

Abstract
Animal models have been tremendously useful to translational research, but there is a need to maximize their predictive value to human disease. This Comment proposes novel strategies that consider evolutionary history and the presence, absence or modification of molecular networks in one species that are being studied in the other.
The visionary physicians Rudolf Virchow and William Osler advanced the ‘One Health’ concept1, according to which human wellness and animal wellness are linked. In line with this paradigm, comparative medicine relies on animal models of disease to enable translational research by providing living systems in which the effect of discrete molecular manipulations can be examined in complex physiological contexts. However, even the best models are, by definition, an approximation and may vary in their fidelity to the induction and progression of human disease2. We believe that the limitations of comparative medicine arise in part from the historical use of reductionist approaches that focused on a limited number of molecules in specific signaling or metabolic pathways. With the explosion of ‘-omics’ technologies, complex molecular profiles and networks can now be assembled and compared between species in healthy or diseased states. These approaches provide a system-wide context for focused studies of disease-associated molecules of interest. The challenge that has emerged is the need for fresh analytical frameworks that consider the complex connectivity of molecular networks in a comparative context.
Evolutionary systems biology is an emerging discipline that aims to delineate how evolution jointly shapes genomes, molecular networks and phenotypes (Fig. 1). Humans and mice derive from a common mammalian ancestor but have evolved independently in distinct biospheres over ~90 million years. This evolutionary process is responsible for the similarities between humans and mice that enable biomedical research and for the differences that must be transcended. Here we introduce some fundamental concepts and technical challenges in evolutionary systems biology. We hope to encourage more systematic efforts to map and analyze systems-level similarities and differences between humans and animal models. Such efforts would shed light onto which pathways are more likely to ‘translate’ between species and would almost certainly reveal surprising evolutionary scenarios while providing context for, and helping in the prediction, interpretation and leveraging of, differences that arise from species’ independent evolutionary histories.
Fig. 1. evolutionary systems biology.
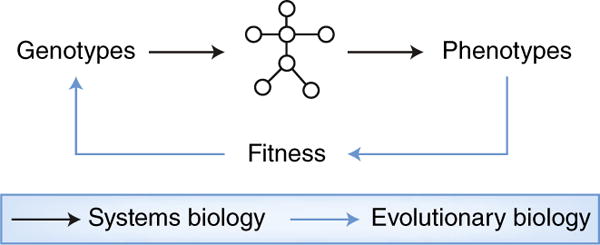
Systems biology examines how complex molecular networks mediate genotype–phenotype relationships within an organism. Phenotypes feed back on genotypes through their effect on fitness, which influences the probability that genotypes are propagated across generations. Evolutionary systems biology is concerned with the joint evolution of genotypes, networks and phenotypes. An evolutionary systems-biology approach would shed light on the cross-species translation problem by considering that molecular networks in two extant species derive from a common ancestor but have diverged due in part to exposure to different environments and selective pressures.
Employing an evolutionary systems-biology approach would probably benefit all areas of medicine, but among these, immunological connectivity presents a remarkable opportunity. Immunological networks are initiated when innate cells are activated by ‘danger signals’ that lead to the production of mediators that recruit and activate other leukocytes. Subsequently, antigen-specific adaptive responses emerge that entail scores of ligands, cytokines, metabolites, receptors, signaling pathways and integrated transcriptional responses. The vast number of cell types and molecular pathways involved in innate and adaptive immune responses present many similarities as well as important differences between mice and humans, as reviewed elsewhere3. We propose that the ideal approach to comparing animal models to immunity in humans will consider the evolutionary history of the genomes, epigenomes and molecular networks that mediate phenotypes in the animal’s respective biospheres — an evolutionary systems-biology approach.
The illusion of similarity
According to a survey of over 7,000 clinical trials performed in the past decade, 90% of treatment regimens fail to progress from phase I to approval4. We suggest that a number of these failures are due to an ‘illusion of similarity’ that masks the inability to predict to what extent animal models inform human medicine (Table 1). Consider, for example, inflammatory bowel diseases (IBDs), including Crohn’s disease (CD) and ulcerative colitis (UC), which arise due to exaggerated host responses to the microbiota in genetically susceptible hosts. In most mouse models of IBD, it appears as disease in the colon and resembles UC more than CD, which classically affects the ileum. Examination of the inflammation in models of IBD reveals that it has several similarities to the inflammation of human disease, including both innate immune responses and adaptive immune responses. For example, expression of the proinflammatory cytokine TNF is increased in diseased humans and most animal models of IBD, and the use of antibody to TNF is often an effective therapeutic strategy in both species. However, these similarities distract from key differences. For example, the fibrotic lesions that affect approximately one third of human patients affected with CD for over 10 years have been difficult to mimic in animal models.
Table 1.
The challenges created by the illusion of similarity
| The paradox | Mouse | Human |
|---|---|---|
| Fibrosis and IBD | Fibrotic lesions such as those seen in CD are rarely observed in mouse models of IBD | Fibrosis and strictures are a common occurrence in CD |
| IL-17 and IBD | IL-17 has been shown to be deleterious or protective | Antibody to IL-17 exacerbates inflammatory bowel disease |
| Phenotype of immune cells | T cells from mice exposed to conventional microbiota resemble T cells from neonatal humans | Mature T cell phenotype in humans observed only in mice colonized with complex microbiota |
| Clinical signs of infections | Mice fail to develop the clinical signs seen in humans after infection with H. pylori, C. difficile or influenza virus | H. pylori, C. difficile and influenza virus each produce a distinct disease in humans |
| LPS | Mice relatively resistant to LPS | Humans very sensitive to LPS |
In these representative examples of differences between animal models and human disease, some heterogeneity could reflect technical limitations of genetic deletions that might not mimic specific alleles, whereas others might reflect environmental differences that affect microbiota or fundamental differences with evolutionary roots. H. pylori, Helicobacter pylori; C. difficile, Clostridium difficile; LPS, lipopolysaccharide.
The molecular distinctions between UC and CD were initially based on cytokine profiles in animal models, which led to the notion that the TH1 subset of helper T cells is associated with CD and the TH2 subset is associated with UC. Subsequently, human genome-wide association studies demonstrated that the immunopathogenesis is more complex. Significant mutations in TH2 cell–related genes are not readily associated with disease in humans, while many other loci, including genes encoding components of the IL-17 pathway, are linked to both CD and UC5. The gene encoding IL-17A is conserved between human and rodents, and its expression is increased in the colon of humans with IBD and in many mouse models of colitis. Some studies have linked IL-17 to the pathogenesis of colitis in mice, but other reports suggest it is protective6, a paradoxical observation that perhaps was overlooked until treatment with antibody to IL-17 was shown to exacerbate IBD in humans6. Thus, an apparent similarity in the expression of a gene in animal models and human disease provided tragically limited insight into the complexities of biology and disease. These errors are perhaps a posteriori attributable to the use of a reductionist approach. Could such failures in drug development have been predicted with an evolutionary systems-biology approach?
Principles of molecular evolution
Evolution is the process through which populations and species change over successive generations. Natural selection is what makes individual organisms with certain phenotypes reproduce more successfully than others in their environments, shaping the frequency of these phenotypes in the population. Many intrinsic (for example, neuroendocrine) factors and extrinsic (for example, biosphere) factors impart selective pressures on the phenotypic expression of different genotypes. Consider how host–microbe interactions are species specific. Mice resemble humans in being omnivorous, but in the wild, they are exposed to diverse microbial populations and they are also coprophagic. In contrast, humans live in conditions that range from less hygienic to perhaps overly sanitary. The immune systems of mice and humans are thus evolving under very different selective pressures. However, the plasticity of immunological responses to varying biospheres and microbiota is thought to be a highly evolved trait in animals7. Perhaps as a result of this evolved plasticity, aspects of the immune system of mice in the laboratory more closely resemble those of neonatal humans, while co-housing laboratory mice with field mice ‘humanizes’ the transcriptional profile of memory T cell populations to mimic that of such populations in adult humans8, and humanizing mice with the microbiota of diseased humans can model human disease9.
A given genotype may confer both beneficial phenotypes and deleterious phenotypes, depending on the environment. For example, several alleles encoding molecules that provide protection against bacterial infection are associated with autoimmune diseases, including lupus and celiac disease10. Phenotypic expression of a given genotype also depends on the overall genome context, whether within species (predispositions) or between species. It is not uncommon that amino-acid substitutions associated with disease in humans correspond to the product of the wild-type allele of the orthologous gene in another species, as in autoimmune lymphoproliferative syndrome11.
Often the effect of a specific phenotype on fitness is too subtle for natural selection to take hold rapidly. The frequencies of the underlying alleles are then essentially independent of the conferred phenotype, and their evolution can be modeled via the laws of random sampling. This phenomenon is known as ‘genetic drift’. When such nearly neutral mutations are governed by genetic drift, their fate depends on the size of the inbreeding population, with weakly deleterious alleles expected to be purged more efficiently in larger populations than in smaller ones12. This leads to distributions of within-species genetic variability that are vastly different between mice and humans, with human protein-coding genes exhibiting, for example, a higher ratio of divergence at nonsynonymous sites relative to synonymous sites than their counterparts in mice13.
Variation in protein-sequences often results from genetic drift, but in rare yet significant occasions, this reflects positive selection for a beneficial change. The latter is thought to be at play for many immune-system-related genes14. Phagocytosis receptors, signaling proteins and proteins involved in microbial defense tend to have very rapidly evolving protein sequences14. Antimicrobial peptides such as defensins exhibit highly variable copy numbers3. The rapid evolution of the immune-system genome is akin to a molecular ‘arms race’ between pathogens and their hosts. How do these evolution-driven changes in genecopy-numbers and protein sequences affect cellular pathways and phenotypes differently in different species?
Modeling the evolution of molecular networks
A central tenet of systems biology is that genotype–phenotype relationships are mediated by dynamic ‘interactome’ networks that reflect interactions between gene products and other biomolecules, including those of environmental origin. In these network representations, gene products and biomolecules are depicted as ‘nodes’, while their pairwise relationships are depicted as ‘edges’ (lines) that represent either physical interactions or functional associations. Network edges are mediated by the underlying sequences and structures of linked nodes. The array of genetic similarities and differences between animal models and humans thus transposes to an evolutionary rewiring of their interactome networks (Fig. 2) that, we argue, can be mapped and modeled for an improved perspective on evolutionary medicine.
Fig. 2. Evolutionary network modeling.
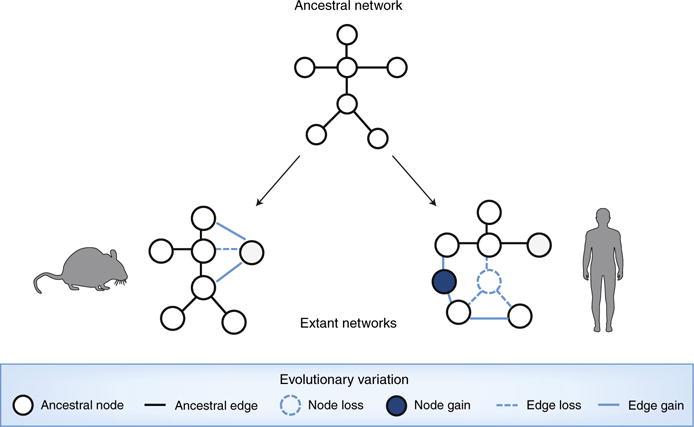
Through the gain or loss of nodes or edges, ancestral networks evolve to different topologies in distinct species.
Independently of evolutionary considerations, network-based modeling has provided powerful insights into the effect of genetic variation on medically relevant networks. For example, Mendelian mutations may result in node losses (nonsense mutations) or, alternatively, may trigger interaction-specific ‘edgetic’ (‘edge-specific genetic’) perturbations, including the removal or addition of specific edges while others remain unperturbed15 (Fig. 2). We encourage adapting these concepts and associated techniques to the study of how evolutionarily driven genetic variation yields node losses and gains, as well as edgetic perturbations. Through careful and unbiased mapping of medically relevant networks in mice and humans, ancestral networks could be reconstructed for a process of interest to identify relevant gains and losses of nodes and edges. The edges of orthologous nodes could be compared, for example, to determine if they have retained an ancestral function or not. This framework could also be quantitative rather than binary and could consider edgetic changes in edge strength or condition-dependent dynamics. When edgetic rewiring is found, structural modeling could be coupled with evolutionary modeling to determine whether the sequence changes responsible for the edge perturbations have occurred through drift or in response to selective pressures. Deploying edgetic mapping and modeling undoubtedly represents a substantial effort relative to more traditional practices, but the reward of providing context and molecular mechanisms associated with a disease process across species would expose new information that has not been examined thus far.
Network-based approaches promise to reveal surprising evolutionary scenarios. For example, orthologs with very similar sequences might have vastly different interaction partners, since network edges are mediated by the underlying sequences and structures of both linked nodes. This is the case for many regulatory interactions, in which transcription factors are highly conserved but the DNA motifs to which they bind are highly variable in sequence and location within the large intergenic regions of mammalian genomes16. Interestingly, despite vast differences in protein–DNA binding events, the actual expression levels of genes can be well correlated between mouse and human17. This suggests that among a large amount of network rewiring, compensatory changes might lead to functional conservation. Cellular networks are, by nature, modular: groups of nodes are more closely connected with each other than with other nodes. Such network modules can correspond to protein complexes or pathways or to genes co-regulated by the same transcription factors, for example. We are tempted to speculate that evolutionarily driven genetic variations with edgetic consequences affect the composition and wiring of cellular systems and might contribute to species-specific gains and losses of entire modules. Future research into evolutionary edgetics will undoubtedly elucidate the mechanistic similarities and differences between immune-system-mediated disease modules across species.
Evolution complicates comparative studies
Despite their great theoretical promise, evolutionary systems-biology approaches are currently hampered by experimental and analytical challenges. It is not uncommon for scientists to arrive at drastically different conclusions when investigating the simple question of whether ‘-omics’ profiles or networks are conserved or divergent across species. For example, studies of transcript levels and splice-variant structures in human and mouse immune-system-related cell types and conditions have reported both great levels of similarity and vast differences18–20. Even when the underlying datasets are shared, conflicting conclusions can be drawn due to the use of different analytical methods18,20. Indeed, when it comes to comparative ‘-omics’ analyses, the devil is in the details.
The difficulty stems from the fact that technical, biological and evolutionary sources of variation are at play simultaneously (Fig. 3). First, technical sensitivity and specificity complicate quantitative comparisons in virtually all ‘-omics’ assays. For example, samples sequenced with different read depths will seem to be distinct even if they are in fact identical. Statistical analyses require that many choices be made (for example, P-value cutoffs, etc.), often somewhat arbitrarily, that typically cannot be generalized across all genes and conditions considered. As statistical analyses are intrinsically dependent on technical sensitivity and specificity, they can rarely be compared across datasets that were not generated in parallel, which complicates comparative meta-analyses. These issues plague all comparative studies, whether they involve different species or not. A current goal of bioinformatics research is to develop refined methodologies that tease apart the technical and biological differences between ‘-omics’ datasets.
Fig. 3. Experimental and analytical factors affect the appearance of observed networks.
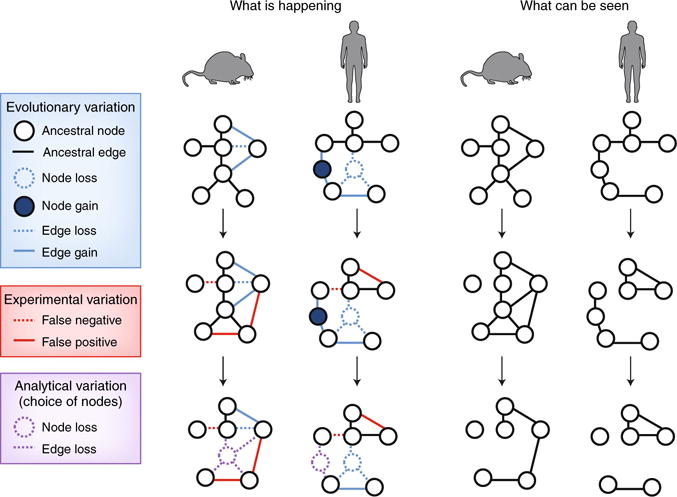
As described in Fig. 2, networks can entail distinct nodes and edges in comparisons of species. In addition, several challenges affect the evaluation of these networks, with misleading results. For example, variation in experimental measurements can lead to false-positive or false-negative edges (middle), or analyses might be restricted to shared ancestral nodes (bottom). The challenge is to estimate properties of the real networks based only on the observed networks.
In the case of cross-species comparisons, an additional complication is that the objects whose expression or interaction patterns are being compared are simply not the same, for reasons related to evolution. The human genome and mouse genome each contain ~20,000 protein-coding genes, among which ~14,000 can be mapped between the two species as ‘one-to-one orthologs’ with, on average, ~80% sequence identity21; that is, phylogenetic reconstructions predict that ~14,000 genes in the human and mouse genomes share a direct common ancestor. However, the sequence similarities and predicted shared ancestries of these genes mask important differences, including, for example, the fact that > 20% of essential human genes have non-essential mouse orthologs22. About 2,000 human and mouse genes resulted from gene duplications that occurred in the past 90 million years, which has rendered the copy numbers of ancestral genes different between the species. This probably has functional consequences, since retained changes in gene copy number are often associated with rapid functional changes23. Despite the fact that most human and mouse genes share common ancestry, their sequence and copy numbers have changed such that they are no longer always functionally equivalent.
In addition, 4,000–5,000 human and mouse protein-coding genes lack evidence of shared ancestry. Whether they diverged so far in sequence space that they have become unrecognizable between the two species or whether they emerged de novo since the split between rodents and primates24, 20% of human and mouse genes are simply not shared. Their expression, interaction patterns and activities might underlie species-specific phenotypes or, alternatively, might mediate ancestral functions in novel ways. In either case, they must participate in molecular networks that are species specific, and including them in comparative analyses presents a fundamental challenge.
In order to pair biological responses across species and compare them, choices must be made, such as whether to consider only one-to-one orthologs to the exclusion of the rest of the genome, or whether to impose a given sequence-similarity threshold to designate a pair of equivalent genes. Such choices weaken comparative analyses by masking the aspects of the systems-level context that are most probably harboring critical differences among species. Interpretations are often dependent on these technical choices. For example, quantitative comparisons of transcription-factor occupancy across species can yield drastically different results depending on the sequence-similarity threshold chosen to determine which portions of the genomes are considered in the analysis25. Thus, comparative studies require broader quantitative consideration than ‘conservation versus divergence’ and must encompass careful examination of the various sources of technical, biological and evolutionary variation at play.
Improving translation of biology between species
The aim of this Comment was to convey the opportunities presented by evolutionary systems biology in the context of comparative immunology. These opportunities will not only improve the power of ‘translating’ between humans and models but also lead to answers to many long-standing and fascinating fundamental questions. How can it be determined if two orthologous genes have conserved functions? How do new genes acquire new functions? Which genetic changes trigger the rewiring of molecular interactions? How do novel cellular machinery and molecular pathways evolve? These and other questions can be addressed in the context of animal models of disease by focusing on four areas of research development. The first is coherent data generation, with systematic mapping of molecular networks and ‘-omics’ surveys for animals and human, in both healthy and diseased tissues, with matching protocols in parallel. The second area is refinement of comparative ‘-omics’ practices, with the development of analytical frameworks for meaningful assessment of cross-species differences, beyond ‘conservation versus divergence’. The third is evolutionary reconstruction, with models for the natural spread of molecular traits relevant to disease predisposition, together with the strength of drift and/or selective pressure acting on sequences and networks of interest. And fourth, critical environmental factors (such as dietary, microbial, emotional and physiological stressors, environmental context relevant to the human life stage of interest) should be included in the analysis.
Those four research axes have the potential to identify the mechanistic underpinnings of cross-species differences through the use of evolutionary network modeling. In some cases, molecular distinctions might hide functional similarities that would allow animal models to be immediately useful. It is likely that some pathways in humans are not represented in mice and therefore understanding of their function or the identification of drug targets will be limited. This is why it will be important to incorporate an evolutionary approach into future experimental designs that consider the plasticity and complexity of molecular networks. Such an approach can delineate biological function within species, and, relevant to the challenge of translation from mice to men, help make sense, both retrospectively and prospectively, of the disparities between them. ❐
Acknowledgments
We thank I. Pessah, M. Nicotra, T. Benos and E. Gomez for suggestions, and the reviewers for the comments (whose wording was, in some cases, included). Supported by the US National Institutes of Health (GM 108865 for A.R.C.; and AI AI079145 for P.B.E.), The Wayne and Gladys Valley Foundation, Takeda California and the Chiba University-UC San Diego Program in Mucosal Immunology, Allergy and Vaccines. P.B.E. holds a joint appointment in the Department of Immunology, Chiba University, Chiba, Japan.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stroud C, et al. Clin Transl Med. 2016;5:26. doi: 10.1186/s40169-016-0107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolker JA. BioEssays. 2017 doi: 10.1002/bies.201700089. [DOI]
- 3.Mestas J, Hughes CC. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 4.Thomas DW, Burns J, Audette J, Carroll A, Dow-Hygelund C, Hay M. Clinical Development Success Rates 2006–2015. 2015 https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf.
- 5.Abraham C, Cho J. Inflamm Bowel Dis. 2009;15:1090–1100. doi: 10.1002/ibd.20894. [DOI] [PubMed] [Google Scholar]
- 6.Targan SR, et al. Am J Gastroenterol. 2016;111:1599–1607. doi: 10.1038/ajg.2016.298. [DOI] [PubMed] [Google Scholar]
- 7.Canas CA, Canas F. Autoimmune Dis. 2012;2012:784315. doi: 10.1155/2012/784315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beura LK, et al. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridaura VK, et al. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos PS, Shedlock AM, Langefeld CD. J Hum Genet. 2015;60:657–664. doi: 10.1038/jhg.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, et al. Genome Biol. 2004;5:R47. doi: 10.1186/gb-2004-5-7-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohta T. Annu Rev Ecol Syst. 1992;23:263. [Google Scholar]
- 13.Kosiol C, et al. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh RS, Jianping X, Kulathinal RJ, editors. Rapidly Evolving Genes and Genetic Systems. Oxford University Press; Oxford, UK: 2012. [Google Scholar]
- 15.Zhong Q, et al. Mol Syst Biol. 2009;5:321. doi: 10.1038/msb.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odom DT, et al. Nat Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breschi A, Gingeras TR, Guigo R. Nat Rev Genet. 2017;18:425–440. doi: 10.1038/nrg.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seok J, et al. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shay T, et al. Proc Natl Acad Sci USA. 2013;110:2946–2951. doi: 10.1073/pnas.1222738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takao K, Miyakawa T. Proc Natl Acad Sci USA. 2014;112:1167–1172. doi: 10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrero J, et al. Database. 2016 doi: 10.1093/database/bav096. [DOI]
- 22.Gharib WH, Robinson-Rechavi M. Brief Bioinform. 2011;12:436–441. doi: 10.1093/bib/bbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conant GC, Wolfe KH. Nat Rev Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 24.Chen S, Krinsky BH, Long M. Nat Rev Genet. 2013;14:645–660. doi: 10.1038/nrg3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvunis AR, et al. eLife. 2015;4 doi: 10.7554/eLife.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]