothesize that Draxin would act before Dact1/2 as the primary regulator of these Wnt signaling events within premigratory neural crest.
Because cranial and trunk neural crest express different axial-level specific regulatory factors (Simoes-Costa and Bronner, 2016), it is unsurprising that the mechanisms of neural crest EMT and migration may vary between the different axial levels (Theveneau and Mayor, 2012). In particular, whereas canonical Wnt is important for cranial neural crest EMT, BMP signaling integrates with Wnt to regulate trunk neural crest delamination (Burstyn-Cohen et al., 2004; Kalcheim and Burstyn-Cohen, 2005). Furthermore, a myriad of extrinsic cues pattern neural crest migration and migratory pathways at specific axial levels, although directionality appears to be commonly regulated by noncanonical Wnt/PCP signaling (Carmona-Fontaine et al., 2008a; Theveneau and Mayor, 2012). Thus, it is interesting to note that whereas Draxin is expressed in a coordinated pulse between neural crest specification and EMT in the cranial region, its pattern and timing of expression in the trunk appears to change later in development, when it is expressed in the roof plate and the somites, where it is presumed to influence directionality of migratory trunk neural crest cells (Su et al., 2009). Parsing how Draxin influences signaling in these different contexts may provide insights into the control of neural crest EMT and migration at different axial levels.
In summary, our results elucidate a mechanism by which Draxin functions as a molecular rheostat of canonical Wnt signaling to control the timing of cranial neural crest EMT. Early expression of Draxin is required to prevent premature cranial neural crest delamination, and its inhibition of Wnt must be abrogated for complete neural crest mesenchymalization and migration. This novel role for Draxin as an inhibitor of EMT may have implications for other cell types that undergo EMT, such as metastatic tumor cells, in both development and disease states.
Materials and methods
Experimental model and subject details
Chicken embryos (Gallus gallus) were obtained commercially and incubated at 37°C to reach the desired Hamburger–Hamilton stage (Hamburger and Hamilton, 1951) as indicated. DF-1 fibroblasts were obtained from ATCC (CRL-12203) and maintained at 37°C/5% CO2 in DMEM supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin-streptomycin.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed as described (Simões-Costa et al., 2015). Briefly, embryos were fixed overnight at 4°C in PBS, pH 7.4, containing 4% PFA, dehydrated, and stored in methanol at −20°C before processing. To process for chromogenic in situ hybridization, embryos were rehydrated with DEPC-PBS/0.1% Tween-20 (PBT), treated with 10 µg/ml Proteinase K for 8 min at room temperature, washed successively with 2 mg/ml glycine/PBT and PBT, then postfixed with 4% PFA/0.2% glutaraldehyde at room temperature. After several washes of PBT, embryos were incubated for at least 3 h before addition of probe at 70°C in hybridization solution (Hyb) containing 50% formamide, 1.3× SSC, pH 5, 5 mM EDTA, pH 8, 200 µg/ml yeast tRNA, 0.2% Tween-20, 0.5% CHAPS, and 100 µg/ml heparin. Embryos were incubated with purified Draxin probe diluted in Hyb (1:300) for at least 16 h at 70°C. After probe hybridization, embryos were washed several times in Hyb at 70°C, then in MABT (1× maleic acid buffer/0.1% Tween-20) at room temperature. Embryos were blocked in 20% sheep serum/2% Boehringer blocking reagent (BBR) in MABT for several hours before addition of anti-digoxigenin–AP antibody (1:2,000; Fab fragments; 11093274910; Roche). After overnight antibody incubation at 4°C, embryos were washed extensively with MABT before chromogenic detection by NBT/BCIP.
To process for fluorescent in situ hybridization, embryos were rehydrated with PBT and then treated with 1% hydrogen peroxide/PBT for 20 min at room temperature. After several washes of PBT, embryos were incubated for at least 3 h before addition of probe at 68°C in Hyb. Embryos were incubated with purified Draxin probe diluted in Hyb (1:300) at least 16 h at 68°C. After hybridization, embryos were washed several times in Hyb at 68°C and then in KTBT (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM KCl, and 0.2% Tween-20) at room temperature. Embryos were blocked in 20% sheep serum/2% BBR in KTBT for several hours before addition of anti-digoxigenin–POD antibody (1:1,000; Fab fragments; 11207733910; Roche). After overnight antibody incubation at 4°C, embryos were washed extensively with KTBT before fluorescent tyramide signal amplification (TSA Plus Cyanine 3 System; Perkin Elmer).
For both chromogenic and fluorescent in situ hybridization, digoxigenin-labeled antisense RNA probe was synthesized for Draxin (GenBank accession: NM_001142848.2; nucleotides 898–1,626) using S6 RNA polymerase (Promega) and purified with Illustra ProbeQuant G-50 Micro Columns (GE Healthcare).
Whole-mount immunohistochemistry
Whole-mount immunohistochemistry was performed as described (Rogers et al., 2013). Briefly, embryos were fixed for 20 min in 4% PFA in sodium phosphate buffer. Washes and blocking buffer (10% donkey serum) used TBSTx (0.5 M Tris-HCl/1.5 M NaCl/10 mM CaCl2/0.5% Triton X-100). Embryos were incubated in primary and secondary antibodies for 2 d each at 4°C before imaging. Primary reagents used are listed in Table S1. Species-specific secondary antibodies were labeled with Alexa Fluor 350, 488, 568, and 647 (Invitrogen).
Electroporations
Chick embryos were electroporated ex ovo at HH4 as described (Sauka-Spengler and Barembaum, 2008; Simões-Costa et al., 2015). Briefly, embryos were extracted from the eggs using filter paper rings and maintained ventral side up in Ringer’s solution until electroporation. Specified reagents (constructs, MOs, etc.) were injected between the vitelline membrane and epiblast as indicated in the text. Embryos were subsequently electroporated using platinum electrodes (five pulses, 6.0 V, 30-ms duration at 100-ms intervals) and cultured at 37°C in fresh albumin supplemented with 1% penicillin-streptomycin to the desired Hamburger–Hamilton stage. Embryos cultured in this manner occasionally develop with a “curved” phenotype; this is likely an artifact of tension sometimes produced by the filter paper and does not directly change neural crest development.
Draxin loss of function
Draxin knockdown was performed by CRISPR/Cas9- or antisense MO electroporation. For CRISPR/Cas9 experiments, Draxin single-guide RNA (gRNA [sgRNA]) was designed to target the protospacer-adjacent motif site adjacent to the start codon using the CRISPR design tool (http://crispr.mit.edu) and produced using the short oligonucleotide method as described (Talbot and Amacher, 2014). Briefly, a short guide oligonucleotide was designed containing a clamp and T7 promoter sequence, a Draxin target sequence, and a sequence overlapping a guide-constant oligonucleotide. PCR was performed using AccuPrime Taq Polymerase (Thermo Fisher Scientific) with the short guide oligonucleotide, the guide-constant oligonucleotide, and two invariant gRNA primers. A single band at 120 bp was gel-extracted using the MinElute gel extraction kit (Qiagen), then used as template for gRNA transcription using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs). For control sgRNA, the short guide oligonucleotide contained the clamp and T7 sequence, a control sequence designed to have no targets within the chick genome (Gandhi et al., 2017), then the guide-constant oligonucleotide overlapping sequence. Primer sequences (IDT) are listed in Table S2. Before electroporation, control or Draxin sgRNA was mixed with recombinant Cas9 (M0646; New England Biolabs), heated at 37°C for 15 min, and then diluted with 10 mM Tris-HCl, pH 8.5, so that sgRNA and protein were each at a final concentration of 0.5 µg/µl for injection.
For MO experiments, a translation-blocking antisense MO (Gene Tools; Table S2) was designed to span the Draxin start codon starting from nucleotide −5 to +20 (Draxin MO). For control MO (control MO) experiments, the standard control MO was used (Gene Tools). MOs were FITC labeled and coinjected at 1 mM concentration with pCIG carrier DNA (Megason and McMahon, 2002) to improve electroporation efficiency.
Expression vectors
The Draxin-FLAG expression vector was made by first PCR amplifying from HH9 cDNA the full-length coding region of Draxin (GenBank accession no. NM_001142848.2) using forward (5′-AAACTCGAGGCCACCAGGATTATGGCAGCTTCTTCCACC-3′) and reverse (5′-AAAGCTAGCCTATTACTTGTCATCGTCGTCCTTGTAGTCAACATTAATGAATGATCCCTGCTCTCCAT-3′) primers to insert an upstream Kozak sequence and C-terminal FLAG epitope tag. Draxin-FLAG coding sequence was then cloned into the pCI-H2B-RFP expression vector (Betancur et al., 2010b). The Draxin-ΔCRD expression vector was made by PCR amplifying the truncated form from the Draxin-FLAG expression vector (forward, 5′-AAACTCGAGGCCACCAGGATTATGGCAGCTTCTTCCACC-3′, and reverse, 5′-AAAGCTAGCCTATTACTTGTCATCGTCGTCCTTGTAGTCCGGCTCTCCTTCTGCCGATG-3′) and then cloning the fragment into pCI-H2B-RFP. The coding region for the CRD-FLAG construct was synthesized as a GeneArt Strings DNA fragment (Invitrogen) to include the CRD of Draxin-FLAG as well as the amino terminal signal peptide and carboxy terminal FLAG. This fragment was then used as a template for PCR amplification (forward, 5′-AAACTCGAGGCCACCAGGATTATGGCAGCTTCTTCCACC-3′, and reverse, 5′-AAAGCTAGCCTATTACTTGTCATCGTCGTCCTTGTAGTCACAGCGCCCTTTCCTTCGG-3′) before cloning into pCI-H2B-RFP. The CRD-ΔSP construct was made by PCR amplifying the CRD from the Draxin-FLAG expression vector (forward, 5′-AAACTCGAGGCCACAACCTGTGACCACCACCTTGACTGCC-3′, and reverse, 5′-AAAGCTAGCCTATTACTTGTCATCGTCGTCCTTGTAGTCACAGCGCCCTTTCCTTCGG-3′) and then cloning the fragment into pCI-H2B-RFP.
To generate the NC1-Δ90βcat construct, the coding for the amino terminal-truncated β-catenin was PCR amplified from a pCAG-Δ90GFP construct (Wrobel et al., 2007) using 5′-AAAGCTAGCGCCACCATGGCTCAGAGGGTCCGAG-3′ and 5′-CGGACATGTATGGCGACCGGTGTCCCAGCAGGTCAGTATCAAACCAGGCC-3′ forward and reverse primers, respectively. This Δ90βcat fragment was then cloned into NC1.1M3 construct upstream of EGFP such that Δ90βcat was directly conjugated to EGFP for visualization (Simões-Costa et al., 2012).
Cryosectioning
For cross sections, embryos were postfixed overnight at 4°C in 4% PFA in sodium phosphate buffer after whole-mount immunohistochemistry or 4% PFA/0.2% glutaraldehyde in PBS after in situ hybridization. Embryos were then washed with several changes of 5% sucrose in PBS at room temperature and then stored in 15% sucrose in PBS overnight at 4°C. Embryos were subsequently incubated in 7.5% gelatin for 4–16 h at 37°C and then frozen at −80°C. Transverse frozen sections were cut at 20-µm thickness and allowed to dry overnight at room temperature. Gelatin was removed by incubating slides in PBS at 37°C for 20 min, and coverslips were mounted with Fluoromount-G (SouthernBiotech).
DF-1 cell transfection and processing
Chicken DF-1 fibroblasts were plated in a 100-mm culture dish containing an 18-mm round coverslip and grown to 90% confluence before transfection. Cells were transfected with Draxin-FLAG using Lipofectamine 3000 (Invitrogen), scaled according to the manufacturer’s specifications. After overnight transfection, cells were washed with PBS and incubated in fresh medium overnight. The coverslip was removed, fixed in 4% PFA in sodium phosphate buffer for 20 min at room temperature, and washed with TBST; nuclei were stained with DAPI (Invitrogen); and the coverslip was mounted with Fluoromount-G before imaging to confirm transfection, indicated by H2B-RFP fluorescence. Conditioned medium was collected, centrifuged to remove cellular debris, and concentrated with Amicon Ultra-4 Centrifugal Filter Units 10,000–molecular weight cutoff (EMD Millipore), and protein was extracted with 8 M urea/2.5% SDS. Plated cells were washed with PBS, and then cellular proteins were extracted with 8 M urea/2.5% SDS. To confirm secretion of Draxin, conditioned medium and cellular protein lysates were analyzed by Western blot.
Western blots
Embryo lysates were prepared by homogenization in 8 M urea/2.5% SDS followed by 15-min incubation at 65°C. Samples were loaded (10 µg total protein per well for embryo lysates; 5% of volume for DF-1 cells and conditioned media) on Bolt 4–12% Bis-Tris Plus gels (Invitrogen), and electrophoresis was run according to manufacturer specifications. Transfer to nitrocellulose was performed in 25 mM Tris/0.2 M glycine/20% methanol for 1 h at 100 V at 4°C. After transfer, blots were blocked in 5% BSA/TBSTw (TBS, pH 7.6, and 0.1% Tween-20) for 1 h at room temperature and then probed with primary antibodies (Table S1) diluted in blocking solution overnight at 4°C. Washes were performed with TBSTw, and peroxidase-labeled species-specific secondary antibodies (KPL/SeraCare) were diluted in 5% milk/TBSTw and incubated for 1 h at room temperature. Bands were visualized by chemiluminescence (ECL Prime Western Blotting System; GE Healthcare) and quantified using ImageJ (64; National Institutes of Health). To probe for reference proteins (e.g., RFP and ribosomal S6) after visualization of GFP, blots were stripped after developing with 2% SDS/0.1M β-mercaptoethanol in 62.5 mM Tris-HCl, pH 6.8, for 30 min at 50°C.
Microscope image acquisition
All whole-mount and cross-section images were acquired at room temperature using a Zeiss Imager.M2 with an ApoTome.2 module, Axiocam 506 color and monochromatic cameras, and Zen 2 Blue software (ZEISS). Whole-mount images were acquired with a Plan Apochromat 10× objective/0.45 NA (ZEISS), and embryos were imaged in TBSTx. Cross sections were imaged using a Plan Apochromat 20× objective/0.8 NA (ZEISS), and embryos were imaged in Fluoromount-G (SouthernBiotech). Images were pseudocolored and minimally processed for brightness and contrast using Adobe Photoshop CC.
Image analysis
Relative maximum migration distance
Image analysis was performed in ImageJ. For each whole-mount image, the line tool was used to draw and measure a straight line from the midline to the farthest point of Pax7+ neural crest migration distance. Measurements were made for the unelectroporated (left) and electroporated (right) sides at the same axial level, and then the electroporated length was divided by the unelectroporated length to calculate the relative maximum migration distance. Six measurements for each side were taken throughout the level of the midbrain at ∼50-µm intervals, and the relative maximum migration distances were averaged for individual embryos. Data points (circles) represent the mean relative maximum migration distance for single embryos.
Relative cell counts
Image analysis was performed on transverse cross sections using Fiji (ImageJ; Schindelin et al., 2012). Images were made binary and segmented using the watershed function, regions of interest (ROIs; e.g. Pax7+ cells) were drawn separately on both the electroporated and unelectroporated sides, and then the Analyze Particles function was used to count fluorescently labeled cells within each ROI. The relative cell counts were determined by dividing the number of fluorescently labeled cells counted on the electroporated side by the unelectroporated side for each section and averaging three nonadjacent sections per embryo. Data are represented as the mean relative cell counts averaged among data from three individual embryos.
Relative fluorescence intensity
Image analysis was performed on transverse cross sections using ImageJ. Images were background subtracted using a 25-pixel rolling ball radius, ROIs were drawn separately around the cell ROIs on both the electroporated and unelectroporated sides, and then the integrated density was measured within each ROI. The relative intensity was determined by dividing the integrated density on the electroporated side by that on the unelectroporated side for each section and averaging three nonadjacent sections per embryo. Data are represented as the mean relative intensity averaged among data from three individual embryos.
Western blots
Image analysis was performed in ImageJ on scanned images of Western blots. Lanes were outlined using the rectangular selection tool, and bands were plotted using the Plot Lanes function. Areas under the peaks were selected using the wand tool and measured as percentages using the Label Peaks function. GFP and RFP bands were normalized by dividing their percentages by their respective ribosomal S6 band percentages and then averaged. The mean normalized percentage for GFP was then divided by the mean normalized percentage for its respective RFP to control for differences in electroporation efficiency. Data are represented as the mean relative intensity averaged across three technical replicates (n = 15 pooled half embryos for each experiment).
Statistical analysis
Statistical analyses were performed using Prism (7; GraphPad Software). P values are indicated in the text, and a cutoff of P < 0.05 was used to determine significance. For all assays comparing control pCI-H2B-GFP with Draxin-FLAG, significance was assessed using an unpaired two-tailed t test. For all other assays of multiple comparisons, P values were calculated using one-way ANOVA with Tukey’s post hoc multiple comparisons test to compare individual mutant/rescue experiments with pCI-H2B-RFP and Draxin-FLAG. Data are presented as mean values, with error bars indicating SEM. Number of embryos/samples and replicates are indicated in figure legends and/or text. Data distribution was assumed to be normal but was not formally tested.
Online supplemental material
Fig. S1 shows comparison of Draxin knockdown methods. Fig. S2 shows validation of the Draxin-FLAG construct used in this study. Fig. S3 shows amino acid sequence alignments among several species for Draxin and compares chick Draxin against human Dkk-1. Fig. S4 shows validation of the NC1-Δ90βcat as an activator of canonical Wnt signaling. Table S1 contains a list of reagents used in this study. Table S2 contains a list of primers used in this study.
Supplementary Material
Acknowledgments
We thank Dr. M. Simões-Costa (Cornell University, Ithaca, NY) for providing the NC1.1M3:EGFP construct, Dr. X. He (Boston Children's Hospital, Harvard Medical School, Boston, MA) for providing the human LRP5 plasmid, and Dr. S. Chapman (Clemson University, Clemson, SC) for providing the chick Dkk-1 probe template. We also thank Dr. M. Piacentino and S. Wilbert for technical assistance, Shashank Gandhi and Dr. Ruth Williams for helpful CRISPR suggestions, Drs. M. Martik, R. Uribe, and M. Piacentino for critical manuscript comments, and M. Maline for illustrations.
This work was supported by a National Institutes of Health grant (R01DE024157 to M.E. Bronner) and a Ruth L. Kirschstein National Research Service Award (F32DE026355 to E.J. Hutchins).
The authors declare no competing financial interests.
Author contributions: E.J. Hutchins and M.E. Bronner conceived and designed the experimental approach. E.J. Hutchins performed the experiments and analyzed the data. E.J. Hutchins and M.E. Bronner wrote the manuscript.
References
- Alvares L.E., Winterbottom F.L., Jorge E.C., Rodrigues Sobreira D., Xavier-Neto J., Schubert F.R., and Dietrich S.. 2009. Chicken dapper genes are versatile markers for mesodermal tissues, embryonic muscle stem cells, neural crest cells, and neurogenic placodes. Dev. Dyn. 238:1166–1178. 10.1002/dvdy.21950 [DOI] [PubMed] [Google Scholar]
- Basch M.L., Bronner-Fraser M., and García-Castro M.I.. 2006. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature. 441:218–222. 10.1038/nature04684 [DOI] [PubMed] [Google Scholar]
- Betancur P., Bronner-Fraser M., and Sauka-Spengler T.. 2010a Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 26:581–603. 10.1146/annurev.cellbio.042308.113245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur P., Bronner-Fraser M., and Sauka-Spengler T.. 2010b Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. USA. 107:3570–3575. 10.1073/pnas.0906596107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M. 1986. Analysis of the early stages of trunk neural crest migration in avian embryos using monoclonal antibody HNK-1. Dev. Biol. 115:44–55. 10.1016/0012-1606(86)90226-5 [DOI] [PubMed] [Google Scholar]
- Burstyn-Cohen T., Stanleigh J., Sela-Donenfeld D., and Kalcheim C.. 2004. Canonical Wnt activity regulates trunk neural crest delamination linking BMP/noggin signaling with G1/S transition. Development. 131:5327–5339. 10.1242/dev.01424 [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C., Matthews H., and Mayor R.. 2008a Directional cell migration in vivo: Wnt at the crest. Cell Adhes. Migr. 2:240–242. 10.4161/cam.2.4.6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C., Matthews H.K., Kuriyama S., Moreno M., Dunn G.A., Parsons M., Stern C.D., and Mayor R.. 2008b Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 456:957–961. 10.1038/nature07441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M., Chaboissier M.C., Mynett A., Hirst E., Schedl A., and Briscoe J.. 2005. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev. Cell. 8:179–192. 10.1016/j.devcel.2004.12.010 [DOI] [PubMed] [Google Scholar]
- De Calisto J., Araya C., Marchant L., Riaz C.F., and Mayor R.. 2005. Essential role of non-canonical Wnt signalling in neural crest migration. Development. 132:2587–2597. 10.1242/dev.01857 [DOI] [PubMed] [Google Scholar]
- del Barrio M.G., and Nieto M.A.. 2002. Overexpression of Snail family members highlights their ability to promote chick neural crest formation. Development. 129:1583–1593. [DOI] [PubMed] [Google Scholar]
- Dickinson M.E., Selleck M.A., McMahon A.P., and Bronner-Fraser M.. 1995. Dorsalization of the neural tube by the non-neural ectoderm. Development. 121:2099–2106. [DOI] [PubMed] [Google Scholar]
- Ferrer-Vaquer A., Piliszek A., Tian G., Aho R.J., Dufort D., and Hadjantonakis A.K.. 2010. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ß-catenin signaling in the mouse. BMC Dev. Biol. 10:121 10.1186/1471-213X-10-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S., and Bronner M.E.. 2018. Insights into neural crest development from studies of avian embryos. Int. J. Dev. Biol. 62:183–194. 10.1387/ijdb.180038sg [DOI] [PubMed] [Google Scholar]
- Gandhi S., Piacentino M.L., Vieceli F.M., and Bronner M.E.. 2017. Optimization of CRISPR/Cas9 genome editing for loss-of-function in the early chick embryo. Dev. Biol. 432:86–97. 10.1016/j.ydbio.2017.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Castro M.I., Marcelle C., and Bronner-Fraser M.. 2002. Ectodermal Wnt function as a neural crest inducer. Science. 297:848–851. [DOI] [PubMed] [Google Scholar]
- Giovannone D., Ortega B., Reyes M., El-Ghali N., Rabadi M., Sao S., and de Bellard M.E.. 2015. Chicken trunk neural crest migration visualized with HNK1. Acta Histochem. 117:255–266. 10.1016/j.acthis.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S.A., Simoes-Costa M., and Bronner M.E.. 2015. Evolution of vertebrates as viewed from the crest. Nature. 520:474–482. 10.1038/nature14436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V., and Hamilton H.L.. 1951. A series of normal stages in the development of the chick embryo. J. Morphol. 88:49–92. 10.1002/jmor.1050880104 [DOI] [PubMed] [Google Scholar]
- Hari L., Miescher I., Shakhova O., Suter U., Chin L., Taketo M., Richardson W.D., Kessaris N., and Sommer L.. 2012. Temporal control of neural crest lineage generation by Wnt/β-catenin signaling. Development. 139:2107–2117. 10.1242/dev.073064 [DOI] [PubMed] [Google Scholar]
- Kalcheim C., and Burstyn-Cohen T.. 2005. Early stages of neural crest ontogeny: Formation and regulation of cell delamination. Int. J. Dev. Biol. 49:105–116. 10.1387/ijdb.041949ck [DOI] [PubMed] [Google Scholar]
- LaBonne C., and Bronner-Fraser M.. 1998. Neural crest induction in Xenopus: Evidence for a two-signal model. Development. 125:2403–2414. [DOI] [PubMed] [Google Scholar]
- Liu J.A., Wu M.H., Yan C.H., Chau B.K., So H., Ng A., Chan A., Cheah K.S., Briscoe J., and Cheung M.. 2013. Phosphorylation of Sox9 is required for neural crest delamination and is regulated downstream of BMP and canonical Wnt signaling. Proc. Natl. Acad. Sci. USA. 110:2882–2887. 10.1073/pnas.1211747110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald B.T., Semenov M.V., Huang H., and He X.. 2011. Dissecting molecular differences between Wnt coreceptors LRP5 and LRP6. PLoS One. 6:e23537 10.1371/journal.pone.0023537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj E., Künneke L., Loresch E., Grund A., Melchert J., Pieler T., Aspelmeier T., and Borchers A.. 2016. Controlled levels of canonical Wnt signaling are required for neural crest migration. Dev. Biol. 417:77–90. 10.1016/j.ydbio.2016.06.022 [DOI] [PubMed] [Google Scholar]
- Martik M.L., and Bronner M.E.. 2017. Regulatory logic underlying diversification of the neural crest. Trends Genet. 33:715–727. 10.1016/j.tig.2017.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews H.K., Marchant L., Carmona-Fontaine C., Kuriyama S., Larraín J., Holt M.R., Parsons M., and Mayor R.. 2008. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 135:1771–1780. 10.1242/dev.017350 [DOI] [PubMed] [Google Scholar]
- Megason S.G., and McMahon A.P.. 2002. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 129:2087–2098. [DOI] [PubMed] [Google Scholar]
- Miyake A., Takahashi Y., Miwa H., Shimada A., Konishi M., and Itoh N.. 2009. Neucrin is a novel neural-specific secreted antagonist to canonical Wnt signaling. Biochem. Biophys. Res. Commun. 390:1051–1055. 10.1016/j.bbrc.2009.10.113 [DOI] [PubMed] [Google Scholar]
- Miyake A., Nihno S., Murakoshi Y., Satsuka A., Nakayama Y., and Itoh N.. 2012. Neucrin, a novel secreted antagonist of canonical Wnt signaling, plays roles in developing neural tissues in zebrafish. Mech. Dev. 128:577–590. 10.1016/j.mod.2012.01.001 [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq A.H., Wang E., and Harland R.. 2005. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell. 8:167–178. 10.1016/j.devcel.2004.12.017 [DOI] [PubMed] [Google Scholar]
- Nakagawa S., and Takeichi M.. 1995. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development. 121:1321–1332. [DOI] [PubMed] [Google Scholar]
- Niehrs C. 2006. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 25:7469–7481. 10.1038/sj.onc.1210054 [DOI] [PubMed] [Google Scholar]
- Nieto M.A., Sargent M.G., Wilkinson D.G., and Cooke J.. 1994. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 264:835–839. 10.1126/science.7513443 [DOI] [PubMed] [Google Scholar]
- Rabadán M.A., Herrera A., Fanlo L., Usieto S., Carmona-Fontaine C., Barriga E.H., Mayor R., Pons S., and Martí E.. 2016. Delamination of neural crest cells requires transient and reversible Wnt inhibition mediated by Dact1/2. Development. 143:2194–2205. 10.1242/dev.134981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C.D., Saxena A., and Bronner M.E.. 2013. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell Biol. 203:835–847. 10.1083/jcb.201305050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauka-Spengler T., and Barembaum M.. 2008. Gain- and loss-of-function approaches in the chick embryo. Methods Cell Biol. 87:237–256. 10.1016/S0091-679X(08)00212-4 [DOI] [PubMed] [Google Scholar]
- Scarpa E., Szabó A., Bibonne A., Theveneau E., Parsons M., and Mayor R.. 2015. Cadherin switch during EMT in neural crest cells leads to contact inhibition of locomotion via repolarization of forces. Dev. Cell. 34:421–434. 10.1016/j.devcel.2015.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. . 2012. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M., and Bronner M.E.. 2016. Reprogramming of avian neural crest axial identity and cell fate. Science. 352:1570–1573. 10.1126/science.aaf2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M., and Bronner M.E.. 2015. Establishing neural crest identity: A gene regulatory recipe. Development. 142:242–257. 10.1242/dev.105445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M.S., McKeown S.J., Tan-Cabugao J., Sauka-Spengler T., and Bronner M.E.. 2012. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet. 8:e1003142 10.1371/journal.pgen.1003142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M., Tan-Cabugao J., Antoshechkin I., Sauka-Spengler T., and Bronner M.E.. 2014. Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res. 24:281–290. 10.1101/gr.161182.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M., Stone M., and Bronner M.E.. 2015. Axud1 integrates Wnt signaling and transcriptional inputs to drive neural crest formation. Dev. Cell. 34:544–554. 10.1016/j.devcel.2015.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon B., and Mayor R.. 2012. Early neural crest induction requires an initial inhibition of Wnt signals. Dev. Biol. 365:196–207. 10.1016/j.ydbio.2012.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon B., Araya C., Linker C., Kuriyama S., and Mayor R.. 2009. Differential requirements of BMP and Wnt signalling during gastrulation and neurulation define two steps in neural crest induction. Development. 136:771–779. 10.1242/dev.029017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl-Mazzulla P.H., and Bronner M.E.. 2012. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J. Cell Biol. 198:999–1010. 10.1083/jcb.201203098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Naser I.B., Islam S.M., Zhang S., Ahmed G., Chen S., Shinmyo Y., Kawakami M., Yamamura K., and Tanaka H.. 2009. Draxin, an axon guidance protein, affects chick trunk neural crest migration. Dev. Growth Differ. 51:787–796. 10.1111/j.1440-169X.2009.01137.x [DOI] [PubMed] [Google Scholar]
- Talbot J.C., and Amacher S.L.. 2014. A streamlined CRISPR pipeline to reliably generate zebrafish frameshifting alleles. Zebrafish. 11:583–585. 10.1089/zeb.2014.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill L.A., Coles E.G., and Bronner-Fraser M.. 2007. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development. 134:1481–1490. 10.1242/dev.02834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E., and Mayor R.. 2012. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366:34–54. 10.1016/j.ydbio.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Théveneau E., Duband J.L., and Altabef M.. 2007. Ets-1 confers cranial features on neural crest delamination. PLoS One. 2:e1142 10.1371/journal.pone.0001142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribulo C., Aybar M.J., Nguyen V.H., Mullins M.C., and Mayor R.. 2003. Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development. 130:6441–6452. 10.1242/dev.00878 [DOI] [PubMed] [Google Scholar]
- Willems B., Tao S., Yu T., Huysseune A., Witten P.E., and Winkler C.. 2015. The Wnt co-receptor Lrp5 is required for cranial neural crest cell migration in zebrafish. PLoS One. 10:e0131768 10.1371/journal.pone.0131768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel C.N., Mutch C.A., Swaminathan S., Taketo M.M., and Chenn A.. 2007. Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev. Biol. 309:285–297. 10.1016/j.ydbio.2007.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Saint-Jeannet J.P., and Klein P.S.. 2003. Wnt-frizzled signaling in neural crest formation. Trends Neurosci. 26:40–45. 10.1016/S0166-2236(02)00011-5 [DOI] [PubMed] [Google Scholar]
- Wu J., Yang J., and Klein P.S.. 2005. Neural crest induction by the canonical Wnt pathway can be dissociated from anterior-posterior neural patterning in Xenopus. Dev. Biol. 279:220–232. 10.1016/j.ydbio.2004.12.016 [DOI] [PubMed] [Google Scholar]
- Yanfeng W., Saint-Jeannet J.P., and Klein P.S.. 2003. Wnt-frizzled signaling in the induction and differentiation of the neural crest. BioEssays. 25:317–325. 10.1002/bies.10255 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.