

Astrocytes promote synapse formation during development via secreted factors including thrombospondin family proteins, which act through the neuronal calcium channel subunit α2δ-1. Risher et al. demonstrate that this process requires signaling via the Rho GTPase Rac1 to facilitate the maturation of dendritic spine synapses in the cortex.
Abstract
Astrocytes control excitatory synaptogenesis by secreting thrombospondins (TSPs), which function via their neuronal receptor, the calcium channel subunit α2δ-1. α2δ-1 is a drug target for epilepsy and neuropathic pain; thus the TSP–α2δ-1 interaction is implicated in both synaptic development and disease pathogenesis. However, the mechanism by which this interaction promotes synaptogenesis and the requirement for α2δ-1 for connectivity of the developing mammalian brain are unknown. In this study, we show that global or cell-specific loss of α2δ-1 yields profound deficits in excitatory synapse numbers, ultrastructure, and activity and severely stunts spinogenesis in the mouse cortex. Postsynaptic but not presynaptic α2δ-1 is required and sufficient for TSP-induced synaptogenesis in vitro and spine formation in vivo, but an α2δ-1 mutant linked to autism cannot rescue these synaptogenesis defects. Finally, we reveal that TSP–α2δ-1 interactions control synaptogenesis postsynaptically via Rac1, suggesting potential molecular mechanisms that underlie both synaptic development and pathology.
Introduction
The controlled development of neuronal networks is crucial for proper function of the central nervous system (CNS). Thus, the formation of CNS synapses, the smallest cell biological units of neural circuits, is tightly regulated (Scheiffele, 2003; West and Greenberg, 2011), but the manner in which this occurs is poorly understood. Research in the last two decades revealed that astrocytes, the most abundant glial cell in the brain, promote the establishment of synaptic connectivity (Clarke and Barres, 2013). Astrocyte-secreted factors strongly enhance excitatory synapse formation in vitro and in vivo (Baldwin and Eroglu, 2017; Bosworth and Allen, 2017). The thrombospondin (TSP) family of extracellular matrix proteins were the first astrocyte-secreted synaptogenic factors to be identified via purified retinal ganglion cell (RGC) neuron cultures. Transgenic mice lacking two of the TSP isoforms (TSP1 and TSP2), which are expressed by astrocytes during early postnatal development, show decreased excitatory synaptic density in the cortex (Christopherson et al., 2005).
TSPs exert their synaptogenic effects via binding to their neuronal receptor, the calcium channel subunit α2δ-1 (Eroglu et al., 2009), also known as the gabapentin receptor (Gee et al., 1996). α2δ-1 (Cacna2d1) is highly expressed by neurons throughout the CNS (Cole et al., 2005), with enrichment in cortical and hippocampal pyramidal neurons. It is predicted to be a type I membrane protein consisting of an entirely extracellular α2 portion and a δ part that spans extracellular (ECD), transmembrane (TM), and cytosolic regions (Davies et al., 2007). Both α2 and δ are translated from the same transcript but are proteolytically cleaved into two mature polypeptides (De Jongh et al., 1990; Jay et al., 1991; Douglas et al., 2006). α2δ-1, via its Von Willebrand factor A (VWF-A) protein–protein interaction domain, interacts with the synaptogenic fragment of TSP2 containing epidermal growth factor–like repeats to promote synapse formation (Eroglu et al., 2009).
α2δ proteins are involved in the trafficking and function of voltage-gated calcium channels (VGCCs; particularly the high voltage L-type channels) and influence presynaptic release probability and homeostasis (Geisler et al., 2015). However, despite the importance of VGCCs to synaptic transmission and plasticity (Catterall and Few, 2008; Li et al., 2016), manipulation of channel expression or function does not affect TSP-induced synaptogenesis (Eroglu et al., 2009). Furthermore, α2δ proteins were shown to have functions independent of their roles in regulation of Ca2+ channels (Kurshan et al., 2009). Therefore, we previously proposed that α2δ-1 acts as a surface receptor for TSP and, after TSP binding to the VWF-A domain of α2δ-1, triggers a downstream synaptogenic signaling cascade that promotes the formation of excitatory synapses (Risher and Eroglu, 2012). α2δ-1 is also the receptor for the drug gabapentin, which is used for the treatment of epilepsy and neuropathic pain (Gee et al., 1996; Field et al., 2006). Gabapentin interferes with the binding of TSP to α2δ-1 and thereby inhibits synaptogenesis (Eroglu et al., 2009), suggesting that impairments in astrocyte signaling may underlie aberrant connectivity that drives neurological disease.
In this study, we have determined that dendritic α2δ-1 is crucial for the establishment of proper cortical synapse connectivity. In the absence of α2δ-1, intracortical excitatory synaptogenesis, synaptic function, and spinogenesis are greatly diminished. Furthermore, we found the small Rho GTPase, Ras-related C3 botulinum toxin substrate 1 (Rac1), to be a key component of the synaptogenic signaling cascade downstream of TSP–α2δ-1.
Results
TSP receptor α2δ-1 is critical for cortical synaptic development
We observed via Western blot from mouse cortical and hippocampal lysates that α2δ-1 expression increases rapidly between postnatal days 5 (P5) and P10 (Fig. 1 A), corresponding with a period in which excitatory synapse formation is initiated (Semple et al., 2013). To determine whether α2δ-1 is required for proper synaptic connectivity in the cortex, we used a mouse line lacking α2δ-1 (i.e., α2δ-1 knockout [KO]; Fig. S1, A and B; Park et al., 2016). No α2δ-1 protein is detected in synaptosome lysates isolated from the α2δ-1 KO mice (Fig. S1 C). We focused our analysis on the Layer I (L1) synaptic zone (S/Z) of primary visual cortex (V1; Fig. 1 B), which receives dendritic projections from excitatory pyramidal neurons in cortical layers II and III (Fig. 1 B), and which has the highest α2δ-1 expression in the CNS (Cole et al., 2005). We immunostained brain sections with pre- and postsynaptic marker pairs that label the two main classes of excitatory synapses in the cortex: (1) intracortical connections positive for presynaptic vesicular glutamate transporter 1 (VGluT1) and (2) sensory pathway VGluT2+ inputs from thalamus (Fig. 1 C). Colocalization of VGluT1 with postsynaptic PSD95 is severely decreased (−58.1%) in α2δ-1 KO mice compared with littermate WT mice at P21 (Fig. 1 D), indicating that loss of α2δ-1 disrupts intracortical synaptic connectivity. By contrast, thalamocortical synapses, quantified as the colocalization of VGluT2/PSD95, do not differ between WT and α2δ-1 KO at P21 (Fig. 1 E). The colocalization of VGluT1/PSD95 puncta is not merely by chance, since randomization of puncta by rotating the channels out of alignment by 90° nearly abolished occurrence of colocalized puncta in both the WT and KO (Fig. S1, D and E). These results suggest that α2δ-1 is specifically required for the formation of intracortical synapses in V1.
Figure 1.

Impaired synaptic connectivity in α2δ-1–deficient cortex. (A) Top: Western blot of α2δ-1 expression from WT cortex and hippocampus from postnatal day (PND) 1 to adult. Tubulin: loading control. Bottom: α2δ-1 expression as fold change from P1 (n = 3 mice per age). (B) Diagram of a mouse coronal brain slice including area V1. Layer I is the S/Z where IHC analyses were performed. Dendrites from this region are primarily from neurons whose cell bodies reside in LII/III. (C) Schematic of excitatory synaptic input to area V1. (D) Top: IHC images of pre- (VGluT1), post- (PSD95), and colocalized (white arrowheads) synaptic puncta from V1 of P21 WT and α2δ-1 KO mice. Bottom: Intracortical synapse quantification as percentage of WT (n = 3 mice per genotype). One-way ANOVA with Tukey’s multiple comparisons post hoc test. (E) Top: Thalamocortical synapse staining shown with pre- (VGluT2), post- (PSD95), and colocalized (white arrowheads) synaptic puncta from V1 of P21 WT and α2δ-1 KO mice. Bottom: Quantification of thalamocortical synapses as percentage of WT (n = 3 mice per genotype). One-way ANOVA. Error bars represent SEM. (F) IHC staining from area V1 at P40 showing VGluT1/PSD95 intracortical synapses in WT and α2δ-1 KO (n = 3 mice per genotype). Nested ANOVA. (G) Representative camera lucida drawings from LII/III pyramidal neurons in P21 WT and α2δ-1 KO. (H) Sholl analysis results show morphological complexity in P21 WT and α2δ-1 KO neurons (n = 4 neurons/mouse; three mice/genotype). ††, P = 0.00023; ANCOVA. (I) Total number of intersections measured via Sholl analysis (left) and total length of the dendritic arbor (right) compared between P21 WT and α2δ-1 KO neurons. Unpaired two-tailed t test. Error bars represent SEM. Bars: (D, E, F) 2 µm; (G) 50 µm. ***, P < 0.0001.
Area S1 somatosensory cortex also showed a strong decrease in the number of intracortical synapses, with no change in thalamocortical synapses (Fig. S1, F and G). The deficit in intracortical synapses becomes apparent between postnatal weeks 2 and 3, coinciding with peak α2δ-1 expression (Fig. 1 A): we found no significant differences between genotypes at P14 V1, but the synaptic growth that typically takes place between P14 and P21 failed to occur in the α2δ-1 KO mice (Fig. S1 H). We also observed a significant deficit in intracortical synapses in the KO at P40, indicating that the synaptic phenotype observed in α2δ-1 KOs is not merely a developmental delay (Fig. 1 F).
The striking decrease in the number of intracortical synapses was not caused by a decrease in neurons (determined by NeuN+ cell density and cortical thickness of V1 at P21; Fig. S1, I–K). However, overall neuronal complexity was significantly decreased in the KOs as quantified by Sholl analysis of Golgi-Cox–stained LII/III pyramidal neurons (Fig. 1, G and H). There was also a trend toward a decrease in total dendrite length (Fig. 1 I). Taken together, these results show that α2δ-1 is also required for dendritic elaboration, which is tightly linked with excitatory synaptogenesis during development (Cline and Haas, 2008).
We next tested whether the impaired synaptic connectivity in the α2δ-1 KO resulted in disrupted synaptic function. We performed electrophysiological recordings of miniature excitatory postsynaptic currents (EPSCs; mEPSCs) from LII/III pyramidal neurons in P21 V1 of littermate α2δ-1 WT and KO mice (Fig. 2 A). In accordance with our histological findings, mEPSCs from α2δ-1 KO cortices showed a strong reduction in frequency and a small but significant decrease in amplitude, reflecting highly decreased synaptic density and mildly decreased synaptic strength, respectively (frequency: WT, 2.534 ± 0.3843 Hz; KO, 1.046 ± 0.1668 Hz [−58.72%]; amplitude: WT, 16.02 ± 0.8360 pA; KO, 14.63 ± 0.5270 pA [−8.68%]; Fig. 2, B–D). On the contrary, miniature inhibitory postsynaptic currents (mIPSCs) showed no differences between genotypes (Fig. 2, E–G). We next recorded evoked EPSCs (eEPSCs) to determine whether loss of α2δ-1 affected the glutamate receptor subtype composition of postsynapses. We found a significant decrease in the N-methyl-D-aspartic acid (NMDA)/α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) ratio (Fig. 2, H and I) in line with significantly decreased rise and decay times observed in the mEPSCs (Fig. S2, A and B) as NMDA receptors contribute to the slow component of synaptic waveforms (Lester et al., 1990). Paired-pulse ratio (PPR) recordings showed no difference between the WT and α2δ-1 KO, indicating that α2δ-1 is not required for the control of presynaptic release properties in the cortex (Fig. 2, J and K). Taken together, these results show that α2δ-1 is required for proper excitatory synapse function but dispensable for the function of inhibitory synapses in the developing cortex.
Figure 2.

Lack of α2δ-1 results in decreased excitatory synaptic function. (A) Recordings were made from LII/III pyramidal neurons in V1 at P21. (B) mEPSC traces from WT and α2δ-1 KO pyramidal neurons. (C and D) Frequency (C, left), interevent interval (C, right), and amplitude (D) of mEPSCs from WT and α2δ-1 KO neurons (n = total of 12 cells from three animals/genotype). Left: Two-tailed t test. Right: Kolmogorov–Smirnov test. (E) mIPSC traces from WT and α2δ-1 KO pyramidal neurons. (F and G) Frequency (F, left), interevent interval (F, right), and amplitude (G) of mIPSCs from WT and α2δ-1 KO neurons (n = total of 10 cells from three animals/genotype). Left: Two-tailed t test. Right: Kolmogorov–Smirnov test. (H) Traces of NMDA-only and AMPA-only evoked currents from WT and α2δ-1 KO pyramidal neurons. (I) Quantification of NMDA/AMPA ratio between WT and α2δ-1 KO (n = total of 12 cells from three to four animals/genotype). Two-tailed t test. Error bars represent SEM. (J) Traces from paired pulse recordings from WT and α2δ-1 KO neurons. (K) Comparison of PPR between WT and α2δ-1 KO (n = total of 12 cells from three to four animals/genotype). One-way ANCOVA. Error bars represent SEM. *, P < 0.05; ***, P < 0.0001.
Our immunohistochemical and electrophysiological experiments revealed major changes in excitatory synaptic connectivity of α2δ-1 KO cortex. Based on its up-regulation at P10 (Fig. 1 A), we hypothesized that α2δ-1 is required for formation and/or maturation of dendritic spines. To investigate synaptic connectivity, we used serial section transmission EM (ssEM) to visualize synaptic components including dendrites, filopodia, spines, and postsynaptic densities in 3D (Harris et al., 2006). We focused on secondary and tertiary dendrites within the S/Z of V1 from P21 mice. EM analysis further confirmed the decreased excitatory synaptic density per spine length in the α2δ-1 KOs compared with WT (Fig. 3, A–C). α2δ-1 KO dendrites have far fewer protrusions than WT (Fig. 3, B and C) and regularly display “bulged” regions separated by thin constrictions of the shaft (Fig. 3 B), with the KO having increased frequencies of unusually small and large shaft measurements compared with WT (Fig. S3 A).
Figure 3.

Ultrastructural analysis reveals that α2δ-1 promotes synapse and spine maturation. (A) Electron micrographs from P21 V1 WT and α2δ-1 KO brains. Arrows, excitatory synapses. Bar, 1 µm. (B) ssEM 3D reconstructions of LI dendrites from P21 WT and α2δ-1 KO V1. Red, excitatory postsynaptic densities; blue arrows, mushroom spines; red arrowheads, nonsynaptic filopodia; black star, bulged region of dendritic shaft. Cube, 0.5 µm3. (C–F) Densities calculated from reconstructions of excitatory synapses (C), protrusions (D), mushroom spines (E), and thin spines (F). E and F show the width (W) and length (L) measurements used in spine identification (n = 4 dendrites/animal; three animals per genotype). Two-tailed t test. Error bars represent SEM. (G) Spines in the WT and filopodia in the α2δ-1 KO. (H) Comparison of filopodia (F) and spine (S) densities between the WT and α2δ-1 KO (n = 4 dendrites/animal; three animals per genotype). One-way ANOVA with Tukey’s multiple comparisons post hoc test. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
Dendritic protrusions receive the majority of excitatory synaptic inputs in the CNS, presenting in a variety of morphologies that reflect different stages of synaptic maturation (Harris and Kater, 1994; Irwin et al., 2001). In particular, as synapses mature, dendritic protrusions decrease in length and increase in width, switching from a filopodial morphology into mushroom spines (Grutzendler et al., 2002). Protrusion density was severely reduced in α2δ-1 KO dendrites compared with WT (Fig. 3 D), primarily driven by a decrease in mature mushroom spines as well as intermediate thin spines (Fig. 3, E and F). The majority of protrusions in the α2δ-1 KO tended to be thin and filopodia-like; occasionally harboring excitatory postsynaptic densities but often barren of synaptic contacts (Fig. 3 B, red arrowheads, and Fig. 3 G). Accordingly, overall protrusion length was increased in the α2δ-1 KO (Fig. S3 B). Comparison of spine versus filopodia protrusions revealed a failure of spinogenesis in the KOs, whereas filopodia numbers were unaffected between WT and KO (Fig. 3 H). α2δ-1 KO dendritic protrusions do not undergo the stereotypical filopodia-to-spine transition during excitatory synaptic development, thus revealing a crucial role for α2δ-1 in this process.
α2δ-1 is required cell autonomously for the formation and maturation of dendritic spine synapses
α2δ-1 is highly expressed in regions outside the brain, particularly in skeletal and cardiac muscles (Gong et al., 2001). To investigate whether the synaptic abnormalities in the global KO are caused by cell-autonomous loss of α2δ-1 in neurons and not secondary to its effects elsewhere, we used a transgenic mouse strain with floxed alleles of α2δ-1 (α2δ-1f/f; Park et al., 2016) to delete α2δ-1 by Cre-dependent recombination. These mice also carry Rosa(STOP)loxP-tdTomato alleles (a.k.a. Ai14, hereafter referred to as RTm), which express tdTomato under control of the Rosa locus after Cre-mediated recombination (Fig. 4 A). Cre recombinase was expressed in a sparse population of LII/III cortical pyramidal neurons by in utero electroporation of α2δ-1f/f-RTmf/f pups at embryonic day 15.5 (E15.5; Fig. 4 A). With this approach, we could preferentially knock out α2δ-1 in a small fraction of LII/III neurons that would be RTm+ (Fig. 4 B). Brains were harvested at P21, and RTm+ dendrites were imaged along with their associated pre- and postsynaptic puncta (Fig. 4 C). After reconstructing dendritic structures and synaptic puncta in 3D via Imaris, we observed similar impairments between this cell-specific loss of α2δ-1 (i.e., α2δ-1 conditional KO; cKO) and the global α2δ-1 KO. Total spine density was decreased in cKO dendrites compared with age-matched control (α2δ-1+/+-RTmf/f) dendrites driven largely by a reduction in mature spines (Fig. 4, D and E). Furthermore, both pre- (VGluT1) and postsynaptic (PSD95) markers associated with α2δ-1 cKO dendrites were decreased as was their colocalization (Fig. 4, D and E). Density of the NMDAR subunit NR1 as well as the juxtaposing of NR1 puncta with presynaptic VGluT1+ terminals were also significantly decreased in α2δ-1 cKO dendrites (Fig. 4, F and G). These findings show that α2δ-1 is required cell autonomously for intracortical synapse formation and spinogenesis.
Figure 4.

α2δ-1 promotes synapse and spine development cell autonomously. (A) Schematic for in utero electroporation. (B) Confocal image of P21 V1. Cre+ neurons in LII/III express RTm. Dendrites were imaged in the S/Z. (C) Workflow for analysis with Imaris. (D) 3D confocal images (top) and Imaris reconstructions (bottom) of RTm+ dendrites from WT and α2δ-1 cKO brains. Pre- (VGluT1), post- (PSD95), and colocalized synaptic puncta in close proximity to the dendrite are shown. (E) Density of total spines, mature and intermediate spines, VGluT1, PSD95, and excitatory synapses between WT and α2δ-1 cKO dendrites (four dendrites/animal; three animals per condition). Two-tailed t test. (F) 3D confocal images (top) and Imaris reconstructions (bottom) of RTm+ dendrites from WT and α2δ-1 cKO brains. Pre- (VGluT1), post- (NR1), and colocalized synaptic puncta in close proximity to the dendrite are shown. (G) Density of NR1 and colocalized VGluT1/NR1 synapses of WT and α2δ-1 cKO dendrites (six dendrites/animal; three animals per condition). Two-tailed t test. Error bars represent SEM. Bars: (B) 200 µm; (C) 1 µm; (D and F, top) 2 µm; (D and F, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
Post- but not presynaptic α2δ-1 is required for TSP-induced synaptogenesis
To determine whether TSP promotes cortical synapse formation through α2δ-1 in a manner similar to that we previously showed in RGCs (Eroglu et al., 2009), we isolated cortical neurons with >95% purity from P1 littermate α2δ-1 heterozygous (Het) and KO pups (Fig. 5 A). After 7 d in vitro (DIV7), neurons were treated with TSP2 for an additional 6 d. In DIV13 cultures from α2δ-1 Het mice, TSP2 promoted a significant increase in excitatory synapses, determined as a twofold increase in colocalization of presynaptic VGluT1 and postsynaptic Homer (Fig. 5, B and C). As expected, the synaptogenic effect of TSP2 was blocked by cotreatment with gabapentin (32 µM), which interferes with the binding of TSP to α2δ-1 (Fig. 5, B and C; Eroglu et al., 2009). The same TSP2 treatment did not induce a synaptogenic response in cortical neurons from α2δ-1 KO mice, which did not differ in synapse density from α2δ-1 Het neurons in the absence of TSP2 (Fig. 5, B and C). These results confirm that TSP2 induces intracortical excitatory synapse formation in vitro in an α2δ-1–dependent manner.
Figure 5.

TSP stimulates synaptogenesis via postsynaptic α2δ-1. (A) Mouse cortical neuron purification and TSP2 treatment timeline. (B) Cortical dendrites from α2δ-1 Het or KO mouse pups. Cells were treated with TSP2-containing or deficient growth media as well as the α2δ-1 ligand gabapentin. Colocalized pre- (VGluT1) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (C) Density of excitatory synapses shown as fold change compared with α2δ-1 Het GM only (n = 30 cells per condition; two independent experiments). (D) Mating strategy to generate four genotypes (G1–G4) needed to determine site of action of α2δ-1 for synaptogenesis. (E) Cortical neuron plating scheme for α2δ-1/GFP experiments. (F) GFP+ cortical neuron dendrites from α2δ-1 Het or KO mouse pups. Cells were treated with TSP2-containing or -deficient growth media. Colocalized pre- (Bassoon) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (G) Density of excitatory synapses shown as fold change compared with α2δ-1 Het/Het GM only (n = 30 cells per condition; two independent experiments). One-way ANOVA with Tukey’s multiple comparisons post hoc test. Bars, 5 µm. Error bars represent SEM. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
α2δ-1 is present both pre- and postsynaptically (Taylor and Garrido, 2008; Bauer et al., 2009). To determine whether pre- or postsynaptic α2δ-1 is critical for excitatory synaptogenesis, α2δ-1 Het and KO mice were crossed with a transgenic mouse line expressing GFP in all cells (Hadjantonakis et al., 1998), generating the following littermates: α2δ-1 Het/GFP (tg/0); α2δ-1 Het/GFP (0/0); α2δ-1 KO/GFP (tg/0); and α2δ-1 KO/GFP (0/0; Fig. 5 D). Cortical neurons from P1 pups were plated such that GFP+ neurons comprised 5% of the cells per condition (Fig. 5 E). In this way, when imaging synapses on GFP+ cells, the majority of the presynaptic inputs would come from surrounding GFP− neurons (Fig. 5 E). We tested three different scenarios: (1) control, where both the “presynaptic” (GFP−) and the “postsynaptic” (GFP+) neurons were α2δ-1 Het; (2) postsynaptic necessity, where a GFP+ α2δ-1 KO cell was surrounded by GFP− α2δ-1 Het neurons; and (3) presynaptic necessity, where GFP− α2δ-1 KO neurons synapse onto an GFP+ α2δ-1 Het neuron (Fig. 5 E). After TSP2 treatment (Fig. 5 A), we observed increased excitatory synapses in the control scenario, where all neurons expressed α2δ-1 (Fig. 5, F and G). Postsynaptic necessity was confirmed when TSP2 treatment had no effect on synapse number even though the presynaptic neurons still expressed α2δ-1. Unexpectedly, when α2δ-1 was present only in the postsynaptic cell, TSP-induced synaptogenesis was significantly enhanced even above control (Fig. 5, F and G), showing that presynaptic α2δ-1 is not required and potentially may have an inhibitory role for the synaptogenic function of TSP. These results confirm that postsynaptic α2δ-1 is necessary and sufficient for TSP-induced cortical synapse formation.
Rac1 is required for induction of excitatory synaptogenesis and spinogenesis by TSP–α2δ-1
Previously, we hypothesized that the TSP–α2δ-1 interaction activates a synaptogenic signaling event in neurons (Risher and Eroglu, 2012), but the downstream components of this process were unknown. Our results from knocking out α2δ-1 cell autonomously showed reduced synaptic localization of NR1 (Fig. 4, F and G), and the global α2δ-1 KO had a significantly diminished NMDA/AMPA ratio (Fig. 2, H and I). Therefore, we next used the CRISPR/Cas9 gene silencing approach to test whether deletion of NR1 impairs TSP-induced synapse formation. Small guide RNAs (sgRNAs) targeting α2δ-1 (Cacna2d1) and NR1 (Grin1) were cloned into the pX601 vector expressing an HA-tagged SaCas9, with gene silencing verified with the T7E1 assay, sequencing, and protein expression (Fig. S4, A–D). Rat cortical neurons were purified, transfected with CRISPR constructs, and treated with TSP2 before immunostaining for pre- and postsynaptic markers (Fig. 6 A). As expected, CRISPR-mediated deletion of Cacna2d1 completely abolished TSP2-induced synapse formation. Deletion of Grin1 did likewise (Fig. 6, B and C), revealing a role for NMDAR-mediated signaling in TSP-induced synapse formation.
Figure 6.
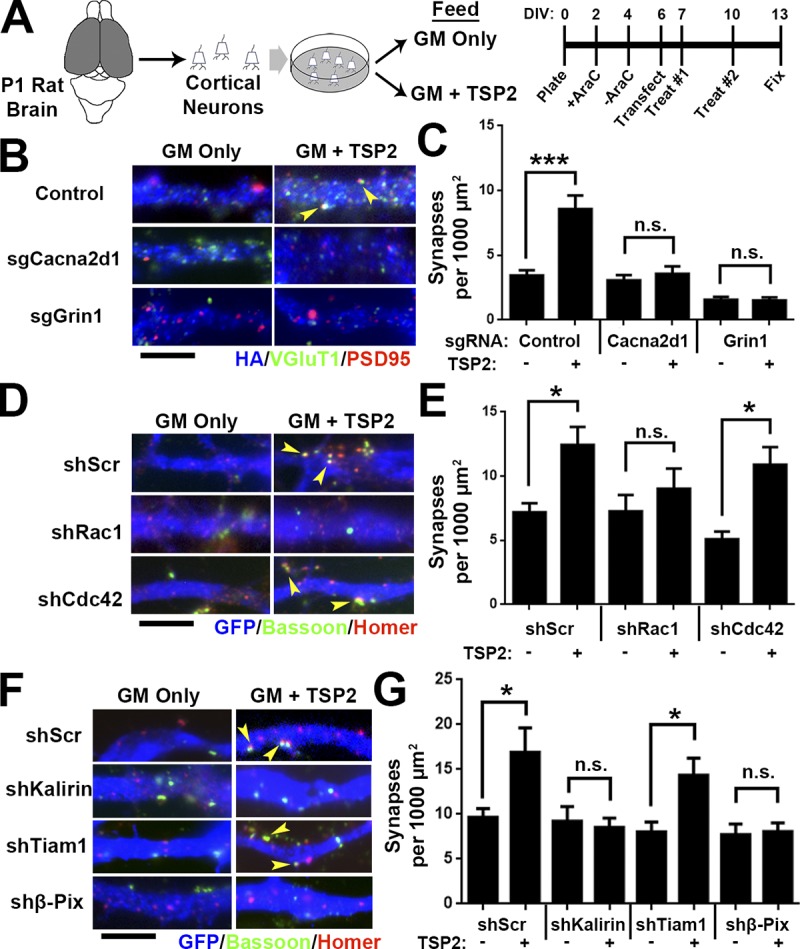
TSP-induced synapse formation requires Rho GTPase Rac1. (A) Rat cortical neuron purification and TSP2 treatment timeline. (B) Rat cortical neurons transfected with a pX601 vector containing saCas9 and sgRNA identified by HA expression. The pX601-only control (top) did not include an sgRNA sequence. Neurons were treated with TSP2-containing or deficient growth media. Colocalized pre- (VGluT1) and postsynaptic (PSD95) puncta reveal sites of excitatory synapses (yellow arrowheads). (C) Excitatory synapse density on HA+ neurons (n = 30 cells per condition; two independent experiments). (D) GFP+ rat cortical neuron dendrites. After transfection with an shRNA-expressing vector (with a scrambled shRNA sequence [shScr] as a control), neurons were treated with TSP2-containing or -deficient growth media. Colocalized pre (Bassoon) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (E) Excitatory synapse density on GFP+ neurons (n = 30 cells per condition; two independent experiments). Error bars represent SEM. (F and G) Same scheme as D and E except using shRNA against the Rho GEFs Kalirin-7, Tiam1, and β-Pix. One-way ANOVA with Tukey’s multiple comparisons post hoc test. Bars, 5 µm. *, P < 0.05; ***, P < 0.0001.
Our analysis of the synaptic connectivity in α2δ-1 KOs (Figs. 1, 2, and 3) showed that α2δ-1 regulates developmental processes in dendrites known to be highly dependent on the actin cytoskeleton (Ethell and Pasquale, 2005; Spence and Soderling, 2015), suggesting the activity of the Rho family of small GTPases. Among these GTPases, Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division control protein 42 (Cdc42) are involved in various stages of synaptic development including dendrite elaboration, filopodia formation, and spine maturation (Tashiro et al., 2000; Scott et al., 2003). Because these processes are all impaired in α2δ-1 KOs (Figs. 1, 2, and 3), we hypothesized that Rac1 and/or Cdc42 may be required downstream of TSP–α2δ-1. We used small hairpin RNAs (shRNAs) to diminish expression of Rac1 or Cdc42 in rat cortical neurons (Fig. S4, E and F). After verifying that these shRNAs are effective in down-regulating Rac1 or Cdc42 expression (Fig. S4, G and H), we transfected cortical neurons with either an shScr (scramble) control, shRac1, or shCdc42, followed by TSP2 treatment (Fig. 6 A). shRNA against Rac1 but not Cdc42 completely prevented the synaptogenic effect of TSP2 (Fig. 6, D and E). In spines, Rac1 is downstream of guanine nucleotide exchange factors (GEFs), which facilitate the GTP binding that converts Rac1 to its active state (Okamoto et al., 2009). We next used shRNA to determine whether the GEFs Kalirin-7, Tiam1, or β-Pix/Cool-1 participate in TSP-induced synapse formation (Fig. S4, E and F; verifications in Fig. S4, I–K). Interfering with the expression of either Kalirin-7 or β-Pix but not Tiam1 prevented TSP2-induced synaptogenesis (Fig. 6, F and G). Taken together, these results show that Rac1 and its upstream GEFs Kalirin-7 and β-Pix are required for TSP-induced synapse formation.
We next asked whether postsynaptic deletion of Rac1 would result in synapse and spine defects similar to the conditional α2δ-1 KO neurons (Fig. 4) using the Rac1f/f mice for Cre-mediated deletion of Rac1 (Glogauer et al., 2003). However, we could not use the in utero electroporation (IUE) approach as we did with α2δ-1 (Fig. 4) because this would inactivate Rac1 in radial glia and newborn neurons and affect neuronal differentiation and migration (Chen et al., 2009; Shoval and Kalcheim, 2012). To circumvent this limitation, we used an ex vivo organotypic slice culture model to delete Rac1 sparsely in cortical slices at a later developmental stage (corresponding with P5) via biolistic transfection with Cre recombinase (Fig. 7 A). Conditional Rac1 deletion resulted in severely decreased spine and synapse density (Fig. 7, B–D) at 19 d ex vivo (DEV; corresponding with P21), reminiscent of the α2δ-1 cKO (Fig. 4, A and B). To test whether α2δ-1 and Rac1 work together to promote excitatory synapse and spine formation, we overexpressed α2δ-1 in Rac1f/f slices and observed significantly increased spine and excitatory synapse densities (Fig. 7, B–D). By contrast, in Rac1f/f neurons cotransfected with Cre, α2δ-1 overexpression had no synaptogenic effect (Fig. 7, B–D). These results show that Rac1 is required downstream of α2δ-1 to promote synaptogenesis and spinogenesis.
Figure 7.

Rac1 promotes synaptic development and spinogenesis downstream of α2δ-1. (A) Schematic for organotypic slice culture/biolistic transfection. GFP+ dendrites (blue) are imaged at high magnification by confocal microscopy to capture spine morphology. (B) 3D confocal images (top) and Imaris reconstructions (bottom) of GFP+ dendrites in the S/Z of organotypic slices at DEV19 (i.e., P21) from Rac1f/f mice. Slices were transfected with cDNAs expressing GFP only or GFP plus Cre, α2δ-1 overexpression (OE), or α2δ-1 overexpression and Cre. Pre- (VGluT1), post- (PSD95), and colocalized (yellow) synaptic puncta in close proximity to the dendrite are shown. (C and D) Spine (C) and excitatory synapse (D) density from Rac1f/f dendrites (n = 12–18 dendrites per construct compiled from two independent experiments). One-way ANOVA with Dunnett’s multiple comparisons post hoc test (using GFP as control). Error bars represent SEM. Bars: (A, main) 50 µm; (A, inset) 5 µm; (B, top) 3 µm; (B, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
Dendritic α2δ-1 bridges extracellular synaptogenic signals with intracellular Rac1 activation
Loss of α2δ-1 results in synapse and spine defects in a cell-autonomous manner (Fig. 4), raising the question whether restoring α2δ-1 expression can reverse this phenotype. To test this, we first confirmed that α2δ-1 KO dendrites in organotypic cortical slices developed significantly fewer spines and excitatory synapses than those from littermate α2δ-1 Het slices (Fig. 8, A and D). Biolistic introduction of α2δ-1 into KO neurons completely rescued spine and synapse density (Fig. 8, B–D), confirming the postsynaptic sufficiency of α2δ-1 for excitatory synapse formation and spinogenesis. To investigate the roles of the extracellular and intracellular regions of α2δ-1, we next tested the sufficiency of two α2δ-1 mutants for rescuing synaptic deficits. The first mutant contains a point mutation in the VWF-A domain of the CACNA2D1 gene of a human patient with autism (Iossifov et al., 2014), changing a highly-conserved arginine 351 residue to threonine (R351T; Fig. 8 B). We verified by heterologous expression in HEK293 cells that α2δ-1 R351T mutant protein expresses efficiently and migrates in SDS-PAGE at the same size as native α2δ-1 (Fig. S5, A and B). However, surface staining in HEK293 cells transfected with α2δ-1 R351T revealed that this mutant cannot reach the plasma membrane surface and/or cannot be stabilized there but is rather localized to a membranous organelle just beneath the outer plasma membrane (Fig. S5 C). Thus, this mutation precludes the ability of α2δ-1 to interact with extracellular ligands such as astrocyte-secreted TSPs or other secreted or cell surface molecules. In the second α2δ-1 mutant, we replaced the TM portion of α2δ-1 with a glycosylphosphotidylinositol (GPI) anchor to generate a protein (α2δ-1 ΔTM), which is effectively produced and transported to the cell surface yet incapable of interacting through its TM and cytoplasmic regions (Figs. 8 B and S5, A–C). When expressed in cortical neurons, the ΔTM mutant localized primarily to dendritic spines, whereas the full-length α2δ-1 was found both on spines and dendritic shafts; α2δ-1 R351T showed little dendritic expression (Fig. S5 D). Neither α2δ-1 mutant was capable of rescuing spinogenesis or synaptogenesis phenotypes observed in organotypic slices from the α2δ-1 KOs (Fig. 8, C and D). These results show that extracellular interactions of α2δ-1, such as its binding to TSP, must be coupled with critical interactions established with the TM and intracellular regions of α2δ-1 to control synapse and spine formation.
Figure 8.

α2δ-1 and Rac1 work in concert to rescue synapses and spines in α2δ-1–null cortex. (A) 3D confocal images (top) and Imaris reconstructions (bottom) of GFP+ dendrites in the S/Z of DEV19 (i.e., P21) organotypic slices from α2δ-1 Het or KO mice. Slices were transfected with GFP to visualize dendritic morphology. Pre- (VGluT1), post- (PSD95), and colocalized (yellow) synaptic puncta in close proximity to the dendrite are shown. (B) α2δ-1–variant constructs used in the rescue experiment. (C) Same format as A but with all images taken from α2δ-1 KO mice. Slices were transfected with GFP plus either α2δ-1 (overexpression construct; OE), α2δ-1 R351T, or α2δ-1 ΔTM. (D) Spine (top) and excitatory synapse (bottom) density from α2δ-1 Het or KO dendrites. n = 12–24 dendrites per construct compiled from two independent experiments. (E) Same format as C, but slices were transfected with either fast-cycling (FC) Rac1 or FC Rac1 plus α2δ-1 ΔTM. (F) Spine (top) and excitatory synapse (bottom) density from α2δ-1 KO dendrites. n = 12–24 dendrites per construct compiled from two independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test (using KO as control). Error bars represent SEM. Bars: (A, C, and E, top) 3 µm; (A, C, and E, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
We next tested whether we could bypass the requirement for α2δ-1 for synaptogenesis by enhancing Rac1 activation. We used a fast-cycling Rac1 mutant (FC Rac1) that allows Rac1 to quickly cycle between its active and inactive states without the need of upstream activation via GEFs (Lin et al., 1999). Biolistic transfection of FC Rac1 rescued the spinogenesis phenotype in α2δ-1 KO cortical slices, but these spines did not fully recover excitatory contacts (Fig. 8, E and F). We speculated that the inability of Rac1 activation to rescue excitatory synapses was caused by the lack of critical interactions that α2δ-1 establishes through its extracellular regions. To test this, we coexpressed the α2δ-1 ΔTM mutant with FC Rac1 and fully restored both spine and synapse numbers in α2δ-1 KO dendrites (Fig. 8, E and F), indicating that extracellular interactions established by α2δ-1 coupled with Rac1 activation are necessary and sufficient for excitatory synapse formation. Collectively, these results show that the binding of astrocytic TSP to neuronal α2δ-1 triggers the activation of Rac1, promoting synapse formation and maturation in the developing cortex.
Discussion
Most excitatory synapses in the cortex are made onto dendritic spines, which undergo a complex morphological transformation that occurs in parallel with synaptic development (Harris and Kater, 1994). During this transition, immature dendritic filopodia, which are proposed to seek out appropriate axonal partners, give way to dendritic spines. Dendritic spines are more stable protrusions, containing an abundance of synaptic scaffolding proteins, neurotransmitter receptors, and an elaborate actin cytoskeleton (Grutzendler et al., 2002; Korobova and Svitkina, 2010). In this study, we show that α2δ-1, the neuronal receptor for astrocyte-secreted TSPs, is critical for the formation and maturation of cortical spine synapses. We found that these functions of α2δ-1 are cell autonomous to neurons, requiring the postsynaptic expression of α2δ-1.
Establishment of cortical connectivity by α2δ-1
In the cortical S/Z, glutamatergic excitatory synapses fall under two main categories, intracortical and thalamocortical (Nakamura et al., 2005). Previously we found that astrocyte-secreted hevin controls the formation and maturation of thalamocortical synapses by bridging presynaptic neurexin-1α and postsynaptic neuroligin-1 (Risher et al., 2014; Singh et al., 2016). In this study, we show that α2δ-1, the receptor for astrocyte-secreted synaptogenic TSPs, plays a predominant role in the formation of intracortical synapses and their localization onto dendritic spines. In agreement with our findings, TSP1/2 double null mice were shown to have a ∼30% reduction in their excitatory cortical synapses, determined by the colocalization of presynaptic protein bassoon and postsynaptic SAP102 (Christopherson et al., 2005). In the same study, the numbers of SV2+ axonal terminals were found to be significantly reduced in TSP1/2 KO mice as early as P8, whereas we found no significant changes in the numbers of excitatory synapses at ages earlier than P21. These results may indicate distinct roles of TSPs in axonal branching and elaboration versus their roles in synaptogenesis through signaling via postsynaptic α2δ-1.
Although α2δ-1 is not required for thalamocortical synaptogenesis, we previously found that overexpression of α2δ-1 under the Thy1 promoter in transgenic mice in a subset of cortical neurons dramatically increases VGluT2+ thalamocortical synapses at P21 (Eroglu et al., 2009). Moreover, in RGC cultures, TSPs induce the formation of excitatory synapses between these neurons, which exclusively express VGluT2 (Fujiyama et al., 2003; Christopherson et al., 2005). Taken together with our results, these findings indicate that an increase in α2δ-1 abundance and its subsequent activation by TSPs is sufficient to increase the overall excitatory synapse numbers regardless of the presynaptic axonal partner identity.
Mechanisms of TSP–α2δ-1–induced synapse and spine growth
Based on our findings, we propose in this study a two-step model for the synaptogenic capabilities of α2δ-1 (Fig. 9): (1) TSP binding to the extracellular domain of postsynaptic α2δ-1 on probing filopodia facilitates clustering of pre- and postsynaptic proteins to nascent synaptic sites, bringing these compartments in close apposition; and (2) the TM region and the C-terminal tail of α2δ-1 trigger intracellular signaling events such as the recruitment of GEFs Kalirin-7 and β-Pix to nascent synaptic sites within filopodia, inducing activation of Rac1 and reorganization of the actin cytoskeleton to promote spine stabilization and growth.
Figure 9.
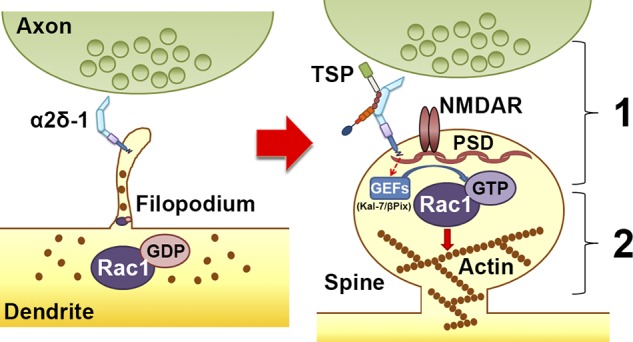
Model for α2δ-1’s dual role in promoting synapse and spine development. Early on, α2δ-1 is present on filopodia seeking contact with axonal partners. Rac1 is predominantly bound to GDP, rendering it inactive. After TSP binding, (1) α2δ-1 at the postsynaptic surface brings together pre- and postsynaptic components to form synapses; and (2) the C terminus of α2δ-1 triggers intracellular signaling via GEFs to stimulate GTP binding to Rac1, promoting actin reorganization to facilitate spine maturation.
In this study, we showed that overexpression of α2δ-1 fails to increase spine or synapse density when Rac1 function is abolished. Intriguingly, we were able to bypass the need for α2δ-1 in spinogenesis by directly stimulating Rac1, but Rac1 activation alone was unable to cluster pre- and postsynaptic machinery in the absence of the α2δ-1 ECD. Similarly, α2δ-1 ECD by itself could not induce synapse formation even though it was capable of localizing to dendritic spines. These results show that for synapse-containing spines to emerge, Rac1 activation should coincide with axon–dendrite matching and adhesion events. By virtue of its extracellular TSP-binding domain, its position within the plasma membrane, and its short C-terminal cytoplasmic tail, α2δ-1 is ideally situated to coordinate the occurrence of synaptogenesis and spinogenesis by recruiting important components of the synaptogenic signaling complex.
What comprises this synaptogenic signaling complex? We previously found that blocking or overexpressing the α1-subunits of the postsynaptic L-type calcium channels Cav1.2 and Cav1.3, the canonical interaction partners for α2δ-1 (Cantí et al., 2005), did not affect TSP-induced synaptogenesis (Eroglu et al., 2009). In this study, we instead show that disruption of the obligatory NMDA receptor subunit, NR1, abolishes the synaptogenic ability of TSP. We also observed cell autonomous loss of synaptic NR1 in the α2δ-1 cKO dendrites as well as a decreased NMDA/AMPA ratio in the α2δ-1 global KOs. These results are in line with previous findings that TSP induces the formation of silent synapses containing NMDA but not AMPA receptors (Christopherson et al., 2005). The binding of TSP to α2δ-1 may serve as an initiating event to recruit and stabilize NMDA receptors on the postsynaptic surface, which then coordinates the downstream signaling events that lead to spinogenesis. α2δ-1 may recruit NMDA receptors (NMDARs) to the synapses by a direct interaction through its C-terminal tail (Chen et al., 2018), which we found to be required for the synaptogenic activity of α2δ-1. Alternatively, TSP–α2δ-1 interaction could recruit NMDARs through an intermediary such as the neuroligin class of cell adhesion molecules, known interactors of NR1 (Budreck et al., 2013). Interestingly, TSPs have previously also been shown to interact with neuroligin family proteins (Xu et al., 2010). Furthermore, α2δ family members have recently been found to bind directly to neurexins, the primary presynaptic interaction partners of neuroligins (Tsetsenis et al., 2014; Tong et al., 2017). Possible cis-interactions between presynaptic α2δ-1 and neurexins would also provide an explanation for the enhanced TSP-induced synaptogenesis we observed when α2δ-1 was absent presynaptically. Presynaptic α2δ-1 may bind neurexins and prevent them from coupling transynaptically with neuroligins, which is important to stabilize newly formed synapses (Graf et al., 2004).
Although it is well established that dendritic protrusions undergo a maturational shift from highly motile filopodia to more stable spines, many of the molecular players underlying this transition have yet to be elucidated. The fact that both filopodia and spines are actin-rich structures means that cytoskeletal regulation is a critical mechanism underlying this morphological shift (Bilousova et al., 2006; Johnson and Ouimet, 2006). Small Rho GTPase proteins such as Rac1, RhoA, and Cdc42 differentially promote or inhibit cytoskeletal dynamics (Hall, 1998). Interestingly, TSP1 was previously shown to induce cytoskeletal reorganization in nonneuronal cells in a Rac1/Cdc42-dependent manner (Adams and Schwartz, 2000). One study showed that WT neurons cultured with TSP1-null astrocytes fail to develop mature spines (Garcia et al., 2010), indicative of problems with actin regulation. GEFs interact with NMDARs and other signaling molecules triggered by synaptic activity (Xie et al., 2007); in particular, Kalirin-7 interacts with NR1 at excitatory synapses and promotes spine enlargement via Rac1 (Xie et al., 2007; Ma et al., 2008), providing a potential link between α2δ-1 and Rac1 activation. Another GEF located in spines is β-Pix (Cool-1), which acts downstream of NMDAR to promote Rac1 activation (Saneyoshi et al., 2008). In this study, shRNA-facilitated knockdown of either Kalirin-7 or β-Pix was sufficient to abolish TSP-induced synaptogenesis in a similar manner to knocking down Rac1 function directly.
Contributions of α2δ-1 dysfunction to disease
α2δ-1 is linked to neurological disorders including epilepsy, neuropathic pain, intellectual disability, and autism spectrum disorder (Newton et al., 2001; Iossifov et al., 2014; Vergult et al., 2015). The antiepilepsy, antineuropathic pain drug gabapentin (Neurontin) binds to α2δ-1, and by doing so, it prevents TSP-induced synaptogenesis (Gee et al., 1996; Field et al., 2006; Eroglu et al., 2009). These findings had significant implications for the role of α2δ-1 in the development of maladaptive circuitry, likely by facilitating the wiring of inappropriate synaptic connections (Li et al., 2006, 2014). As we show in this study, the opposite situation is problematic as well, with too few synapses being formed and/or maintained as lack of α2δ-1 leads to a severe decrease in intracortical synapses. Reduced intracortical connectivity has been found in some patients with autism and was proposed to underlie the cognitive challenges faced by those individuals (Just et al., 2007). Although the mechanism of cortical synaptic impairment in those specific patients is unclear, we found that a previously identified autism-linked point mutation in α2δ-1 (Iossifov et al., 2014) diminished the effects of α2δ-1 on synaptic development and spine maturation. Our findings indicate that this single amino acid change in the extracellular VWF-A domain of α2δ-1 is sufficient to disrupt the localization of α2δ-1 to the cell surface, rendering it unavailable for binding TSP. Taken together, these findings underscore the importance of α2δ-1 for proper cortical development.
Our finding that Rac1 is downstream of TSP–α2δ-1 in mediating synapse and spine growth also provides a novel potential mechanistic insight into psychiatric illness and intellectual disability, where aberrant Rac1 signaling have been strongly implicated (Golden et al., 2013; Zeidán-Chuliá et al., 2013; Tejada-Simon, 2015). Rac1 impairments have been suggested as a mechanism underlying structural abnormalities in fragile X syndrome (Bongmba et al., 2011) as well as aberrant synaptic transmission in schizophrenia (Hayashi-Takagi et al., 2010). We have now elucidated a series of events in cortical development and synaptic growth wherein astrocytes can control neuronal Rac1 signaling via TSP–α2δ-1. Going forward, it will be essential to understand whether modulation of the TSP–α2δ-1–Rac1 pathway could provide a novel avenue for therapeutic approaches that aim to correct aberrant wiring of cortical circuits.
Materials and methods
Animal studies
All experiments were conducted in accordance with the Institutional Animal Care and Use Committee guidelines (protocol numbers A173-14-07 and A147-17-06).
Western blotting
C57/Bl6 WT mice (Jackson Laboratory) were perfused with PBS intracardially to clear blood before the brains were removed. Cortex and hippocampus were dissected out and homogenized in ice-cold solubilization buffer (25 mM Tris, pH 7.2, 150 mM NaCl, 1 mM CaCl2, and 1 mM MgCl2) containing 0.5% NP-40 (Thermo Fisher Scientific) and protease inhibitors (Roche). The protein concentrations of the lysates were determined using micro BCA protein assay kit (Pierce). Samples for SDS-PAGE were prepared at a 1 µg protein/µl concentration using 5× SDS-PAGE buffer (Pierce). 10 µg protein was loaded into each well. Samples were resolved by SDS-PAGE on 4–15% polyacrylamide gels (Bio-Rad) and transferred onto an Immobilon-FL PVDF membrane (EMD Millipore).
Blots were blocked in 50% fluorescent blocking buffer in PBS (MB-070; Rockland) containing 0.01% Tween-20 for 1 h at RT. Blots were incubated with primary antibody dilution in blocking buffer: mouse anti-DHP receptor, α2 subunit, 1:500 (D219; Sigma-Aldrich); rabbit anti–β-tubulin, 1:1,000 (926-42211; Li-Cor) overnight at 4°C. Fluorescently labeled secondary antibodies (Li-Cor) were diluted (1:5,000) in the same buffer as primary antibodies, and Western blots were incubated with secondary antibodies for 1 h at RT in the dark. Detection was performed using the Li-Cor Odyssey System.
Generation of α2δ-1 KO mice
Cavα2δ-1 cKO (α2δ-1f/f) mice were generated as described by Park et al. (2016) by flanking exon 6 of the Cavα2δ-1 gene (MGI; ID 88295; Mouse Genomics Informatics) with loxP sites. Homozygous Cavα2δ-1f/f mice were then crossed with a germline Cre driver (Sox2-Cre; Jackson Laboratory) to generate homozygous α2δ-1 KO mice. Genotypes were confirmed via PCR using F1 (WT forward, 5′-TCTCAGTTACAAGACTATGTGG-3′), F3 (KO forward, 5′-GGCTGTGTCCTTATTTATGG-3′), and LAF-Test (reverse, 5′-AGTAGGAGAAGGTACAATCGGC-3′) primers (Integrated DNA Technologies).
Cortical synaptosome preparation
Cortices were rapidly dissected from the brains of P21 α2δ-1 WT or KO littermate animals (n = 3 of each genotype) and homogenized using a Teflon glass homogenizer in 1.3 ml ice-cold homogenization buffer containing 320 mM sucrose, 4 mM Hepes, pH 7.4, 1 mM EGTA, phosphate inhibitors (5 mM NaF and 1 mM Na3VO4), and protease inhibitors (Roche). The homogenate was centrifuged at 700 g for 10 min at 4°C. The supernatant (S1) was centrifuged again at 12,000 g for 15 min to obtain the crude synaptosome fraction (P2). For separating synaptic cytosol-containing vesicles (LS1) and synaptic membrane (LP1), the pellet (P2) was lysed hypoosmotically and centrifuged at 35,000 g for 20 min. Pellet LP1 was resuspended in homogenization buffer containing 1% Triton X-100 (Roche). Protein concentration was determined by Micro BCA protein assay kit, and Western blotting was performed as described above using the mouse anti-DHP receptor, α2 subunit primary antibody (1:500; D219; Sigma-Aldrich), mouse anti–β-actin (1:1,000; ab8226; Abcam) and Li-Cor detection methods.
Immunohistochemistry and synaptic puncta imaging
For synaptic puncta analysis of mouse V1, α2δ-1 Het and KO mice on a C57/Bl6 background and littermate age-matched WT controls were stained with pre- (VGluT1 or VGluT2) and postsynaptic (PSD95) marker pairs as described previously (Kucukdereli et al., 2011; Risher et al., 2014; Singh et al., 2016). Three animals/genotype/age were perfused intracardially with TBS (TBS, 25 mM Tris-base, 135 mM NaCl, and 3 mM KCl, pH 7.6) supplemented with 7.5 µM heparin followed with 4% PFA (Electron Microscopy Sciences) in TBS. The brains were then removed and fixed with 4% PFA in TBS at 4°C overnight. The brains were cryoprotected with 30% sucrose in TBS overnight and were then embedded in a 2:1 mixture of 30% sucrose in TBS:OCT (Tissue-Tek). Brains were cryosectioned (coronal) at 20 µm using a Leica CM3050S. Three independent coronal sections per each mouse containing either V1 visual cortex (bregma, −2.5 to −3.2 mm; interaural, 1.3 to 0.6 mm; Franklin and Paxinos, 2001) or S1 somatosensory cortex (bregma, −1.0 to −2.0 mm; interaural, 2.86 to 1.86 mm) were used for analyses. Sections were washed and permeabilized in TBS with 0.2% Triton X-100 (TBST; Roche) three times at RT. Sections were blocked in 5% normal goat serum (NGS) in TBST for 1 h at RT. Primary antibodies (guinea pig anti-VGluT1 1:3,500 [AB5905; EMD Millipore], guinea pig anti-VGluT2 1:7,500 [135 404; Synaptic Systems], and rabbit anti-PSD95 1:300 [51-6900; Invitrogen]) were diluted in 5% NGS containing TBST. Sections were incubated overnight at 4°C with primary antibodies. Secondary Alexa fluor–conjugated antibodies (Invitrogen) were added (1:200 in TBST with 5% NGS) for 2 h at RT. Slides were mounted in Vectashield with DAPI (H-1200; Vector Laboratories), and images were acquired on a Leica SP5 confocal laser-scanning microscope.
5-µm-thick confocal z stacks (optical section depth, 0.33 µm; 15 sections/z stack; imaged area/scan = 20,945 µm2) of the S/Z were imaged with a 63× oil objective (1.4 NA) on a Leica SP5 confocal laser-scanning microscope using Leica Application Suite Advanced Fluorescence (LAS AF) software. Maximum projections of three consecutive optical sections (corresponding with 1 µm total depth) were generated from the original z stack. These maximum projections were then used for quantification of synaptic puncta in ImageJ (National Institutes of Health/; see the Quantification and statistical analysis section).
For cell staining, three P21 α2δ-1 KO and littermate WT mice were perfused and sectioned as described previously for synaptic staining. Sections containing V1 visual cortex (bregma, −2.5 to −3.2 mm; interaural, 1.3 to 0.6 mm; Franklin and Paxinos, 2001) were washed and permeabilized in TBS with 0.2% Triton X-100 (TBST; Roche) three times at RT. Sections were blocked in 5% normal donkey serum (NDS) in TBST for 1 h at RT. Primary antibodies (mouse anti-NeuN clone A60 1:1,000; MAB377; EMD Millipore; rabbit anti-Iba1 1:500; 019-19741; Wako; goat anti–aldolase C 1:250; sc-12066; Santa Cruz Biotechnology, Inc.) were diluted in 5% NDS containing 0.5% TBST. Sections were incubated overnight at 4°C with primary antibodies. Secondary Alexa fluor–conjugated antibodies (Invitrogen) were added (1:200 in TBST with 5% NDS) for 2 h at RT. Slides were mounted in Vectashield with DAPI (Vector Laboratories), and adjacent z stacks spanning the area between the pial surface and the corpus callosum were acquired with a 63× oil objective (1.4 NA) on a Leica SP5 confocal laser-scanning microscope using LAS AF software. Images were stitched together, and cells were quantified in ImageJ.
Golgi-cox staining and neuronal morphology
Golgi-cox staining was performed on P21 α2δ-1 KO and littermate WT control mice (n = 3 mice per genotype) as described in the FD Rapid GolgiStain Kit (FD NeuroTechnologies). Dye-impregnated brains were embedded in Tissue Freezing Medium (Triangle Biomedical) and rapidly frozen on ethanol pretreated with dry ice. Brains were cryosectioned coronally at 100-µm thickness and mounted on gelatin-coated microscope slides (LabScientific). Sections were stained according to the directions provided by the manufacturer.
Three independent coronal sections per each mouse, which contain the V1 visual cortex (bregma, −2.5 to −3.2 mm; interaural, 1.3 to 0.6 mm; Franklin and Paxinos, 2001) were acquired with a 100× oil objective (1.4 NA) on a Zeiss AxioImager D2 microscope. Layer II/III pyramidal neurons were identified by their distance from pia and their distinct morphologies.
Whole-cell patch-clamp recording
Brains from α2δ-1 KO mice and littermate age-matched WT controls were removed quickly into ice-cold solution bubbled with 95% O2, 5% CO2 containing the following (in mM): 194 sucrose, 30 NaCl, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose. After 5 min, the brain was blocked, and coronal slices containing area V1 were taken at 250 µm. During the recovery period (30 min), the slices were placed at 35.5°C with oxygenated artificial cerebrospinal fluid (aCSF) solution containing the following: 124 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 26 mM NaHCO3, 1.2 mM NaH2PO4, and 10 mM d-glucose, with pH adjusted to 7.4 with HCl and osmolality set to ∼320 mOsm. Pipettes (2.5–5 MΩ) contained the following: 120 mM cesium methane sulfonate, 5 mM NaCl, 10 mM tetraethylammonium chloride, 10 mM Hepes, 4 mM lidocaine N-ethyl bromide, 1.1 mM EGTA, 4 mM magnesium ATP, and 0.3 mM sodium GTP, with pH adjusted to 7.2 with CsOH and osmolality set to ∼300 mOsm.
All recordings were performed with a MultiClamp 700B amplifier (Molecular Devices). Signals were filtered at 10 kHz and digitized at 20 kHz with a Digidata 1440A digitizer (Molecular Devices). During the recordings, the slice was maintained under continuous perfusion of aCSF at 28–29°C with a 2–3-ml/min flow rate. In the whole-cell configuration (series resistance <25 MΩ), we recorded mEPSCs, mIPSCs, NMDA/AMPA ratio, and PPR of evoked EPSCs from layer II/III pyramidal neurons. mEPSCs were recorded with 1 µM tetrodotoxin and 50 µM picrotoxin in the bath solution in voltage-clamp mode (cells held at −70 mV). mIPSCs were recorded with 1 µM tetrodotoxin and 50 µM 2-amino-5-phosphonopentanoic acid (APV) and 50 µM 6,7-dinitroquinoxaline-2,3-dione in the bath solution at −70 mV. For evoked EPSCs, a small concentric bipolar electrode (FHC) was placed ∼100–200 µm to the recording electrode, and a Master 8 stimulator (AMPI) was used to control the stimulation parameters. To obtain the NMDA/AMPA ratio, peak amplitude of ESPCs at −70 mV in the presence of 50 µM picrotoxin was used as the AMPA component, and peak amplitude of EPSCs measured at +40 mV and 50 ms after the stimulation artifact was used as the NMDA component.
ssEM
For ssEM analysis of mouse V1, P21 α2δ-1 KO mice and their littermate WT controls (three mice per genotype/age) were first transcardially perfused with warm PBS solution to clear out blood cells, and then with warm (37°C) 2% PFA, 2.5% glutaraldehyde (EMS), 2 mM CaCl2, and 4 mM MgCl2 in 0.1 M cacodylate buffer (EMS; pH 7.4) under tribromoethanol (Sigma-Aldrich) anesthesia. 400-µm-thick coronal sections per each mouse, which contain the V1 visual cortex (Bregma −2.5 to −3.2 mm, interaural 1.3 to 0.6 mm; Franklin and Paxinos, 2001) were cut with a tissue chopper (Stoelting) and area V1 was dissected out with a #11 scalpel blade. V1 slices were immersed in 2% glutaraldehyde, 2 mM CaCl2, and 4 mM MgCl2 in 0.1 M cacodylate buffer, pH 7.4, and fixed overnight at 4°C. At the Duke Electron Microscopy Service core facility, slices were rinsed 3 × 5 min in 0.1 M phosphate buffer (PB) and postfixed in 1% OsO4 (Sigma-Aldrich) while heating in a microwave (2 min on, 2 min off, and then 2 min on at 70% power with vacuum). After rinsing 2 × 5 min with 0.1 M PB, they were dehydrated in ethanol/acetone series enhanced with 40 s microwave processing. They were next incubated in 50:50 acetone:epoxy overnight at RT. After two changes of straight Epon 3× 3 min in the microwave, slices were left to stand for 30 min and then embedded in 100% Epon resin at 60°C for 48 h. Ultrathin serial sections (45–50 nm) were cut from a small trapezoid positioned 50–100 µm below the pial surface, corresponding with the S/Z (a.k.a. layer I), which contains the dendrites of layer II/III neurons. Serial sectioning, processing, and photography were performed by the Electron Microscopy Core at Augusta University following a protocol adapted from Harris et al. (2006). Briefly, a diamond trim tool was used to cut ribbons of serial thin sections, which were retrieved on piloform-coated slot grids. Grids were stained with a series of solutions including filtered uranyl acetate and Reynold’s lead citrate, washed, and allowed to dry overnight. Stained grids were then loaded into a gimbal that was inserted into the rotational holder of a JEOL 1200 EX electron microscope. Using a cross sectioned dendrite of ∼1-µm diameter, a series of photographs was taken at 10,000× magnification of consecutive sections on the ribbons, taking care to maintain the same field of view/orientation from section to section. The images were then imported into Reconstruct software for alignment, tracing, and analysis (Fiala, 2005).
IUE and 3D analysis of confocally imaged synaptic structures
α2δ-1f/f mice were crossed with the B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J strain (Jackson Laboratory) to generate the α2δ-1f/f/RTmf/f mice that were used for IUE. All electroporations were performed at E15.5 to target neocortical layer 2/3 pyramidal neurons. Dams were sedated with continuously vaporized isoflurane, and abdominal incisions were performed to expose both uterine horns. 1 µg DNA plasmid containing pCAG-Cre with loading dye was injected into one lateral ventricle of each embryo using a pulled glass pipette. pCAG-Cre was a gift from C. Cepko (Harvard Medical School, Boston, MA; 13775; Addgene; Matsuda and Cepko, 2007). Five 50-ms pulses of 60 V spaced 950 ms apart were applied with tweezertrodes (positive paddle against the skull over the injection site, negative paddle across the body away from the placenta) using the BTX ECM 830 (Harvard Apparatus). PBS was applied to embryos and dam to prevent drying. After electroporation, the uterine horns were returned to the abdominal cavity, and the peritoneum, anterior muscle, and skin were sutured separately. The dam was then placed on a heating pad to recover and was monitored daily after the surgery.
Electroporated brains were harvested and cryoprotected at P21 after 4% PFA fixation. Sections (20 µm) were cut on a cryostat (Leica Biosystems) and stained for immunohistochemistry (IHC) using primary antibodies against RFP (1:200; rat; 5f8-100; ChromoTek), VGluT1 (1:500; guinea pig; AB5905; EMD Millipore), PSD95 (1:300; rabbit; 51-6900; Invitrogen), and NR1 (1:500; mouse; 75-272; Neuromab) followed by Alexa fluor–conjugated secondary antibodies (Invitrogen). RTm-expressing secondary/tertiary dendrites in the S/Z, along with surrounding pre- and postsynaptic puncta, were imaged with a 63× oil objective (1.4 NA) on a Zeiss 780 inverted confocal microscope using Zen Black software with 8× optical zoom at 0.13 µm optical section thickness. Z stacks were deconvolved (auto background; 20 signal-to-noise; 40 iterations; brick mode auto; optimized iteration mode) with Huygens image processing software (Scientific Volume Imaging) and then imported into Imaris (Bitplane) for analysis.
Cortical cell culture and synapse assay
Cortical neurons were purified from male P1 Sprague-Dawley rats (Charles River) or male α2δ-1 Het/KO pups (± EGFP, obtained by crossing the C57/Bl6 α2δ-1 mice with the FVB.Cg-Tg(CAG-EGFP)B5Nagy/J transgenic line from Jackson Laboratory; Hadjantonakis et al., 1998) by sequential immunopanning as follows. After dissection, cortices were digested for 45 min in papain (∼7.5 units/ml; Worthington). Papain digestion was then inhibited in sequential low/high concentrations of ovomucoid inhibitor (Worthington), and the resultant digested tissue was passaged through a 20-µm Nitex mesh filter (Sefar). The cell solutions then underwent negative immunopanning (to remove nontarget cells and debris) on Bandeiraea Simplicifolia Lectin I (Vector Laboratories)–coated Petri dishes (×2), AffiniPure goat anti–mouse IgG + IgM (H+L; 115-005-044, Jackson ImmunoResearch Laboratories)-coated dish, and AffiniPure goat anti–rat IgG (H+L; 112-005-044, Jackson ImmunoResearch Laboratories)-coated dish. A round of positive panning using mouse antineural cell adhesion molecule L1 (ASCS4; Developmental Studies Hybridoma Bank) to purify rat neurons or rat antineural cell adhesion molecule L1 antibody, clone 324 (MAB5272; EMD Millipore) for mouse neurons was used to isolate neurons from other cell types (predominantly astrocytes) to >95% purity. Adherent cells were washed 4× with DPBS (Gibco), collected with DPBS supplemented with BSA and insulin (Sigma-Aldrich), pelleted with centrifugation, and finally resuspended in serum-free growth medium containing Neurobasal, B27 supplement, 2 mM l-glutamine, 100 U/ml penicillin/streptomycin, 1 mM Na pyruvate (Gibco), 50 ng/ml brain-derived neurotrophic factor, 10 ng/ml ciliary neurotrophic factor (PeproTech), and 4.2 µg/ml forskolin (Sigma-Aldrich). Primocin (100 µg/ml; Invivogen) was added to the growth medium to prevent contamination of mouse neurons. Cells were plated at a density of either 40,000/well (nontransfection) or 75,000/well (transfection) on poly-d-lysine (PDL; Sigma-Aldrich) and laminin (Trevigen)-coated coverslips (Christopherson et al., 2005; Kucukdereli et al., 2011). Neurons were cultured for 2 d at 37°C/10% CO2 and then were treated for 48 h with AraC (Sigma-Aldrich) to kill any contaminating mitotic cells (i.e., astroglia). Neurons then were cultured with 500 ng/ml TSP2 or TSP2-free growth medium for an additional 9 d. Recombinant TSP2 protein was purified from CHO cells expressing mouse TSP2 following protocols described by Oganesian et al. (2008). Briefly, TSP2+ CHO cells (Genzyme) were grown to confluence in RPMI medium containing 10% FBS, 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, 1 mM Na pyruvate (Gibco), 5 µg/ml insulin (Sigma-Aldrich), and 0.1% β-mercaptoethanol (Sigma-Aldrich). Cells were switched to serum-free medium for 48–72 h. After collection of media, TSP2 was purified via affinity chromatography with HiTRAP heparin HP (GE Healthcare) according to the manufacturer’s protocols.
Synapse assay of cortical cultures follows a procedure modified from Kucukdereli et al. (2011). Briefly, DIV13 cells were fixed for 7 min with 4% PFA in PBS, washed three times in PBS, and blocked in 200 µl antibody blocking buffer (50% NGS and 0.2% Triton X-100) at RT for 30 min. After blocking, coverslips were washed three times in PBS and then incubated at 4°C overnight in 10% NGS antibody buffer containing primary antibodies against presynaptic VGluT1 (1:1,000; guinea pig; AB5905; EMD Millipore) and postsynaptic PSD95 (1:500; mouse; MA1-045; Thermo Fisher Scientific) or presynaptic Bassoon (1:500; mouse; VAM-PS003F; Enzo/Assay Designs) and postsynaptic Homer1 (1:500; rabbit; 160 002; Synaptic Systems). After another 3× wash in PBS, coverslips were incubated at RT for 90 min in 10% NGS antibody buffer containing Alexa fluor–conjugated secondary antibodies diluted 1:500. Coverslips were washed three times in PBS, mounted onto glass slides with Vectashield with DAPI, and imaged on an AxioImager M1 (Zeiss) with a 63× oil objective (1.4 NA). Image capture was performed with a Zeiss AxioCam MR3 camera with AxioVision software.
CRISPR/Cas9 constructs
sgRNA sequences against the rat CACNA2D1 and GRIN1 genes were generated using the DESKGEN Cloud tool (Illumina) using SaCas9 as the nuclease. Target sequences chosen (CACNA2D1, 5′-GGGAATACTCACGTCAGCAAG-3′; and GRIN1, 5′-AGCTGTGTAGGAGACAGGGGT-3′) were found within exons and had a minimum activity score of 45 (Doench et al., 2014). Custom oligonucleotides were generated (CACNA2D1 forward, 5′-CACCGGGAATACTCACGTCAGCAAG-3′; and CACNA2D1 reverse, 5′-AAACCTTGCTGACGTGAGTATTCCC-3′; and GRIN1 forward, 5′-CACCGAGCTGTGTAGGAGACAGGGGT-3′; and GRIN1 reverse, 5′-AAACACCCCTGTCTCCTACACAGCTC-3′; IDT) and cloned into the pX601 SaCas9 vector according to a protocol modified from Ran et al. (2013), substituting BsaI instead of BbsI for digestion of the pX601 vector. pX601-AAV-CMV::NLS-SaCas9-NLS-3×HA-bGHpA;U6::BsaI-sgRNA was a gift from F. Zhang (Massachusetts Institute of Technology, Cambridge, MA; 61591; Addgene). The subsequent sgRNA-containing pX601 vectors were then verified by transfecting into cultured rat lung fibroblasts. Briefly, lung fibroblasts were isolated from neonatal Sprague-Dawley rat pups according to standard protocols (Chang et al., 2016) via trypsin/collagenase-aided enzymatic digestion and cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco), 100 U/ml penicillin/streptomycin, 2 mM glutamine, and 1 mM Na pyruvate. Fibroblasts were grown to ∼80% confluence on 10-cm tissue culture dishes at 37°C/5% CO2, at which point they were trypsinized and passaged to 800,000 per dish. The next day, fibroblasts were transfected overnight with the pX601 vectors with XtremeGENE (Roche). The next morning, transfection medium was aspirated and replaced with 10 ml fresh growth medium. 72 h later, plates were washed 2× with PBS, trypsinized, then spun at 1,300 rpm for 10 min. Pellets were resuspended in 200 µL PBS, and genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen). After Phusion PCR amplification (New England Biolabs) of the sgRNA target sites (including adjacent PAM sequence) using custom oligonucleotides (CACNA2D1 forward, 5′-TCCCAAGACGACCCTGTCACTC-3′; and CACNA2D1 reverse, 5′-GAGCACTTGGCTTGATGGCA-3′; and GRIN1 forward, 5′-TGCACATAGAAACCCTGCTTCC-3′; and GRIN1 reverse, 5′-GTGAGGATGCTGATGGTGCA-3′; IDT), CRISPR/Cas9-cut DNA was verified with the T7E1 assay (Kim et al., 2009). In this assay, double-strand breaks in the DNA resulting from Cas9 cleavage of sgRNA sites introduce mutational insertions/deletions into the host sequence. The resulting DNA mismatches are recognized and cleaved by T7 endonuclease 1 (T7E1), producing bands of varying sizes (depending on the sgRNA target sites chosen) when run on a DNA agarose gel. Further verification was performed via sequencing of PCR amplicons after cloning into the Zero Blunt TOPO vector according to the manufacturer’s protocols (Invitrogen) and by Western blot after transduction of rat primary cortical neurons as described below.
shRNA constructs
pLKO.1 Puro plasmids containing shRNAs against mouse/rat, Rac1 (TRCN0000055188; 5′-GCTTGATCTTAGGGATGATAA-3′), Cdc42 (TRCN0000071684; 5′-CTGTCCAAAGACTCCTTTCTT-3′), Kalirin-7 (TRCN0000024365; 5′-CCCTTCTTAGATGAGAGCAAA-3′), Tiam1 (TRCN0000042595; 5′-CGGAATTTGGTGTCGGATATT-3′), and Cool-1/β-Pix (TRCN0000110028; 5′-GCCCTCCCAAAGGGTTCGATA-3′), were obtained from the RNAi Consortium (TRC) via GE Healthcare. CAG-EGFP was cloned into TRC shRNA constructs by digesting pLenLox-shNL1-CAG-EGFP (Chih et al., 2006) with Kpn1 and SpeI to release CAG-EGFP and inserting it in place of the puromycin ORF between Kpn1 and Spe1 sites in pLKO.1 puro.
Cortical neuron transfection
Cultured rat neurons were isolated as described above. At DIV 6, neurons in 24-well dishes were transfected with pLKO.1 shRNA plasmids using Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, 250 ng DNA was diluted in OptiMEM (Gibco), Plus Reagent, and LTX (1:2 DNA to LTX) and incubated at RT for 20 min. Half of the neuronal growth medium was saved to a separate dish and replaced with fresh medium. The transfection solution was added to the cultured neurons and incubated at 37°C for 2 h. Cells were washed 3× with warmed DPBS and returned with the saved plus fresh growth medium (1:1). After 24-h recovery, neurons were cultured with 500 ng/ml TSP2 or TSP2-free growth medium for an additional 6 d.
Cortical astrocyte isolation and culture
Cortices were harvested from P1 Sprague-Dawley rat pups and digested with papain followed by trituration in low and high ovomucoid solutions. Cells were filtered through a 20-µm Nitex mesh filter and resuspended in astrocyte growth medium (AGM; DMEM, 10% FBS, 10 µM hydrocortisone, penicillin/streptomycin, glutamate, 5 µg/ml insulin, Na pyruvate, and 5 µg/ml N-acetyl-l-cysteine). 15–20 million cells were plated on PDL-coated 75-mm2 flasks and incubated at 37°C/10% CO2. AGM was removed on DIV 3 and replaced with DPBS. Flasks were then vigorously shaken by hand for 15 s, leaving behind the adherent monolayer of astroglia. DPBS was then replaced with fresh AGM. AraC was added to the flasks on DIV 5 for 48 h to minimize the number of dividing cells. On DIV 7, astrocytes were passaged into 6-well dishes with 0.05% trypsin (Gibco) in preparation for transduction with lentivirus.
Lentiviral production and transduction
Lentiviruses encapsulating shRNA targeting vectors were produced to test knockdown efficiency of genes of interest in cultured primary astrocytes. To produce lentivirus, HEK293T cells were simultaneously transfected with a GFP-tagged shRNA targeting pLKO.1 plasmid (see above), an envelope plasmid (VSVG), and a packaging plasmid (ΔR8.2) with XtremeGENE (Roche). The next morning, the medium was switched to AGM for lentiviral conditioning. Medium containing lentivirus was collected on days 2 and 3 after transfection, centrifuged (11 min at 200 g) to remove dead cells and debris, and passed through a 0.45-µm filter to remove additional debris while leaving virus particles intact. Supernatant was tested for the presence of lentiviral particles using Lenti-X GoStix (Takara Bio Inc.), aliquoted, and stored at −80°C until use.
To test the knockdown efficiency of shRNA targeting vectors with puromycin selection, primary rat astrocyte cultures (DIV 8) in 6-well dishes were treated with 500 µl lentivirus supplemented with 2 µg polybrene per well. Puromycin (1 µg/ml) was added to the transduced cultures on DIV 10 and DIV 12 to select for the transduced cells. The cells were lysed on DIV 15 for protein extraction and subsequent Western blot analysis.
Organotypic slice culture and biolistic transfection
P2 Rac1f/f (Rac1tm1Djk/J, Jackson Laboratory; Glogauer et al., 2003) mouse pups, α2δ-1 Het/KO (backcrossed to CD1 background) mouse pups, or Sprague-Dawley rat pups were deeply anesthetized with isoflurane and decapitated; cortices were rapidly dissected into medium containing the following: 25 mM Hepes, 2 mM NaHCO3, 248 mM sucrose, 10 mM glucose, 4 mM KCl, 5 mM MgCl2, and 1 mM CaCl2. Then, 350-µm slices were cut with a tissue chopper (Ted Pella) and transferred to the surface of membrane inserts (PICM0RG50; EMD Millipore) placed in culture medium containing the following: 1 mM l-glutamine, 1 mM CaCl2, 2 mM MgSO4, 12.9 mM d-glucose, 5.2 mM NaHCO3, 30 mM Hepes, 0.001 mM insulin, 0.53 mM ascorbic acid, and 8.4 g/liter 20% heat-inactivated horse serum and 80% Hepes-based MEM. The pH was adjusted to 7.35 with 1 N NaOH, and osmolarity was adjusted to 320 mOsm. Slice-containing plates were maintained in a 37°C incubator with 5% CO2. 3 d after preparation, cultures were biolistically transfected with a Helios gene gun (Bio-Rad) following protocols described by Woods and Zito (2008). Briefly, plasmid DNA (∼3 µg per bullet) was combined with a well-vortexed and sonicated mixture of gold microcarriers (Bio-Rad) and 50 mM spermidine. 1 M CaCl2 was added while vortexing, and then the DNA/gold/spermidine mixture was incubated at RT for 20 min. The mixture was then centrifuged, the supernatant was aspirated, and the pellet was washed 3× with 1 ml of 100% EtOh to remove all traces of water. The pellet was then combined with 0.05 mg/ml polyvinylpyrrolidone and syringe-loaded into a 2-foot length of predried Tefzel tubing. The tubing was then cut into ∼1-cm “bullets” stored in a scintillation with desiccation pellets at −20°C until the time of transfection.
Slices were either transfected with bullets containing pβA-GFP alone or pβA-GFP together with Cre recombinase and/or variants of Rac1 or α2δ-1 (see below). Slices were then fixed with 4% PFA/4% sucrose at DIV 19 in PBS for 15 min at 37°C. Slices were permeabilized overnight at 4°C with 1% Triton X-100 in PBS and then blocked for 3 h with 0.25% Triton X-100 and 5% NGS in PBS at RT. Samples were then incubated with primary antibodies against GFP (1:1,000; chicken; AB16901; EMD Millipore), PSD95 (1:500; rabbit; 51-6900; Invitrogen), VGluT1 (1:1,000; guinea pig; AB5905; EMD Millipore), and/or FLAG (1:500; mouse; F1804; Sigma-Aldrich) for 3 d at 4°C. After washing three times for 1 h each with PBS-T (PBS containing 0.25% Triton X-100), slices were incubated with Alexa fluor–conjugated secondary antibodies (Thermo Fisher Scientific) for 2 d at 4°C. Slices were washed three times as before, mounted onto glass slides using Vectashield with DAPI, and imaged on an upright SP5 confocal microscope with resonant scanner using LAS AF software (Leica Microsystems). Images were acquired with a 63× oil objective (1.4 NA) with 8× optical zoom at 0.13-µm optical section thickness. Z stacks were deconvolved (auto background; 20 signal-to-noise; 40 iterations; brick mode auto; optimized iteration mode) with Huygens image processing software (Scientific Volume Imaging) and then imported into Imaris (Bitplane) for analysis.
cDNA constructs
Plasmid overexpressing α2δ-1, based on a construct obtained from D. Lipscombe (Brown University, Providence, RI), was a gift from Z.D. Luo (University of California, Irvine, Irvine, CA). α2δ-1 R351T, an autism-linked mutation found in a human patient as reported by Iossifov et al. (2014), was created by generating a single point mutation in the α2δ-1 expression plasmid using the QuikChange II XL Site Directed Mutagenesis Kit (Agilent Technologies) per manufacturer’s protocols. α2δ-1 ΔTM was created by subcloning a GPI anchor sequence into the α2δ-1 expression plasmid, replacing the latter’s TM domain. All three α2δ-1 variants were additionally subcloned to include a FLAG-tag sequence at the N terminus immediately before the start codon (α2δ-1 does not contain a true secretion sequence that is typically found in type I membrane proteins). An expression construct for Rac1 F28L, a fast-cycling Rac1 mutant, was created by using the QuikChange Mutagenesis Kit on WT human Rac1 (cDNA Resource Center).
HEK cell culture, lysate preparation, and immunocytochemical staining
HEK293 cells were cultured in DMEM supplemented with 10% FBS (Gibco), 100 U/ml penicillin/streptomycin, 2 mM glutamine, and 1 mM sodium pyruvate. Cells were incubated at 37°C/5% CO2 and regularly passaged every 3 d. Cells were then trypsinized and plated at a density of 30,000 per well into a 24-well plate containing PDL-coated coverslips (for immunocytochemistry) or 150,000 per well into a 6-well tissue culture plate (for Western blotting). After 24 h, cells were cotransfected (XtremeGENE) with different cDNA constructs including a membrane-targeted mCherry (CAAX). The next morning, the transfection medium was replaced with fresh growth medium, and cells were incubated for an additional 48 h before lysate preparation/immunocytochemistry staining.
For lysate preparation, cells were treated with RIPA buffer (Sigma-Aldrich) plus protease inhibitors (Roche) on ice for 15 min to lyse cells. Cells were then spun at max speed at 4°C for 10 min, and supernatant was collected. Protein concentration was determined by BCA assay kit, and Western blot protocol was performed as described above. Primary antibodies used include: mouse anti-DHP receptor, α2 subunit, 1:500 (D219; Sigma-Aldrich); rabbit anti-FLAG, 1:1,000 (F7425; Sigma-Aldrich); and mouse anti-GAPDH, 1:1,000 (ab9484; Abcam).
For immunocytochemistry, after PBS wash (×2), cells were fixed with ice-cold 4% PFA in PBS at RT for 5 min. After another PBS wash (×3), half of the cells were permeabilized with 0.2% Triton X-100 (Roche) in PBS at RT for 15 min, shaking; the remaining half received only PBS. After washing (PBS ×3), cells were blocked in a buffer containing 50% NGS at RT for 30 min on a shaker followed by primary antibody for 3 h (10% NGS; FLAG, 1:1,000, mouse, F1804; Sigma-Aldrich; RFP, 1:1,000, rabbit, ab62341; Abcam). After washing (PBS ×3), cells were treated with Alexa fluor–conjugated secondary antibodies at RT for 1 h on a shaker, washed for a final time, then mounted onto glass slides with Vectashield mounting medium. High-magnification images were taken on an Olympus FluoView confocal microscope with a 60× silicone oil objective (1.3 NA) using FV31S-SW software. For transfected nonpermeabilized cells, endogenous mCherry signal could be visualized but not the anti-RFP antibody; transfected permeabilized cells displayed both mCherry and anti-RFP expression (Fig. S5 C).
Quantification and statistical analysis
All data are presented as mean ± SEM unless otherwise specified. Statistical analyses were performed in Microsoft Excel, Statistica (Statsoft), or GraphPad Prism. Figure legend descriptions include values of n for each sample and respective P values.
Parametric tests were used after determining normality for all datasets via D’Agostino-Pearson omnibus K2 test (GraphPad Prism). Statistical analyses were conducted using either unpaired t tests (Fig. 1 H; Fig. 2, C, D, F, G, and I; Fig. 3, C–F; and Fig. 4, E and G), one-way (Fig. 1, C and D; Fig. 3 H; Fig. 5, C and G; Fig. 6, C, E, and G; Fig. 7, C and D; and Fig. 8, D and F) or nested (Fig. 1 E) ANOVAs, analyses of covariance (ANCOVAs; Figs. 1 G and 2 K), or Kolmogorov–Smirnov tests (Fig. 2, C, D, F, and G) as indicated in the respective figure legends. For one-way ANOVAs, followup Tukey’s or Dunnett’s multiple comparisons post hoc tests were conducted as stated. Significance was shown as *, P < 0.05; **, P < 0.001; and ***, P < 0.0001. Values that did not attain significance were either not noted or shown as ns.
IHC synaptic puncta analysis
Analyses were performed blind as to genotype. The Puncta Analyzer plugin that was developed by B. Wark for ImageJ (version ImageJ 1.29; Ippolito and Eroglu, 2010; Risher et al., 2014) was used to count the number of colocalized, pre-, and postsynaptic puncta. This quantification method is based on the fact that pre- and postsynaptic proteins (such as VGluT1 and PSD95) are not within the same cellular compartments and would appear colocalized only at synapses because of their close proximity. Previous studies showed that this quantification method yields an accurate estimation of the number of synapses in vitro and in vivo, which were previously confirmed by other methods such as EM and electrophysiology by us and others (Christopherson et al., 2005; Eroglu et al., 2009; Kucukdereli et al., 2011). Details of the quantification method have been described previously (Ippolito and Eroglu, 2010). Briefly, 1-µm-thick maximum projections are separated into red and green channels, background subtracted (rolling ball radius = 50), and thresholded to detect discrete puncta without introducing noise. The Puncta Analyzer plugin then uses an algorithm to detect the number of puncta that are in close alignment across the two channels, yielding quantified colocalized puncta. To calculate percentage of WT colocalization, colocalized puncta values for WT were averaged, then all image values (WT and KO) were converted to percentage of the calculated WT average.
Golgi-cox dendritic outgrowth and branching
For quantification of neurite outgrowth and branching, a total of 24 V1 layer 2–3 neurons (four neurons per anima; three animals per condition) were selected for dendritic tracing from α2δ-1 null and littermate WT controls. Tracing was performed with Neurolucida tracing tool (MBF Bioscience). Convex hull analysis was used to measure total dendritic length and area, and Sholl analysis was used to determine dendritic complexity/branching. All analyses were done with NeuroExplorer (MBF Bioscience) software with analyst blinded as to genotype. Statistical ANCOVA took into account a categorical independent variable (i.e., genotype) and a continuous independent variable (i.e., radius of concentric circle generated for Sholl analysis).
Whole-cell electrophysiology
Recordings were performed blinded as to genotype. The amplitudes of mEPSCs over 10 pA by peak detection software in pCLAMP10 (Molecular Devices) were included for this study. The amplitudes of mIPSCs over 20 pA were included. For EPSC experiments, NMDA and AMPA currents measured in these types of analyses are highly variable from cell to cell. Therefore, the ratio between the NMDA and AMPA currents are reported rather than the values for each. PPR was determined by calculating the ratio of the peak amplitude of the second EPSC to that of the first EPSC (at 50-, 100-, and 150-ms interstimulus intervals). Analysis was performed with a custom-written plugin for MatLab (MathWorks).
ssEM
Series consisting of 100–150 consecutive micrographs each were blinded as to condition before analysis. Serial sections were aligned and synaptic structures were traced using Reconstruct software (Fiala, 2005). Section thickness was calculated with the cylindrical method using the diameter of longitudinally sectioned mitochondria (Fiala and Harris, 2001). Dendrites were chosen for analysis on the basis of (a) spanning at least 75 consecutive serial sections, (b) measuring between 0.4–0.8 µm in diameter in cross section (to exclude large, apical dendrites and only include secondary and tertiary dendrites), and (c) having at least one spine (to exclude aspinous dendrites from interneurons; Bourne and Harris, 2011). PSD area was calculated by multiplying the 2D length on each section by average section thickness and the total number of sections on which the PSD appears. For classifying spines, spines with width (W) measurements ≥0.6 µm were considered mushroom spines; measurements of W <0.6 µm, length (L) <2.0 µm, and L:W ratio >1 were considered thin spines. To measure dendritic shaft areas, the cross-sectional dendritic area was taken from 10 evenly distributed points along each dendrite.
Confocal 3D dendrite reconstructions
Deconvolved confocal z stacks were imported into Imaris software. Dendrites were reconstructed in 3D using the FilamentTracer tool with the analyst blinded as to the condition. Briefly, the widest point in the dendritic shaft was used to generate the origin of the filament, with the minimum shaft diameter used to determine the entire dendritic outline. Erroneous connections were manually removed. Spines were automatically detected along the length of the rendered dendrite based on a minimum head width of 0.2 µm and maximum protrusion length of 4 µm. Spines were then categorized on the basis of their neck length, head width, and length:width ratio, with mature spines having either a head width >0.45 µm or a head width > spine length. Discrete pre-, post-, and colocalized synaptic puncta were resolved with the Spots tool after manual thresholding to detect discrete puncta without introducing noise. Puncta within 0.2 µm of dendrites were then isolated using the Find Spots Close to Filaments MatLab algorithm.
Online supplemental material
Fig. S1 shows design and further verification of the α2δ-1–null mouse. Fig. S2 shows faster mEPSC kinetics in α2δ-1 KO neurons. Fig. S3 shows how α2δ-1 KO dendrites have altered morphology. Fig S4 shows CRISPR/saCas9 and shRNA gene-manipulation strategies to elucidate the molecular mechanism of TSP-induced synapse formation. Fig. S5 shows cellular expression and localization of α2δ-1 variant constructs.
Supplementary Material
Acknowledgments
We thank Sara Miller and Neil Medvitz (Duke University Electron Microscopy Service, Durham, NC), Libby Perry and Brendan Marshall (Electron Microscopy Core at Augusta University, Augusta, GA), and Brittney Coleman (University of Alabama–Birmingham, Birmingham, AL) for their excellent technical assistance.
This research was funded by National Institutes of Health/National Institute on Drug Abuse (R01 DA031833) and National Institutes of Health/National Institute of Neurological Disorders and Stroke (NINDS; R01 NS096352) to C. Eroglu, a National Institutes of Health National Research Service Award (1F32NS083283-01A1) to W.C. Risher, and an NINDS award to H.H. Yin (R01 NS094754).
The authors declare no competing financial interests.
Author contributions: W.C. Risher and C. Eroglu designed the study. N. Kim and H.H. Yin designed, analyzed, and interpreted the electrophysiological experiments. W.C. Risher, S. Koh, J.-E. Choi, P. Mitev, E.F. Spence, and L-J. Pilaz conducted the remaining experiments and analyzed the data. D. Wang and G. Feng generated the CACNA2D1 KO mice. W.C. Risher, D.L. Silver, S.H. Soderling, H.H. Yin, and C. Eroglu approved data analyses. W.C. Risher and C. Eroglu wrote the manuscript, which was critically reviewed by all authors.
References
- Adams J.C., and Schwartz M.A.. 2000. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J. Cell Biol. 150:807–822. 10.1083/jcb.150.4.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin K.T., and Eroglu C.. 2017. Molecular mechanisms of astrocyte-induced synaptogenesis. Curr. Opin. Neurobiol. 45:113–120. 10.1016/j.conb.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C.S., Nieto-Rostro M., Rahman W., Tran-Van-Minh A., Ferron L., Douglas L., Kadurin I., Sri Ranjan Y., Fernandez-Alacid L., Millar N.S., et al. 2009. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J. Neurosci. 29:4076–4088. 10.1523/JNEUROSCI.0356-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova T.V., Rusakov D.A., Ethell D.W., and Ethell I.M.. 2006. Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J. Neurochem. 97:44–56. 10.1111/j.1471-4159.2006.03701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongmba O.Y., Martinez L.A., Elhardt M.E., Butler K., and Tejada-Simon M.V.. 2011. Modulation of dendritic spines and synaptic function by Rac1: A possible link to Fragile X syndrome pathology. Brain Res. 1399:79–95. 10.1016/j.brainres.2011.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosworth A.P., and Allen N.J.. 2017. The diverse actions of astrocytes during synaptic development. Curr. Opin. Neurobiol. 47:38–43. 10.1016/j.conb.2017.08.017 [DOI] [PubMed] [Google Scholar]
- Bourne J.N., and Harris K.M.. 2011. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 21:354–373. 10.1002/hipo.20768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budreck E.C., Kwon O.B., Jung J.H., Baudouin S., Thommen A., Kim H.S., Fukazawa Y., Harada H., Tabuchi K., Shigemoto R., et al. 2013. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. USA. 110:725–730. 10.1073/pnas.1214718110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C., Nieto-Rostro M., Foucault I., Heblich F., Wratten J., Richards M.W., Hendrich J., Douglas L., Page K.M., Davies A., and Dolphin A.C.. 2005. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of alpha2delta subunits is key to trafficking voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. USA. 102:11230–11235. 10.1073/pnas.0504183102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A., and Few A.P.. 2008. Calcium channel regulation and presynaptic plasticity. Neuron. 59:882–901. 10.1016/j.neuron.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Chang Y., Guo K., Li Q., Li C., Guo Z., and Li H.. 2016. Multiple directional differentiation difference of neonatal rat fibroblasts from six organs. Cell. Physiol. Biochem. 39:157–171. 10.1159/000445613 [DOI] [PubMed] [Google Scholar]
- Chen J., Li L., Chen S.R., Chen H., Xie J.D., Sirrieh R.E., MacLean D.M., Zhang Y., Zhou M.H., Jayaraman V., and Pan H.L.. 2018. The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Reports. 22:2307–2321. 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Melendez J., Campbell K., Kuan C.Y., and Zheng Y.. 2009. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev. Biol. 325:162–170. 10.1016/j.ydbio.2008.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B., Gollan L., and Scheiffele P.. 2006. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 51:171–178. 10.1016/j.neuron.2006.06.005 [DOI] [PubMed] [Google Scholar]
- Christopherson K.S., Ullian E.M., Stokes C.C., Mullowney C.E., Hell J.W., Agah A., Lawler J., Mosher D.F., Bornstein P., and Barres B.A.. 2005. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 120:421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Clarke L.E., and Barres B.A.. 2013. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 14:311–321. 10.1038/nrn3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline H., and Haas K.. 2008. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: A review of the synaptotrophic hypothesis. J. Physiol. 586:1509–1517. 10.1113/jphysiol.2007.150029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole R.L., Lechner S.M., Williams M.E., Prodanovich P., Bleicher L., Varney M.A., and Gu G.. 2005. Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J. Comp. Neurol. 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- Davies A., Hendrich J., Van Minh A.T., Wratten J., Douglas L., and Dolphin A.C.. 2007. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol. Sci. 28:220–228. 10.1016/j.tips.2007.03.005 [DOI] [PubMed] [Google Scholar]
- De Jongh K.S., Warner C., and Catterall W.A.. 1990. Subunits of purified calcium channels. Alpha 2 and delta are encoded by the same gene. J. Biol. Chem. 265:14738–14741. [PubMed] [Google Scholar]
- Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., and Root D.E.. 2014. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32:1262–1267. 10.1038/nbt.3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas L., Davies A., Wratten J., and Dolphin A.C.. 2006. Do voltage-gated calcium channel alpha2delta subunits require proteolytic processing into alpha2 and delta to be functional? Biochem. Soc. Trans. 34:894–898. 10.1042/BST0340894 [DOI] [PubMed] [Google Scholar]
- Eroglu C., Allen N.J., Susman M.W., O’Rourke N.A., Park C.Y., Ozkan E., Chakraborty C., Mulinyawe S.B., Annis D.S., Huberman A.D., et al. 2009. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell I.M., and Pasquale E.B.. 2005. Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 75:161–205. 10.1016/j.pneurobio.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Fiala J.C. 2005. Reconstruct: A free editor for serial section microscopy. J. Microsc. 218:52–61. 10.1111/j.1365-2818.2005.01466.x [DOI] [PubMed] [Google Scholar]
- Fiala J.C., and Harris K.M.. 2001. Cylindrical diameters method for calibrating section thickness in serial electron microscopy. J. Microsc. 202:468–472. 10.1046/j.1365-2818.2001.00926.x [DOI] [PubMed] [Google Scholar]
- Field M.J., Cox P.J., Stott E., Melrose H., Offord J., Su T.Z., Bramwell S., Corradini L., England S., Winks J., et al. 2006. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc. Natl. Acad. Sci. USA. 103:17537–17542. 10.1073/pnas.0409066103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K.B.J., and Paxinos G.. 2001. The Mouse Brain in Stereotaxic Coordinates. Academic Press, New York. [Google Scholar]
- Fujiyama F., Hioki H., Tomioka R., Taki K., Tamamaki N., Nomura S., Okamoto K., and Kaneko T.. 2003. Changes of immunocytochemical localization of vesicular glutamate transporters in the rat visual system after the retinofugal denervation. J. Comp. Neurol. 465:234–249. 10.1002/cne.10848 [DOI] [PubMed] [Google Scholar]
- Garcia O., Torres M., Helguera P., Coskun P., and Busciglio J.. 2010. A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down’s syndrome. PLoS One. 5:e14200 10.1371/journal.pone.0014200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee N.S., Brown J.P., Dissanayake V.U., Offord J., Thurlow R., and Woodruff G.N.. 1996. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 271:5768–5776. 10.1074/jbc.271.10.5768 [DOI] [PubMed] [Google Scholar]
- Geisler S., Schöpf C.L., and Obermair G.J.. 2015. Emerging evidence for specific neuronal functions of auxiliary calcium channel α2δ subunits. Gen. Physiol. Biophys. 34:105–118. 10.4149/gpb_2014037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogauer M., Marchal C.C., Zhu F., Worku A., Clausen B.E., Foerster I., Marks P., Downey G.P., Dinauer M., and Kwiatkowski D.J.. 2003. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 170:5652–5657. 10.4049/jimmunol.170.11.5652 [DOI] [PubMed] [Google Scholar]
- Golden S.A., Christoffel D.J., Heshmati M., Hodes G.E., Magida J., Davis K., Cahill M.E., Dias C., Ribeiro E., Ables J.L., et al. 2013. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 19:337–344. 10.1038/nm.3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H.C., Hang J., Kohler W., Li L., and Su T.Z.. 2001. Tissue-specific expression and gabapentin-binding properties of calcium channel alpha2delta subunit subtypes. J. Membr. Biol. 184:35–43. 10.1007/s00232-001-0072-7 [DOI] [PubMed] [Google Scholar]
- Graf E.R., Zhang X., Jin S.X., Linhoff M.W., and Craig A.M.. 2004. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 119:1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J., Kasthuri N., and Gan W.B.. 2002. Long-term dendritic spine stability in the adult cortex. Nature. 420:812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Hadjantonakis A.K., Gertsenstein M., Ikawa M., Okabe M., and Nagy A.. 1998. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech. Dev. 76:79–90. 10.1016/S0925-4773(98)00093-8 [DOI] [PubMed] [Google Scholar]
- Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- Harris K.M., and Kater S.B.. 1994. Dendritic spines: Cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17:341–371. 10.1146/annurev.ne.17.030194.002013 [DOI] [PubMed] [Google Scholar]
- Harris K.M., Perry E., Bourne J., Feinberg M., Ostroff L., and Hurlburt J.. 2006. Uniform serial sectioning for transmission electron microscopy. J. Neurosci. 26:12101–12103. 10.1523/JNEUROSCI.3994-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A., Takaki M., Graziane N., Seshadri S., Murdoch H., Dunlop A.J., Makino Y., Seshadri A.J., Ishizuka K., Srivastava D.P., et al. 2010. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 13:327–332. 10.1038/nn.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 515:216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito D.M., and Eroglu C.. 2010. Quantifying synapses: An immunocytochemistry-based assay to quantify synapse number. J. Vis. Exp. (45):2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin S.A., Patel B., Idupulapati M., Harris J.B., Crisostomo R.A., Larsen B.P., Kooy F., Willems P.J., Cras P., Kozlowski P.B., et al. 2001. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 98:161–167. [DOI] [PubMed] [Google Scholar]
- Jay S.D., Sharp A.H., Kahl S.D., Vedvick T.S., Harpold M.M., and Campbell K.P.. 1991. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J. Biol. Chem. 266:3287–3293. [PubMed] [Google Scholar]
- Johnson O.L., and Ouimet C.C.. 2006. A regulatory role for actin in dendritic spine proliferation. Brain Res. 1113:1–9. 10.1016/j.brainres.2006.06.116 [DOI] [PubMed] [Google Scholar]
- Just M.A., Cherkassky V.L., Keller T.A., Kana R.K., and Minshew N.J.. 2007. Functional and anatomical cortical underconnectivity in autism: Evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb. Cortex. 17:951–961. 10.1093/cercor/bhl006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Lee H.J., Kim H., Cho S.W., and Kim J.S.. 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 19:1279–1288. 10.1101/gr.089417.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F., and Svitkina T.. 2010. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell. 21:165–176. 10.1091/mbc.e09-07-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H., Allen N.J., Lee A.T., Feng A., Ozlu M.I., Conatser L.M., Chakraborty C., Workman G., Weaver M., Sage E.H., et al. 2011. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. USA. 108:E440–E449. 10.1073/pnas.1104977108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan P.T., Oztan A., and Schwarz T.L.. 2009. Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat. Neurosci. 12:1415–1423. 10.1038/nn.2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester R.A., Clements J.D., Westbrook G.L., and Jahr C.E.. 1990. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 346:565–567. 10.1038/346565a0 [DOI] [PubMed] [Google Scholar]
- Li B., Tadross M.R., and Tsien R.W.. 2016. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science. 351:863–867. 10.1126/science.aad3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.Y., Zhang X.L., Matthews E.A., Li K.W., Kurwa A., Boroujerdi A., Gross J., Gold M.S., Dickenson A.H., Feng G., and Luo Z.D.. 2006. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 125:20–34. 10.1016/j.pain.2006.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.W., Yu Y.P., Zhou C., Kim D.S., Lin B., Sharp K., Steward O., and Luo Z.D.. 2014. Calcium channel α2δ1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J. Biol. Chem. 289:7025–7037. 10.1074/jbc.M114.548990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R., Cerione R.A., and Manor D.. 1999. Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J. Biol. Chem. 274:23633–23641. 10.1074/jbc.274.33.23633 [DOI] [PubMed] [Google Scholar]
- Ma X.M., Wang Y., Ferraro F., Mains R.E., and Eipper B.A.. 2008. Kalirin-7 is an essential component of both shaft and spine excitatory synapses in hippocampal interneurons. J. Neurosci. 28:711–724. 10.1523/JNEUROSCI.5283-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T., and Cepko C.L.. 2007. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA. 104:1027–1032. 10.1073/pnas.0610155104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K., Hioki H., Fujiyama F., and Kaneko T.. 2005. Postnatal changes of vesicular glutamate transporter (VGluT)1 and VGluT2 immunoreactivities and their colocalization in the mouse forebrain. J. Comp. Neurol. 492:263–288. 10.1002/cne.20705 [DOI] [PubMed] [Google Scholar]
- Newton R.A., Bingham S., Case P.C., Sanger G.J., and Lawson S.N.. 2001. Dorsal root ganglion neurons show increased expression of the calcium channel alpha2delta-1 subunit following partial sciatic nerve injury. Brain Res. Mol. Brain Res. 95:1–8. 10.1016/S0169-328X(01)00188-7 [DOI] [PubMed] [Google Scholar]
- Oganesian A., Armstrong L.C., Migliorini M.M., Strickland D.K., and Bornstein P.. 2008. Thrombospondins use the VLDL receptor and a nonapoptotic pathway to inhibit cell division in microvascular endothelial cells. Mol. Biol. Cell. 19:563–571. 10.1091/mbc.e07-07-0649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K., Bosch M., and Hayashi Y.. 2009. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: A potential molecular identity of a synaptic tag? Physiology (Bethesda). 24:357–366. [DOI] [PubMed] [Google Scholar]
- Park J., Yu Y.P., Zhou C.Y., Li K.W., Wang D., Chang E., Kim D.S., Vo B., Zhang X., Gong N., et al. 2016. Central mechanisms mediating thrombospondin-4-induced pain states. J. Biol. Chem. 291:13335–13348. 10.1074/jbc.M116.723478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., and Zhang F.. 2013. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher W.C., and Eroglu C.. 2012. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 31:170–177. 10.1016/j.matbio.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher W.C., Patel S., Kim I.H., Uezu A., Bhagat S., Wilton D.K., Pilaz L.J., Singh Alvarado J., Calhan O.Y., Silver D.L., et al. 2014. Astrocytes refine cortical connectivity at dendritic spines. eLife. 3:e04047 10.7554/eLife.04047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T., Wayman G., Fortin D., Davare M., Hoshi N., Nozaki N., Natsume T., and Soderling T.R.. 2008. Activity-dependent synaptogenesis: Regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 57:94–107. 10.1016/j.neuron.2007.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P. 2003. Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 26:485–508. 10.1146/annurev.neuro.26.043002.094940 [DOI] [PubMed] [Google Scholar]
- Scott E.K., Reuter J.E., and Luo L.. 2003. Small GTPase Cdc42 is required for multiple aspects of dendritic morphogenesis. J. Neurosci. 23:3118–3123. 10.1523/JNEUROSCI.23-08-03118.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple B.D., Blomgren K., Gimlin K., Ferriero D.M., and Noble-Haeusslein L.J.. 2013. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107:1–16. 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoval I., and Kalcheim C.. 2012. Antagonistic activities of Rho and Rac GTPases underlie the transition from neural crest delamination to migration. Dev. Dyn. 241:1155–1168. 10.1002/dvdy.23799 [DOI] [PubMed] [Google Scholar]
- Singh S.K., Stogsdill J.A., Pulimood N.S., Dingsdale H., Kim Y.H., Pilaz L.J., Kim I.H., Manhaes A.C., Rodrigues W.S. Jr., Pamukcu A., et al. 2016. Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via hevin. Cell. 164:183–196. 10.1016/j.cell.2015.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence E.F., and Soderling S.H.. 2015. Actin out: Regulation of the synaptic cytoskeleton. J. Biol. Chem. 290:28613–28622. 10.1074/jbc.R115.655118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A., Minden A., and Yuste R.. 2000. Regulation of dendritic spine morphology by the rho family of small GTPases: Antagonistic roles of Rac and Rho. Cereb. Cortex. 10:927–938. 10.1093/cercor/10.10.927 [DOI] [PubMed] [Google Scholar]
- Taylor C.P., and Garrido R.. 2008. Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2-delta (alpha2-delta) type 1 protein. Neuroscience. 155:510–521. 10.1016/j.neuroscience.2008.05.053 [DOI] [PubMed] [Google Scholar]
- Tejada-Simon M.V. 2015. Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem. 133:767–779. 10.1111/jnc.13100 [DOI] [PubMed] [Google Scholar]
- Tong X.J., Lopez-Soto E.J., Li L., Liu H., Nedelcu D., Lipscombe D., Hu Z., and Kaplan J.M.. 2017. Retrograde synaptic inhibition is mediated by alpha-neurexin binding to the alpha2delta subunits of N-type calcium channels. Neuron. 95:326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsenis T., Boucard A.A., Araç D., Brunger A.T., and Südhof T.C.. 2014. Direct visualization of trans-synaptic neurexin-neuroligin interactions during synapse formation. J. Neurosci. 34:15083–15096. 10.1523/JNEUROSCI.0348-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergult S., Dheedene A., Meurs A., Faes F., Isidor B., Janssens S., Gautier A., Le Caignec C., and Menten B.. 2015. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur. J. Hum. Genet. 23:628–632. 10.1038/ejhg.2014.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.E., and Greenberg M.E.. 2011. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3:a005744 10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods G., and Zito K.. 2008. Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J. Vis. Exp. (12):675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Srivastava D.P., Photowala H., Kai L., Cahill M.E., Woolfrey K.M., Shum C.Y., Surmeier D.J., and Penzes P.. 2007. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 56:640–656. 10.1016/j.neuron.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Xiao N., and Xia J.. 2010. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat. Neurosci. 13:22–24. 10.1038/nn.2459 [DOI] [PubMed] [Google Scholar]
- Zeidán-Chuliá F., Rybarczyk-Filho J.L., Salmina A.B., de Oliveira B.H., Noda M., and Moreira J.C.. 2013. Exploring the multifactorial nature of autism through computational systems biology: Calcium and the Rho GTPase RAC1 under the spotlight. Neuromolecular Med. 15:364–383. 10.1007/s12017-013-8224-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.