

Abstract
Pol β lyase removal of 5′-dRP from BER intermediates is critical in mammalian BER involving the abasic site. We found that pol β also removes 5′-adenylated dRP from BER intermediates after abortive ligation. The crystal structure of a pol β-DNA complex revealed that the 5′-AMP-dRP group is positioned in the lyase active site. Expression of pol β rescued MMS sensitivity in APTX (hnt3) and FEN1 (rad27) deficient yeast.
In order to address base excision repair (BER) enzyme transactions in the case of abortive ligation on the 5′-deoxyribose phosphate (5′-dRP) containing intermediate, we investigated the structural and kinetic features of DNA polymerase β (pol β) in complementing aprataxin (APTX) activity during abasic site BER. Pol β is a bifunctional enzyme with lyase and nucleotidyl transferase activities1. In single-nucleotide BER involving the natural unmodified abasic site or 5′-dRP group, excision of the dRP group is catalyzed by the pol β lyase activity2. If the 5′-dRP is not removed prior to the ligation step, the DNA ligase reaction is hindered. This may result in abortive ligation with 5′-adenylation of the BER intermediate (i.e., formation of the 5′-AMP-dRP group at a nicked or gapped 5′-terminus). Lysine alterations (i.e., K35A K68A K72A; termed 3KΔ) in the lyase active site eliminate the pol β dRP lyse activity3. A previous crystal structure of a pol β-DNA complex indicates that the 5′-sugar phosphate group is bound in the dRP lyase active site4. During BER, if a 5′-sugar phosphate group is refractory to the pol β lyase activity, the modified sugar phosphate is subjected to repair by FEN1 in long patch (LP) BER sub-pathway5. Saccharomyces cerevisiae (S. cerevisiae), as a model organism, has a similar upstream BER pathway for repair of methylated bases as that in mammalian cells, along with Hnt3 and Rad27 proteins that are APTX and FEN1 homologues, respectively6. We set out to confirm that abortive ligation by both human DNA ligase I and DNA ligase III/XRCC1 complex yielded this 5′-adenylated BER intermediate. Yet, these enzymes had somewhat different ligation efficiencies depending on Mg2+ and ATP concentrations (Supplementary Fig. 1). In this study, we showed that the pol β dRP lyase activity can remove the 5′-AMP-dRP group from substrates that mimic BER intermediates after abortive ligation. The results revealed that pol β removed the 5′-adenylated-dRP from both the gapped (Fig. 1a) and nicked (Fig. 1b) DNA substrates (Supplementary Table 1). Yet, the reaction in both cases was weaker than that for removal of the unmodified 5′-dRP group, and the migration positions of the products after 5′-dRP and 5′-AMP-dRP removal were similar (Fig. 1a,b). We showed that the 3KΔ mutant failed to remove the 5′-adenylated-dRP group from both the gapped and nicked DNA substrates (Supplementary Fig. 2b,c). These results confirmed that the dRP lyase activity of pol β was responsible for the 5′-AMP-dRP removal. In light of the results described here illustrating 5′-AMP-dRP removal by pol β, we obtained a crystal structure of pol β in complex with nicked DNA that represents the 5′-adenylated-dRP-containing BER intermediate (Supplementary Table 2). We used the 5′-adenylated sugar analog tetrahydrofuran (THF). The structure revealed that the 5′-AMP-THF group in the lyase active site (Fig. 2a). Comparison of the positions of the sugar ring and phosphate with the corresponding groups in the previously determined structure4 with a 5′-dRP group revealed that THF groups were in a similar position, and the structures were very similar (RMSD = 0.7 Å). The location of the 5′-AMP-dRP group in the active site was consistent with the observation of lyase removal of the group (Fig. 1a,b). The data also revealed that the THF and phosphate groups (P) were well resolved in the crystal structure, but the base and sugar of the adenosine (rA) may have multiple conformations (Fig. 2b). The rate of removal for the 5′-dRP group was higher than that for removal of the 5′-AMP-dRP group, as will be described below. Based on this new structure, one explanation for the lower activity could be the additional contact made by the 5′-AMP-dRP group with Lys68 (Fig. 2b). The ε-amino group of Lys68 makes contact with the 5′-phosphate of the dRP group, whereas this ε-amino group makes this contact plus an additional (2.6 Å) contact with the phosphate of the AMP moiety of the 5′-AMP-dRP group in the new structure. It is possible that this additional contact could immobilize the AMP-dRP group, and thus prevent or impede a substrate conformational change required for its removal. Therefore, to test this idea, we examined the lyase activity of the K68A mutant. As compared to 5′-AMP-dRP removal by wild type pol β, the alanine substitution for Lys68 had no significant effect on the removal of 5′-AMP-dRP (Supplementary Fig. 2d,e). Thus, these results suggested that the lower activity may be related to an effect of the bulky AMP moiety on the conformational adjustment required for chemistry. In order to further understand the fate of abortive ligation products involving the abasic site BER intermediates, we investigated 5′-AMP removal by APTX from different substrates (Supplementary Table 1). APTX removed the 5′-AMP moiety, while pol β removed the entire 5′-AMP-dRP group from both the gapped and nicked 5′-adenylated-dRP-containing substrates (Supplementary Fig. 3a). APTX removed the 5′-adenylate from the abortive ligation product containing 5′-AMP only, while pol β was not able to remove this group, as expected (Supplementary Fig. 3b). In contrast, APTX removed the 5′-adenylate from both the gapped and nicked 5′-adenylated-THF-containing substrates, while pol β had no activity on these substrates (Supplementary Fig. 3c). The differences in results with the substrates containing the abasic site analogue, THF, and the natural dRP group were consistent with the chemistry of the pol β dRP lyase activity7: the C1’ carbon in the THF analogue does not have aldehydic character required for the formation of the Schiff base intermediate in the lyase reaction. THF, therefore does not support lyase chemistry, but is considered a structural analogue4. This is probably the explanation for the differences in our results and previously reported findings regarding the pol β dRP lyase activity8. We also investigated whether FEN1 could remove the 5′-adenylated-dRP group in a BER intermediate, and if so, whether LP BER could be an alternative BER sub-pathway for removal of the blocking 5′-adenylate group. For this purpose, we measured excision activity of FEN1 using different DNA substrates (Supplementary Table 1). The results revealed that FEN1 removed the 5′-AMP-dRP group plus mainly one nucleotide from the gapped 5′-adenylated-dRP-containing substrate; a small amount of two nucleotide excision product was also produced (Supplementary Fig. 4b). With the nicked 5′-AMP-dRP-containing substrate, the 5′-AMP-dRP group and predominantly two nucleotides were removed; small amounts of three nucleotide excision products also were produced (Supplementary Fig. 4a). In reference reactions, pol β removed the 5′-adenylated-dRP group only (Supplementary Fig. 4a,b). FEN1 was effective on the DNA substrate containing 5′-AMP, and removed 5′-AMP plus predominantly a single nucleotide, while pol β was not active on this substrate (Supplementary Fig. 4c). In order to further understand kinetic features of the enzymes involved in processing the 5′-adenylated BER intermediates, quantitative measurements were obtained for the removal of 5′-AMP-dRP by pol β, flap excision of 5′-AMP-dRP by FEN1, and 5′-AMP removal by APTX using the gapped 5′-adenylated-dRP-containing substrate (Supplementary Table 1). When the kinetic features of pol β and FEN1 were compared, these enzymes exhibited similar rates for processing the blocked 5′-adenylates, i.e., 0.05 s−1 vs 0.03 s−1, respectively. The rate of APTX 5′-AMP hydrolysis from adenylated BER intermediates was slightly faster than the rate of pol β 5′-AMP-dRP lyase removal, i.e., 0.2 s−1 vs 0.05 s−1, respectively (Supplementary Fig. 4d). These measurements supported a model illustrating the fate of the 5′-adenylated BER intermediate as a function of the enzymatic activities of pol β, FEN1 and APTX (Supplementary Fig. 4e). We also were interested in examining the fate of the blocked 5′-adenylates in the yeast S. cerevisiae (Supplementary Table 3). We first investigated FEN1, pol β, and APTX activities in yeast cell extracts. In control reference reactions, we obtained the expected reaction products by purified enzymes (Supplementary Fig. 5a). With cell extract from the wild type yeast strain, we observed excision by FEN1 demonstrating removal of the 5′-adenylated-dRP group plus mainly one nucleotide; in addition to 5′-AMP removal by APTX (Supplementary Fig. 5b). With cell extract from the hnt3Δ strain that contained the single hnt3 gene deletion, we observed similar FEN1 product as for wild type cells along with a small amount of a two nucleotide excision product Supplementary Fig. 5c). For the rad27Δ strain, with the single rad27 gene deletion, we only observed 5′-AMP removal by APTX (Supplementary Fig. 5d). We did not observe these FEN1 and APTX reaction products when the double mutant extract (rad27Δhnt3Δ) was used (Supplementary Fig. 5e). For cell extracts prepared from yeast strains expressing pol β (Supplementary Fig. 5f,g), we observed both 5′-AMP-dRP removal by pol β and 5′-AMP removal by APTX. The result was similar with cell extract was from the strain with the single rad27 gene deletion (rad27Δ::POLB) (Supplementary Fig. 5f). In contrast, we observed only 5′-adenylated-dRP removal by pol β in the cell extract from the rad27Δhnt3Δ::POLB strain that contained the rad27 and hnt3 gene deletion background (Supplementary Fig. 5g). These results demonstrated that FEN1 and pol β could complement the lack of APTX activity for the processing of 5′-adenylated BER intermediate in cell extracts. To examine whether deficiency in APTX in living cells could be complemented by pol β and FEN1, we tested the sensitivity of S. cerevisiae strains to methyl methanesulfonate (MMS) using the spot dilution assay. We failed to observe any hypersensitivity in the hnt3Δ single mutant, as revealed by wild type levels of growth on YPDA plates with or without MMS (Fig. 3a). The lack of hypersensitivity in hnt3Δ yeast cells6 and APTX deficient human cells9 are reported previously. In contrast, we observed strong MMS hypersensitivity in the strain defective in FEN1 (hnt3Δrad27Δ) compared to the hnt3Δ cells. Finally, the results showed that pol β could rescue MMS sensitivity of hnt3Δrad27Δ-null cells on YPDA plates containing 0.5, 0.8 or 1.0 mM MMS (Fig. 3a). To confirm the enzymatic function of pol β responsible for this rescue effect, we constructed the lyase deficient strain (hnt3Δrad27Δ::POLB-3KΔ). While all strains grew on control plates, the pol β dRP lyase deficient strain failed to grow on YPDA containing MMS (Fig. 3b). This result demonstrated that lyase activity was required for the pol β-dependent resistance to MMS in the absence of hnt3 and rad27. Since we used MMS as genotoxic agent that is known to lead to abasic sites, it is likely that Rad27(FEN1) protective effect was via removal of 5′-dRP-containing BER intermediates. The effect of Hnt3(APTX) deletion was mild, but detectable in the system, consistent with published results6 and with the idea that abortive ligation was at play at least to some extent. Therefore, our results were consistent with a straightforward interpretation that Rad27(FEN1) was robust in removing 5′-dRP-containing repair intermediates including the abortive ligation products, and in the absence of Rad27(FEN1), pol β was capable of complementation. Overall, our in vivo and in vitro findings supported a model in which the fate of the 5′-adenylated BER intermediate was determined by three enzymatic processes (Supplementary Fig. 4e). In this model, APTX hydrolyzes the 5′-AMP group and allows for renewed attempts at processing of the repair intermediate, and the 5′-dRP may be channeled into the normal BER pathway for pol β dRP removal and gap-filling. In LP BER, the blocking BER intermediates can be processed via flap excision by FEN1. In an alternative mechanism, pol β could play a role by direct removal of the entire 5′-AMP-dRP blocking group via its dRP lyase activity. The later two cases could impact the fate of the blocking 5′-adenylated BER intermediates in a cell that is deficient in APTX, e.g., as in the neurodegenerative disorder ataxia oculomotor apraxia type 1 (AOA1). It is interesting that studies are consistent with the presence of APTX deficiency back up systems, such that the APTX deficiency alone fails to confer hypersensitivity to BER inducing agents10–12.
Figure 1.
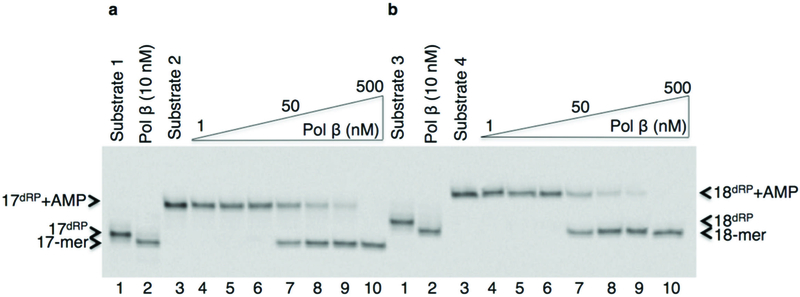
The adenylated sugar phosphate removal by pol β. (a,b) The pol β dRP lyase assay for the gapped (a) and nicked (b) 5′-dRP and 5′-AMP-dRP-containing substrates. Lanes 1 and 3 are minus enzyme controls; lane 2 and lanes 4–10 are the lyase products after 5′-dRP removal from Substrate 1 and 3, and 5′-AMP-dRP removal from Substrate 2 and 4, respectively. The positions of DNA substrates (17dRP and 17dRP+AMP in panel a; 18dRP and 18dRP+AMP in panel b) and pol β lyase products (17-mer in panel a; 18-mer in panel b) are indicated. The 5′-dRP refers to 5′-deoxyribose phosphate group. Original image of the gel can be found in Supplementary Fig. 2a.
Figure 2.
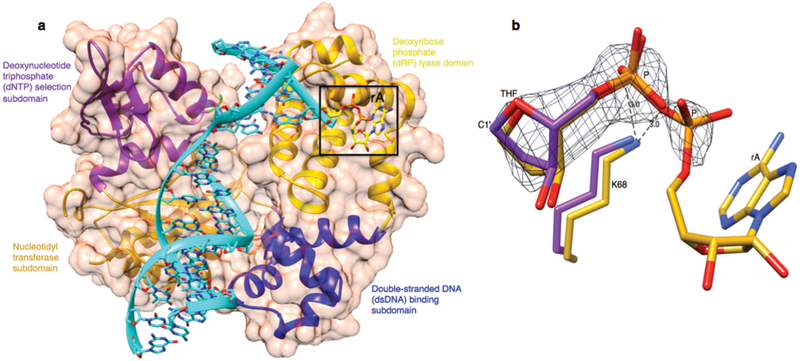
The position of 5′-AMP-dRP group in the pol β lyase active site. (a) The X-ray crystal structure of the pol β binary complex with the 5′-adenylated-THF-containing DNA. Different domains and subdomains of pol β are shown through the surface (light purple) at 50% transparency: amino-terminal dRP lyase domain (yellow), dsDNA binding subdomain (blue), nucleotidyl transferase subdomain (gold), and dNTP selection subdomain (purple). The DNA phosphodiester backbone is shown in cyan. The boxed region represents the pol β dRP lyase active site and also shows a stick model of the 5′-AMP-THF group (yellow). (b) An overlay structure of the adenylated THF (gold) with the non-adenylated THF (purple) containing DNA (PDB ID: 2P66)4. The position of the C1’ atom in THF in the pol β lyase active site is shown. A lysine residue (K68) involved in stabilizing the substrate along with its interactions to substrate phosphate oxygens is shown. A simulated-annealing fo-fc omit map (black) is shown, contoured at 3σ.
Figure 3.
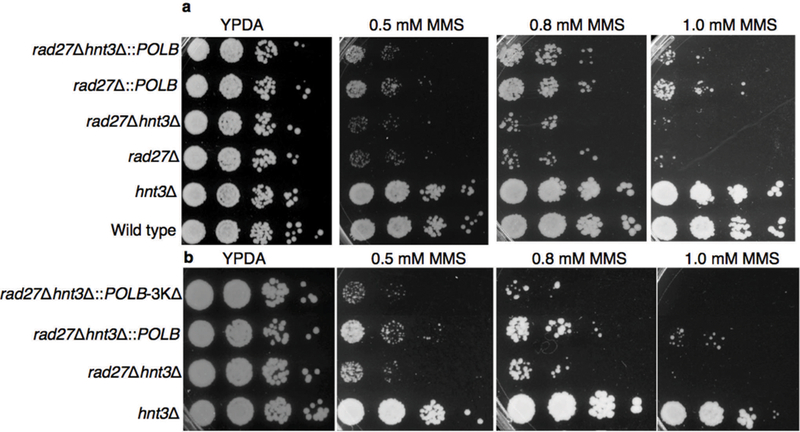
Complementation of Hnt3(APTX) deficiency by pol β in the absence of Rad27(FEN1). (a,b) Spot dilution assay for MMS sensitivity of the yeast strains that contained hnt3Δ (a) and pol β lyase deficient 3KΔ (b) deletions. MMS refers to methyl methanesulfonate.
Accession code.
Coordinates and structure factors for human DNA polymerase β complexed with adenylated tetrahydrofuran (abasic site) containing DNA have been deposited in the Protein Data Bank under accession code 4O9M.
Online Methods
Materials.
Synthetic oligodeoxyribonucleotides with 3′−6-carboxyfluorescein (FAM)-labeled were from Midland Certified Reagent Co. (Midland, TX). Recombinant human wild type pol β, and the pol β mutants 3KΔ and K68A were overexpressed and purified as described3,4. Human FEN1 and UDG with 84 amino acids deleted from the amino-terminus were purified as described7,13. Human DNA ligase I was purified as described14. Recombinant human APTX was from Fitzgerald (North Acton, MA). The 5′-DNA adenylation kit and MMS were from New England BioLabs (Ipswich, MA) and Sigma-Alrich (St. Louis, MO), respectively.
Preparation of DNA substrates.
The nicked and gapped DNA substrates were prepared with and without AMP at the 5′-end of 3′-FAM-labeled oligonucleotides. In order to generate DNA substrates containing 5′-AMP-dRP, 17- or 18-mer oligonucleotides with a normal uracil base or a THF, were adenylated using the 5′-DNA adenylation kit. Similarly, the 18-mer oligonucleotide containing 5′-phosphate was adenylated to generate a DNA substrate with 5′-AMP. The adenylation reaction was performed in a reaction mixture (20 μl) containing 100 pmol oligonucleotide, 50 mM sodium acetate, pH 6.0, 10 mM MgCl2, 5 mM DTT, 0.1 mM EDTA, and 0.1 mM ATP. The reaction was initiated by adding 100 pmol Mth RNA ligase, and the incubation was at 65 °C for 1 h. After heat inactivation of the enzyme at 85 °C for 5 min, the adenylated oligonucleotides were separated from other DNA species by electrophoresis in a 18% polyacrylamide gel containing 8 M urea in 89 mM Tris-HCl, 89 mM boric acid, and 2 mM EDTA, pH 8.8, as described15. All of the DNA substrates used in this study and the sequence information of the oligonucleotides are represented in Supplementary Tables 1 and 4, respectively.
UDG treatment of DNA substrates.
The uracil-containing double-stranded DNA substrates (Substrates 1–4, Supplementary Table 1) were pretreated with UDG as follows: DNA substrate (200 nM) was incubated with UDG (20 nM) at 37 °C for 30 min in a reaction mixture (50 μl) containing 50 mM Hepes, pH 7.4, 20 mM KCl, 0.5 mM EDTA, and 2 mM DTT to generate an AP site. Due to the labile nature of UDG-treated DNA, the DNA substrates were treated just prior to conducting the subsequent incubations.
Ligation incubation.
The ligation reaction incubation was performed using UDG-pretreated double-stranded DNA (Substrate 1, Supplementary Table 1) that had not been adenylated before. The reaction mixture (10 μl) contained DNA substrate (140 nM) and DNA ligase I or DNA ligase III-XRCC1 complex (500 nM), 50 mM NaMOPS, pH 7.5, 1 mM DTT, 0.05 mg/ml BSA, 100 mM NaCl. The ligase reaction mixture also contained different ATP and Mg2+ concentrations as indicated in Supplementary Fig. 1. The incubation was at 37 °C for 1 min. The reaction products were then stabilized by addition of freshly prepared 1 M NaBH4 to a final concentration of 100 mM and incubation was continued for 30 min on ice. The reaction products were then mixed with 10 μl of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromphenol blue, and 0.02% xylene cyanol). After incubation at 95 °C for 3 min, the reaction products were separated by electrophoresis in a 15% polyacrylamide gel containing 8 M urea in 89 mM Tris-HCl, 89 mM boric acid, and 2 mM EDTA, pH 8.8. The gels were scanned on a Typhoon PhosphorImager, and the data were analyzed using ImageQuant software.
The dRP lyase activity assay.
The dRP lyase assay was performed as described previously7 using DNA substrates 1–4 (Supplementary Table 1). Briefly, dRP lyase activity was determined in a reaction mixture (10 μl) that contained 50 mM Hepes, pH 7.5, 20 mM KCl, 0.5 mM EDTA, 2 mM DTT, and 140 nM DNA substrate. The reaction was initiated by addition of pol β (1, 10, 20, 50, 100, 200, or 500 nM) for DNA substrates 2 and 4 containing 5′-AMP-dRP. For DNA substrates 1 and 3 containing 5′-dRP, 10 nM pol β was used. The incubation was at 37 °C for 15 min. The reaction products were stabilized by addition of 100 mM NaBH4, incubated on ice for 30 min, and then separated in a 15% polyacrylamide gel as described above. The dRP lyase assay was performed under similar reaction conditions with the wild-type and pol β mutants (3KΔ and K68A).
DNA deadenylation assay.
The 5′-AMP removal from DNA substrates (Supplementary Table 1) with 5′-AMP-dRP (Substrates 2 and 4), 5′-AMP (Substrate 5), or 5′-AMP-THF (Substrates 6 and 7) by APTX was determined in the reaction mixture (10 μl) that contained 50 mM Hepes, pH 7.5, 20 mM KCl, 0.5 mM EDTA, 2 mM DTT, and 140 nM DNA substrate. For DNA substrates 2, 4, 6 and 7, the reaction was initiated by addition of 100 nM APTX, and the reaction mixture was incubated at 37 °C for 15 min. For the removal of 5′-AMP from APTX substrate (Substrate 5), 10 nM APTX was used. The reaction products were separated in a 15% polyacrylamide gel, the gel was scanned, and the data were analyzed as above.
FEN1 excision activity assay.
FEN1 activity assays were performed using DNA substrates (Supplementary Table 1) containing 5′-AMP-dRP (Substrates 2 and 4) or 5′-AMP (Substrate 5). FEN1 excision activity was determined in the reaction mixture (10 μl) that contained 50 mM HEPES, pH 7.5, 50 mM KCl, 10 mM MgCl2, 0.5 mM EDTA, and 140 nM DNA substrate. The reaction was initiated by addition of FEN1 (1, 20, 50, 100, 200, or 500 nM). In some case, pol β (500 nM) was used instead of FEN1 as a reference. The incubation was at 37 °C for 15 min. The reaction products were separated in a 15% polyacrylamide gel, the gel was scanned, and the data were analyzed as above.
Kinetic measurements.
In order to determine the catalytic rate of pol β, FEN1 and APTX for the processing of the 5′-adenylated BER intermediates, the kinetic analyses for the dRP lyase, FEN1 excision and DNA deadenylation reactions were performed manually under single-turnover conditions using the gapped DNA substrate with 5′-AMP-dRP (Substrate 2, Supplementary Table 1). The reactions were initiated by addition of pol β, FEN1 or APTX (500 nM) to the reaction mixture containing UDG-pretreated DNA substrate (140 nM), 50 mM Hepes, pH 7.5, 20 mM KCl, 0.5 mM EDTA, and 2 mM DTT. The incubation was at 37 °C. Aliquots (9 μl) were withdrawn at the indicated time intervals, transferred into the tubes that contained 1 μl of freshly prepared 100 mM NaBH4, and the mixtures were incubated for 30 min on ice. An equal volume of gel-loading buffer was then added, and the mixture was incubated at 95 °C for 3 min. The reaction products were then separated, gels were scanned and the data were analyzed as above. The data were fitted to a single exponential equation to determine the first-order rate constant (kobs) as described16. All of the kinetic measurements were repeated in triplicate, and the values represent the mean values of three experimental values.
Yeast strain construction.
S. cerevisiae strains (Supplementary Table 3) were isogenic derivatives of strains ALE1000 and ALE1001 (MATα-HML/Rδ leu2–3112 ade5–1 his 7–2 ura3δ trip1–289 BAR+ [(chr II) lys2::Alu-DIR-LEU2-lys2δ5′]). We constructed Hnt3, (APTX homolog in S. cerevisiae), deletion background in the yeast strains VP101 (rad27::CORE), VP102 (rad27::polβ), and MH102 (rad27::POLB-3KΔ) that were previously reported17. The hnt3::kanMX strains (hnt3Δ) were generated by deletion-replacement of Hnt3 via transformation with the PCR product containing the kanMX cassette flanked by 58–60 nucleotides of homology upstream and downstream of the hnt3 ORF. The PCR product was amplified from pFA6a-kanMX using the Forward and Reverse primers (Supplementary Table 4) The replacement of Hnt3 with kanMX in the transformant was verified by PCR analysis and sequencing.
Preparation of yeast cell extracts.
The cell extracts from the yeast strains were prepared as described18.
Enzymatic activity assays in yeast cell extracts.
The dRP lyase, FEN1 excision, and DNA deadenylation activities were investigated in yeast cell extracts as described above. The gapped DNA substrate that contained 5′-AMP-dRP (Substrate 2, Supplementary Table 1) was used without pretreatment with UDG. The reactions were initiated by the addition of 50 μg yeast extract, and incubated at 30 °C for 5, 15, or 30 min.
MMS sensitivity spot dilution assay.
In order to detect DNA damage sensitivity in S. cerevisiae strains, the spot dilution assay was performed as described17. Ten-fold serial dilutions were spotted onto YPDA agar plates containing either 0, 0.5, 0.8 or 1.0 mM MMS. Plates were incubated at 30 °C and photographed 2 or 3 days after plating. All spot dilution assays were repeated in triplicate and performed at least twice on separate days.
Crystallization of pol β-DNA substrate complex.
Crystallography, data collection and structure determination for the binary complex crystals of human pol β with the 5′-adenylated THF containing nucleotide in a nicked DNA were performed as described4,19,20. The sequence information for the 16-mer template, 11-mer primer strand, and 5-mer downstream oligonucleotide containing 5′-AMP-THF is given in Supplementary Table 4.
Supplementary Material
ACKNOWLEDGMENTS
We thank J.K. Horton for critical reading of the manuscript and M. Heacock and D. Shock for helpful discussions. We thank A. Tomkinson (University of New Mexico Cancer Center) for the XRCC1/DNA ligase III complex. This work was supported by the Intramural Research Program of US National Institutes of Health, National Institute of Environmental Health Sciences (Grants Z01 ES050158 and ES050159 to S.H.W.).
Contributor Information
Melike Çağlayan, Laboratory of Structural Biology, National Institute of Environmental Health Sciences, US National Institutes of Health, Research Triangle Park, North Carolina, USA.
Vinod K. Batra, Laboratory of Structural Biology, National Institute of Environmental Health Sciences, US National Institutes of Health, Research Triangle Park, North Carolina, USA
Akira Sassa, Laboratory of Structural Biology, National Institute of Environmental Health Sciences, US National Institutes of Health, Research Triangle Park, North Carolina, USA.
Rajendra Prasad, Laboratory of Structural Biology, National Institute of Environmental Health Sciences, US National Institutes of Health, Research Triangle Park, North Carolina, USA.
Samuel H. Wilson, Laboratory of Structural Biology, National Institute of Environmental Health Sciences, US National Institutes of Health, Research Triangle Park, North Carolina, USA
References
- 1.Kumar A, Abbotts J, Karawya EM & Wilson SH Identification and properties of the catalytic domain of mammalian DNA polymerase β. Biochemistry 29, 7156–7159 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Piersen CE, Prasad R, Wilson SH & Lloyd RS Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. J. Biol. Chem 271, 17811–17815 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Prasad R et al. Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase β as revealed by site-directed mutagenesis. DNA binding and 5′-deoxyribose phosphate lyase activities. J. Biol. Chem 273, 11121–11126 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Prasad R et al. Structural insight into the DNA polymerase β deoxyribose phosphate lyase mechanism. DNA Repair 4, 1347–1357 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Klungland A & Lindahl T Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J 16, 3341–3348 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daleya JM, Wilson TE & Ramotara D Genetic interactions between HNT3/Aprataxin and RAD27/FEN1 suggest parallel pathways for 5′ end processing during base excision repair. DNA Repair 9, 690–699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasad R, Beard WA, Strauss PR & Wilson SH Human DNA polymerase β deoxyribose phosphate lyase: Substrate specificity and catalytic mechanism. J. Biol. Chem 273, 15263–15270 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Rass U, Ahel I & West SC Actions of aprataxin in multiple DNA repair pathways. J. Biol. Chem 282, 9469–9474 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Reynolds JJ et al. Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia 1. Mol. Cell Biol 29, 1354–1362 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gueven N et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum. Mol. Genet 13, 1081–1093 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Mosesso P et al. The novel human gene aprataxin is directly involved in DNA single-strand-break repair. Cell Mol. Life Sci 62, 485–491 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Gueven N et al. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience 145, 1418–1425 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Prasad R, Dianov GL, Bohr VA & Wilson SH FEN1 stimulation of DNA polymerase β mediates an excision step in mammalian long patch base excision repair. J. Biol. Chem 275, 4460–4466 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Chen X et al. Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol 409, 39–52 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Ellington A & Pollard JD Purification of oligonucleotides using denaturing polyacrylamide gel electrophoresis. Curr. Protoc. Mol. Biol 10.1002/0471142727.mb0212s42 (2001). [DOI] [PubMed]
- 16.Prasad R, Shock DD, Beard WA & Wilson SH Substrate channeling in mammalian base excision repair pathways: passing the baton. J. Biol. Chem 285, 40479–40488 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heacock M, Poltoratsky V, Prasad R & Wilson SH Evidence for abasic site sugar phosphate-mediated cyctotoxicity in alkylating agent treated Saccharomyces cerevisiae. PLoS One 7, e47945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Wu X & Friedberg EC The detection and measurement of base and nucleotide excision repair in cell-free extracts of the yeast Saccharomyces cerevisiae. Methods Companion Methods Enzymol 7, 177–186 (1995). [Google Scholar]
- 19.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.