

Abstract
Emerging evidence has shown that FXR activation ameliorates the development of alcoholic liver diseases (ALD) while whole-body deficiency of FXR in mice leads to more severe ALD. However, it’s unknown whether the enhanced susceptibility to ALD development in FXR−/− mice is due to deficiency of hepatic FXR or increased toxicity secondary to increased bile acid (BA) levels. Hepatocyte-specific FXR knockout mice (FXRhep−/−) present similar BA levels compared to wild-type mice, and are therefore a useful model to study a direct role of hepatic FXR in ALD development. FXRhep−/− mice were subject to an ALD model with chronic plus binge drinking of alcohol to determine the effects of hepatic FXR deficiency on ALD development. The FXRhep−/− mice showed an altered expression of genes involved in BA and lipid homeostasis with alcohol treatment. Despite a slightly increased trend in hepatic lipid deposition and collagen accumulation in FXRhep−/− mice, there were no significant differences in the severity of steatosis, inflammation, or fibrosis between WT and FXRhep−/− mice. Therefore, these findings indicate that FXR deficiency in hepatocytes might only play a minor role in ALD development. Deficiency of FXR in other non-hepatic tissues and/or increased BA levels resultant from whole-body FXR deficiency might be responsible for more severe ALD development.
Keywords: ethanol toxicity, tissue specific knockout, lipid metabolism, bile acid metabolism, FXR, alcoholic liver disease
Introduction
Alcoholic liver disease (ALD) is caused by chronic and overt alcohol consumption, and is one of the major causes of liver morbidity and mortality worldwide. The mechanisms underlying ALD pathogenesis are complex and remain elusive. The currently accepted concept is that alcohol consumption causes gut leakage-induced inflammation, alters hepatic metabolism, increases oxidative stress and alters lipid metabolism, which collectively leads to hepatotoxicity [1–4]. In addition to the amount of alcohol intake, environmental and genetic factors play crucial roles in ALD development. These factors include gender, ethnicity, nutritional factors and hepatic co-morbid conditions [3–5].
ALD is recognized as a progressive disease which comprises a wide spectrum of liver pathologies. The morphological changes in ALD range from hepatic steatosis, inflammation, and fibrosis culminating to alcoholic cirrhosis and potentially hepatocellular carcinoma [4–6]. FXR is a ligand-activated transcription factor belonging to the nuclear receptor superfamily, and is essential for regulating lipid and bile acid (BA) homeostasis [7–9]. Moreover, FXR also protects the liver against inflammation [10–12] and chemical-induced liver injury [13, 14]. BAs are endogenous ligands of FXR, and critical for solublization and absorption of dietary lipids and fat-soluble vitamins in the intestine [15]. In addition, BAs function as important signaling molecules and activate various cell signaling pathways [15]. Juxtaposed to their beneficial roles, BAs can also be toxic with high concentrations of causing cell injury, apoptosis [15, 16], and promotion of tumorigenesis in the liver and intestine [15, 17]. Therefore, BA levels in the body need to be tightly controlled.
Emerging evidence has shown that pharmacological FXR agonism attenuates chronic alcohol treatment-induced liver injury and steatosis [18, 19], while whole-body FXR knockout worsens alcohol-induced liver injury [19]. This suggests that FXR plays an important role in ALD development. However, deficiency of FXR in the whole body leads to increased BA levels thereby making it difficult to determine the independent effects of FXR from that of BAs on ALD severity. In order to study the tissue specific roles of FXR in ALD development, hepatocyte-specific FXR knockout (FXRhep−/−) mice were used in this study to determine the contribution of hepatocyte FXR deficiency in the well-developed ALD mouse model, Lieber-DeCarli alcohol-containing diet plus one binge dose [20]. Importantly, the FXRhep−/− mice lack FXR in hepatocytes but show similar BA levels to that of wild type (WT) mice.
Materials and Methods
Animals and treatments
FXRhep−/− mice have been described previously [9]. Briefly, FXRhep−/− mice were generated via cross-breeding between FXR floxed/floxed and albumin Cre (+) mice. FXR floxed/floxed, Cre (−) mice served as WT controls. These mice were maintained in the pathogen-free animal facilities at Rutgers University, under a standard 12 hr:12 hr light/dark cycle with temperature and humidity controlled conditions. All animal protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Rutgers University.
Male WT and FXRhep−/− mice (8-12 week old, n=5 mice/genotype/diet) were fed a Lieber-DeCarli alcohol-containing diet (5% v/v alcohol liquid diet) or isocaloric control liquid diet (Vehicle, alcohol replaced by isocaloric maltose-dextrin) for 10 days before administration of a single dose of alcohol (5 g/kg body weight) or maltose solution (9 g maltose/kg of body weight) via oral gavage [20]. The mice were euthanized 9 hrs after the binge gavage, with liver, intestine and serum collected for further analysis.
Serum biochemical assays
The activities of serum alanine aminotransferase (ALT) and alkaline phosphatase (ALP), as well as levels of serum total triglyceride (TG), total cholesterol (TC), and total BAs were measured using commercially available kits (TG, TC -Pointe Scientific, Canton, MI; BAs -Diazyme Laboratories, Poway, CA).
Liver histopathological examination
Hematoxylin and eosin (H&E) and Picro-Sirius Red staining were performed on fixed livers according to standard methods.
Total RNA isolation and quantitative RT-PCR assay
Total RNA was isolated from frozen tissues by TRIzol reagent (Invitrogen, Carlsbad, CA), and reverse-transcribed according to the manufacturer’s protocols. Quantitative real-time RT-PCR (qPCR) using SYBR green chemistry was used to measure the expression of genes of interest. Expression of each gene was normalized to β-actin mRNA levels using the ΔΔCt method. Primers sequences used for qPCR analysis are provided in Supplementary table 1.
Western blot analysis
Twenty μg lysates were loaded into separate wells and were separated in a 10% SDS-polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes, probed with antibodies against Cyp2e1 (Abcam, Cambridge, MA), and bands visualized using ECL kit (Thermo Scientific, Rockford, IL). Band density was quantified by ImageJ software and normalized to levels of β-actin.
Statistical analysis
The data are expressed as mean ± SD. Difference among groups was assessed by two-way analysis of variance (ANOVA) followed by a post-hoc Student-Newman-Keuls test (p<0.05 was considered significant).
Results
Hepatic FXR deficiency did not worsen alcohol-induced liver injury
Liver to body weight ratio (LW/BW) was slightly increased after alcohol feeding in WT and FXRhep−/− mice (Fig. 1A), however, no significant difference was observed between the two strains. Alcohol consumption decreased BW in WT but not FXRhep−/− mice. Specifically, the BW of WT mice fed the control diet increased 12.9 ± 7.6 % but was decreased by −1.7 ± 5.9 % by the alcohol containing diet. In the FXRhep−/− mice, BW changed by 7.9 ± 6.4% and 4.2 ± 3.5% when fed the control and alcohol containing diet respectively (Fig. 1A).
Figure 1. The effects of hepatic FXR deficiency and alcohol treatment on liver weight (LW), body weight (BW) and serum parameters.
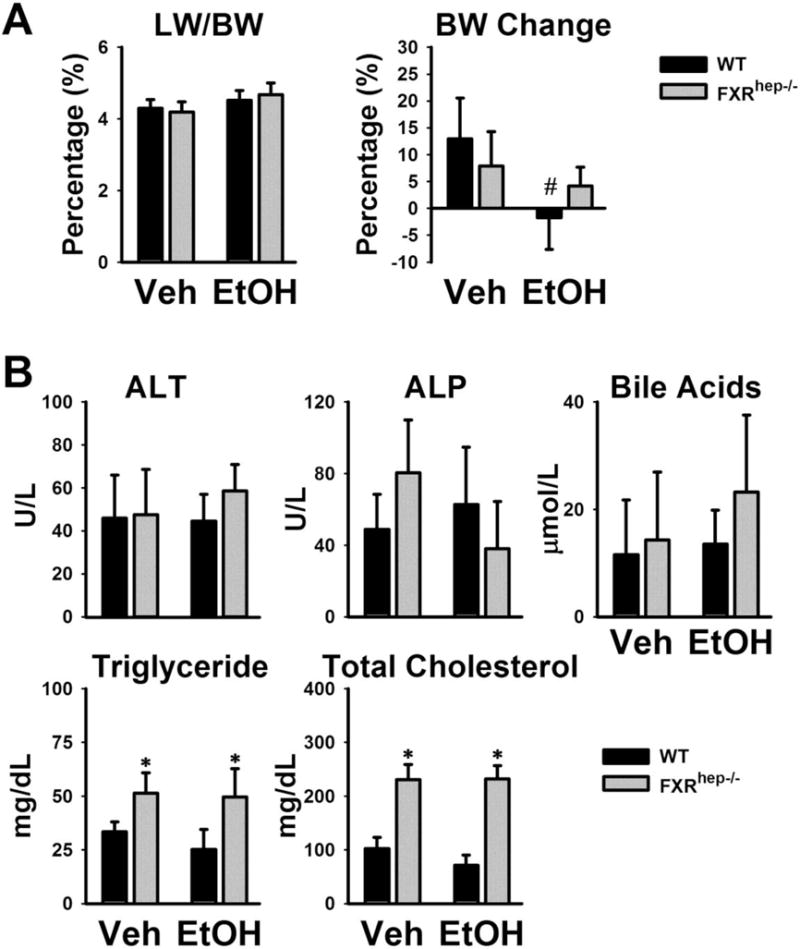
(A) Relative LW/BW ratio (left) and BW gain after alcohol feeding. The values represent the percent changes in BW compared to the starting BW (right). (B) Serum activities of ALT and ALP and serum levels of BAs, TG and TC in mice after being fed with isocaloric vehicle or Lieber-DeCarli alcohol-containing diet (5% v/v). Data are expressed as mean ± SD. * indicates significant difference (p<0.05) between different genotypes (WT vs FXRhep−/−); # indicates significant difference (p<0.05) between treatments (Veh vs EtOH).
Serum ALT activities were measured as an indicator of hepatic cellular injury. Chronic alcohol administration was well tolerated in WT and FXRhep−/− mice with ALT not significantly in either strain. Serum activities of ALP and levels of total BAs were similar in WT and FXRhep−/− mice, and showed no significant increase after alcohol feeding. However, there was a trend for increased BA levels in FXRhep−/− mice after alcohol feeding (Fig. 1B).Alcohol feeding did not alter serum TG or TC levels in either strain albeit both TG and TC levels were higher in FXRhep−/− mice compared to WT (Fig. 1B).
Hepatic FXR deficient mice showed similar severity of alcohol-induced liver steatosis, inflammation and fibrosis
The livers of WT and FXRhep−/− mice appeared normal histologically on control diet (Fig. 2A). Alcohol feeding resulted in mild hepatic microsteatosis in both WT and FXRhep−/− mice with a trend of increased lipid droplets in the livers of FXRhep−/− mice. Scattered foci of inflammatory cell infiltration was present in the livers of mice fed the alcohol containing diet. The degree of infiltrate was similar between WT and FXRhep−/− mice (Fig. 2A). The severity of liver fibrosis was determined by Picro-Sirius Red staining (Fig. 2B). When fed a control diet, collagen (red) was slightly present only in the perivascular region of the portal vein and central vein in WT mice. Slight collagen staining was found in hepatic sinusoids in FXRhep−/− mice. After alcohol feeding, more collagen was accumulated in the hepatic sinusoidal area in WT mice, and a higher deposition of collagen surrounding the portal vein and central vein was found in FXRhep−/− mice. FXRhep−/− mice tended to have slightly more fibrosis than WT mice (Fig. 2B).
Figure 2. The effects of hepatic FXR deficiency on hepatic steatosis, inflammation and fibrosis under alcohol diet.
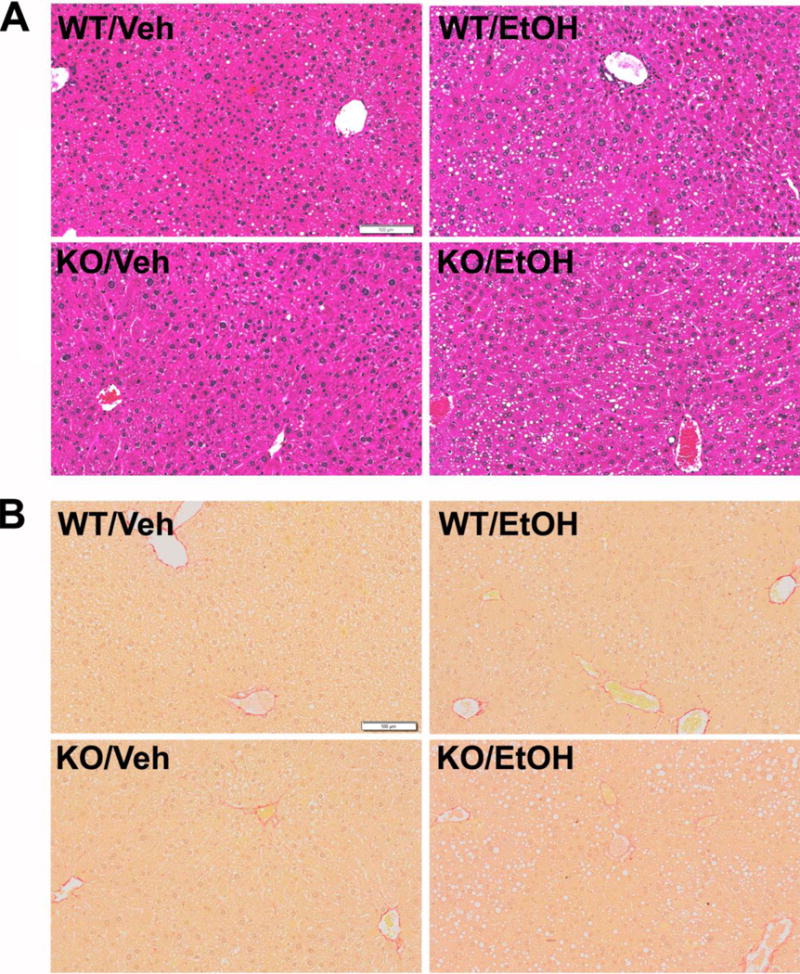
(A) Representative images of H&E stained liver sections after alcohol treatment (200X magnification). The major histopathological change induced by alcohol in mouse livers was microvesicular steatosis. Fat droplet deposition was observed in mice fed the alcohol diet, but not in those fed the control vehicle diet. (B) Representative images of Picro-Sirius Red stained liver sections after alcohol treatment (200X magnification).
The mRNA levels of genes involved in inflammation, lipopolysaccharide (LPS) response, and fibrosis were determined (Fig. 3). The mRNA levels of Tlr4 were not changed regardless of genotype or alcohol treatment. Levels of another LPS receptor, Cd14, were slightly decreased in FXRhep−/− mice on control diet, but increased in WT and FXRhep−/− mice by alcohol feeding (Fig. 3A). The mRNA levels of Myd88 were lower in FXRhep−/− mice on control diet compared to WT mice, but not induced by alcohol feeding either genotype. The mRNA levels of pro-inflammatory cytokines, Tnfα and Il6, were lower in FXRhep−/− mice on control diet, but both were induced to similar levels by alcohol feeding in WT and FXRhep−/− mice. The expression of an inflammatory marker gene Lcn2 tended to be increased in FXRhep−/− mice on control diet, and was induced to a greater extent by alcohol feeding in FXRhep−/− mice than WT mice (Fig. 3A).
Figure 3. Expression of hepatic genes in inflammation and fibrosis.
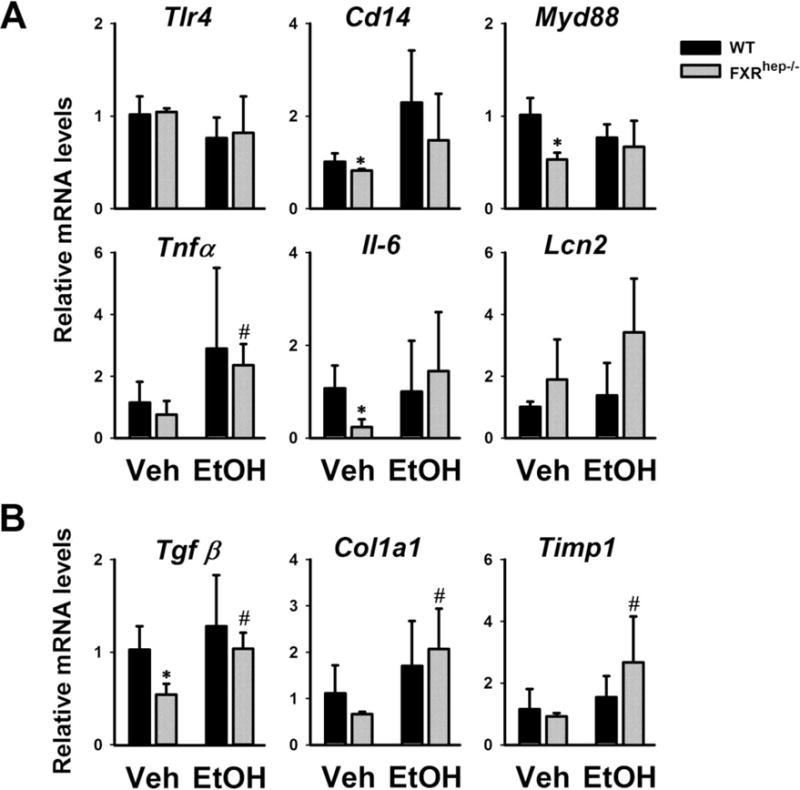
Gene expression of (A) LPS binding proteins (Tlr4, Cd14, Myd88), pro-inflammatory cytokines (Tnfα, Il-6 and Lcn2), and (B) fibrotic mediators (Tgfβ, α-Sma, Col1a1 and Timp1). Data are expressed as mean ± SD. * indicates significant difference (p<0.05) between different genotypes (WT vs FXRhep−/−); # indicates significant difference (p<0.05) between treatments (Veh vs EtOH).
Under the control diet, the mRNA levels of genes in fibrosis, Tgf β, Col1a1 and Timp1, tended to be decreased in FXRhep−/− mice compared to WT mice. Alcohol feeding induced the gene expression of Tgf β, Col1a1 and Timp1 in both strains. However, Col1a1 and Timp1 were induced slightly higher in FXRhep−/− mice (Fig. 3B).
Alcohol feeding disrupted BA homeostasis and lipid metabolism in WT and FXRhep−/− mice
The mRNA expression of hepatic and intestinal genes involved in lipid (Fig. 4) and BA homeostasis (Fig. 5) was measured in WT and FXRhep−/− mice after alcohol treatment. In particular, the mRNA expression of several lipogenic genes, including sterol regulatory element-binding protein 1c (Srebp-1c), fatty acid synthase (Fas) and acyl-CoA synthetase short-chain family member 2 (Acss2), was significantly decreased by alcohol feeding (Fig. 4). Furthermore, the hepatic mRNA expression of a gene involved in fatty acid β-oxidation Cyp4a10, a PPARα target gene, was significantly increased after alcohol treatment (Fig. 4), with a trend of higher induction in FXRhep−/− mice. The mRNA expression of cluster of differentiation 36 (Cd36), which takes up long-chain free fatty acids into the liver, didn’t change in WT mice, but significantly increased by alcohol treatment in FXRhep−/− mice (Fig. 4). Moreover, alcohol didn’t affect the gene expression of microsomal triglyceride transfer protein (Mtp) that is involved in VLDL secretion.
Figure 4. Expression of hepatic genes involved in lipid homeostasis.
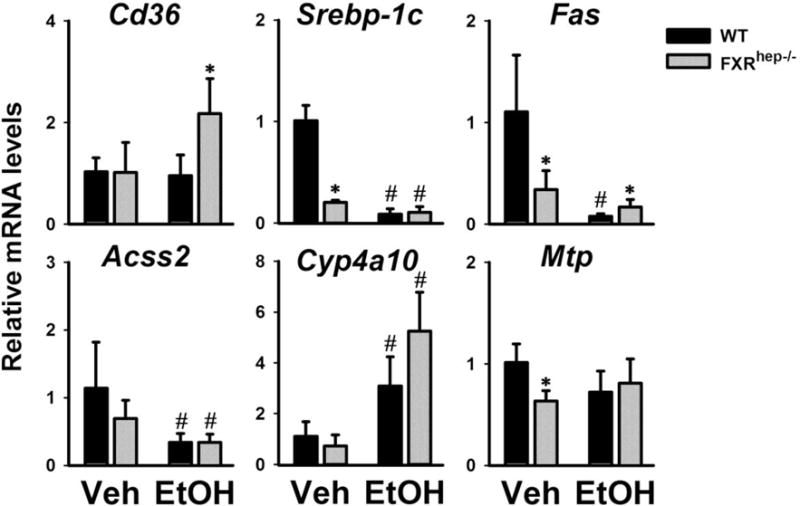
Expression of key hepatic genes contributing to uptake of free fatty acids from the blood (Cd36), de novo lipogenesis (Srebp-1c, Fas and Acss2), fatty acid β-oxidation (Cyp4a10, PPARα target gene) and hepatic VLDL secretion (Mtp). Data are expressed as mean ± SD. * indicates significant difference (p<0.05) between different genotypes (WT vs FXRhep−/−); # indicates significant difference (p<0.05) between treatments (Veh vs EtOH).
Figure 5. Expression of hepatic (A) and intestinal (B) genes involved in BA homeostasis.
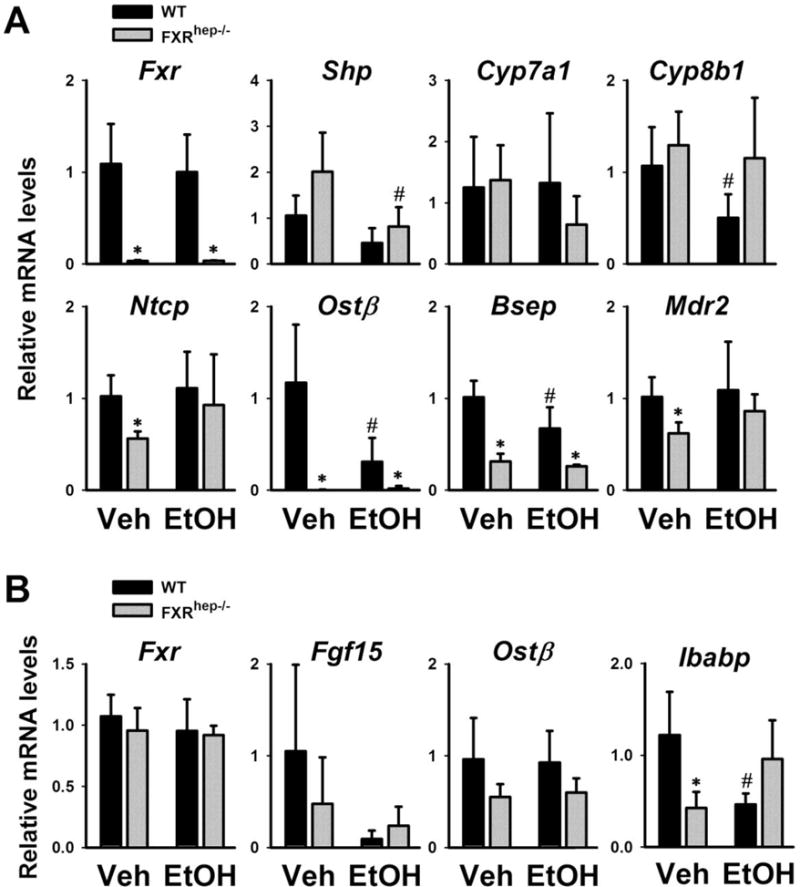
Genes in synthesis (Fxr, Shp, Cyp7a1, Cyp8b1 and Fgf15) and transport (Ntcp, Ost β, Bsep, Mdr2 and Ibabp). Data are expressed as mean ± SD. * indicates significant difference (p<0.05) between different genotypes (WT vs FXRhep−/−); # indicates significant difference (p<0.05) between treatments (Veh vs EtOH).
Hepatic FXR has been shown to only play a minor role in suppressing liver BA synthesis [8, 9, 21], but is critical in the regulation of hepatic BA transport. The mRNA levels of hepatic (Fig. 5A) and intestinal (Fig. 5B) genes in BA homeostasis were determined. On control diet, the effects of hepatic FXR deficiency on the expression of bile acid synthetic genes, Cyp7a1 and Cyp8b1, were very small. However, hepatic FXR deficiency significantly decreased the mRNA levels of transporter genes, Ntcp, Ost β, Bsep and Mdr2 (Fig. 5A). In WT mice, alcohol treatment decreased the mRNA levels of Shp, Ost β, and Bsep, but did not alter those of Ntcp or Mdr2. Furthermore, alcohol tended to have no effect on Cyp7a1 gene expression in WT mice but decreased it in FXRhep−/− mice. Conversely, the opposite was observed regarding Cyp8b1 with expression decreased in WT mice but not FXRhep−/− mice (Fig. 5A). In the ileum, Fgf15 gene expression in both strains tended to be decreased by alcohol feeding (Fig. 5B). Alcohol didn’t affect the gene expression of Ost β in either strain. Ibabp gene expression was decreased in FXRhep−/− mice compared to WT on control diet. Interestingly, alcohol feeding markedly reduced Ibabp expression in WT mice but tended to increase its expression in FXRhep−/− mice (Fig. 5B).
Hepatic FXR deficiency did not alter the expression of genes involved in alcohol metabolism
Alcohol dehydrogenases (ADHs), catalase and cytochrome P450 2E1 (CYP2E1) are involved in the conversion of alcohol to acetaldehyde. Aldehyde dehydrogenases (ALDHs) then convert acetaldehyde to acetate. Acetaldehyde and its metabolites can directly attack proteins, lipids, and many other cellular components resulting in liver toxicity and cellular damage [3]. The protein expression levels of Cyp2e1 were determined by Western blot (Fig 6A). The basal Cyp2e1 protein levels were similar between WT and FXRhep−/− mice. Upon alcohol treatment, Cyp2e1 protein levels were highly induced in both strains to similar levels. Shown in Fig 6B, the mRNA levels of Adh1 and Catalase were decreased in FXRhep−/− mice. Alcohol feeding tended to decrease Adh1 gene expression in WT mice but increased its expression in FXRhep−/− mice. Aldh1 gene expression was not altered by either hepatic FXR deficiency or alcohol feeding. In addition, alcohol feeding decreased Catalase gene expression in both strains similarly.
Figure 6. Alcohol-induced changes in the expression of genes involved in alcohol metabolism.
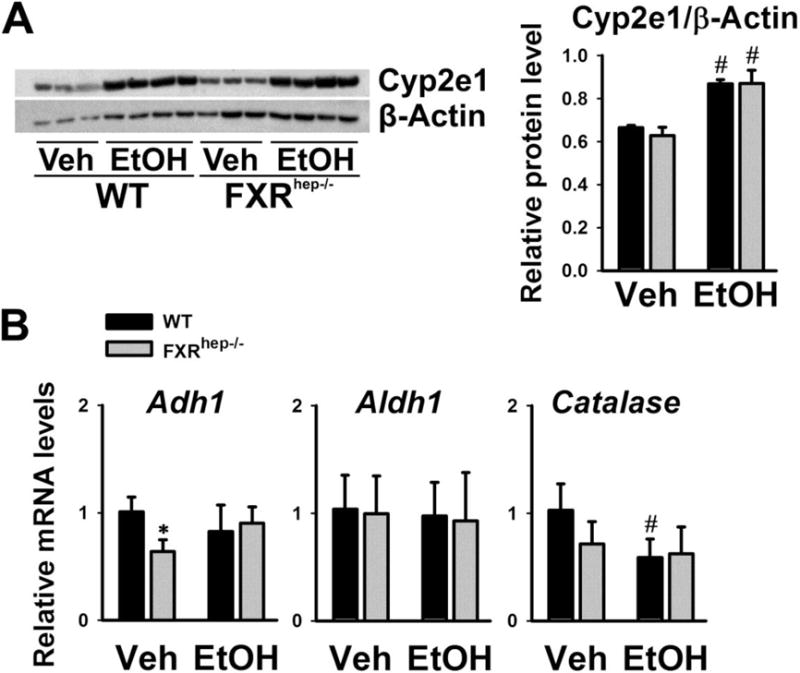
(A) Representative immunoblot of Cyp2e1 protein levels in liver lysates from WT and FXRhep−/− mice after alcohol feeding. Densitometric values are calculated and shown after normalized with internal loading control, β-actin. (B) Hepatic mRNA levels of Adh1, ALDH1 and Catalase in WT and FXRhep−/− mice under alcohol diet. Data are expressed as mean ± SD. * indicates significant difference (p<0.05) between different genotypes (WT vs FXRhep−/−); # indicates significant difference (p<0.05) between treatments (Veh vs EtOH).
Discussion
FXR is a critical sensor for BA homeostasis in the body by regulating BA synthesis, transport and metabolism. In addition to regulating BA biosynthesis, FXR protects the liver from chemical-induced injury and inflammation, and promotes liver regeneration. Activation of FXR has recently been shown to protect against liver injury induced by CCl4 and acetaminophen [13, 14]. Activation of FXR may antagonize NF-κB signaling to decrease proinflammatory cytokine production in the liver and decrease LPS-induced hepatic inflammation [2, 3]. Studies have shown that FXR activation can protect the liver from alcohol-induced liver injury, while deletion of FXR in mice worsens the alcoholic liver disease [18, 19]. All these previously described studies were performed by systemic activation or deficiency of FXR leaving the tissue specific roles of FXR in ALD development unclear. Determining these tissue specific roles of FXR in ALD pathogenesis is of great importance as it is well known that the functions of FXR from different organs vary; for example intestinal FXR playing a major role in suppressing bile acid synthesis [7, 9, 21]. Moreover, systemic FXR alterations, either agonism or whole-body deficiency, are accompanied by changes in BA levels. BAs can cause cell injury, apoptosis, inflammation and tumorigenesis, and therefore alterations of BA levels in studies using systemic FXR models can confound the interpretation of study findings [15].
The FXRhep−/− mice present with a normal BA pool size [9], which makes them useful to study the direct effects of hepatic FXR deficiency on ALD development independent of the effects of increased BA pool size. The current study showed that deletion of the FXR gene only in hepatocytes did not deteriorate alcohol-induced hepatocyte ballooning, inflammation and injury. This was revealed by unchanged liver histopathology, activities of serum ALT and ALP, (Fig. 1 and 2), and expression of genes encoding pro-inflammatory factors (Fig. 3). However, hepatic FXR deficiency disturbed BA homeostasis, and slightly increased hepatic microsteatosis and collagen deposition (Fig. 2), indicating hepatic FXR might play some role in ALD progression. One of the mechanisms may be that the disturbance of BA homeostasis after alcohol treatment may have sensitized the liver to alcohol-induced injury. But more studies are needed to investigate this hypothesis.
In our study, hepatocyte specific FXR deficiency had only minor effects on ALD development. This contrasts to other studies which describe how whole-body FXR deficiency in mice increases ALD severity and systemic activation of FXR attenuates ALD severity [18, 19]. The collective findings from our study and the previous reports therefore indicate that BAs or FXR deficiency in tissues other than the liver might be more important for ALD development. FXR signaling in the intestine is able to protect the integrity of the intestines; intestine integrity and defense against bacteria were reported to be compromised in FXR−/− mice [21, 22]. As alcohol has been demonstrated to induce hepatic oxidative stress and inflammation from gut bacterial-derived LPS [2, 4, 23], this indicates that intestinal FXR deficiency might be a more important mediator for alcohol-induced liver injury. Consistent with this hypothesis are the findings of a recent study which showed that the FXR/FGF15/FGFR4 axis protects the liver from ALD progression through maintaining BA homeostasis and modifying gut microflora [24]. However, additional studies are still needed to clarify the effects and mechanisms of intestinal FXR and BAs in maintaining intestinal integrity against gut leakage and inhibiting gut microbiota overgrowth and translocation.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grants (R01GM104037), VA grant (BX002741), and Rutgers Busch grant.
List of abbreviations
- ADHs
Alcohol dehydrogenases
- ALD
alcoholic liver diseases
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- Acss2
acyl-CoA synthetase short-chain family member 2
- BAs
bile acids
- BW
body weight
- Fas
fatty acid synthase
- FXR
farnesoid X receptor
- H&E
hematoxylin and eosin
- LPS
lipopolysaccharide
- LW
liver weight
- Srebp-1c
sterol regulatory element-binding protein 1c
- TC
total cholesterol
- TG
triglycerides
- WT
wild type
- NAFLD
non-alcoholic fatty liver disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dianzani MU. Lipid peroxidation in ethanol poisoning: a critical reconsideration. Alcohol and alcoholism. 1985;20:161–73. [PubMed] [Google Scholar]
- 2.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and nonalcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. Journal of hepatology. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 3.Bondy SC. Ethanol toxicity and oxidative stress. Toxicology letters. 1992;63:231–41. doi: 10.1016/0378-4274(92)90086-y. [DOI] [PubMed] [Google Scholar]
- 4.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–85. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saberi B, Dadabhai AS, Jang YY, et al. Current Management of Alcoholic Hepatitis and Future Therapies. Journal of clinical and translational hepatology. 2016;4:113–22. doi: 10.14218/JCTH.2016.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams JA, Manley S, Ding WX. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World journal of gastroenterology. 2014;20:12908–33. doi: 10.3748/wjg.v20.i36.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell metabolism. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. Journal of lipid research. 2007;48:2664–72. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Kong B, Wang L, Chiang JY, et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–43. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–72. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 11.Wang YD, Chen WD, Wang M, et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–43. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Liu Q, Wang J, et al. Suppression of interleukin-6-induced C-reactive protein expression by FXR agonists. Biochemical and biophysical research communications. 2009;379:476–9. doi: 10.1016/j.bbrc.2008.12.117. [DOI] [PubMed] [Google Scholar]
- 13.Lee FY, de Aguiar Vallim TQ, Chong HK, et al. Activation of the farnesoid X receptor provides protection against acetaminophen-induced hepatic toxicity. Molecular endocrinology. 2010;24:1626–36. doi: 10.1210/me.2010-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng Z, Wang Y, Wang L, et al. FXR regulates liver repair after CCl4-induced toxic injury. Molecular endocrinology. 2010;24:886–97. doi: 10.1210/me.2009-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li T, Chiang JY. Bile Acid signaling in liver metabolism and diseases. Journal of lipids. 2012;2012:754067. doi: 10.1155/2012/754067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Liu H, Zhang M, et al. Fatty liver diseases, bile acids, and FXR. Acta pharmaceutica Sinica B. 2016;6:409–12. doi: 10.1016/j.apsb.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong B, Zhu Y, Li G, et al. Mice with hepatocyte-specific FXR deficiency are resistant to spontaneous but susceptible to cholic acid-induced hepatocarcinogenesis. American journal of physiology Gastrointestinal and liver physiology. 2016;310:G295–302. doi: 10.1152/ajpgi.00134.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livero FA, Stolf AM, Dreifuss AA, et al. The FXR agonist 6ECDCA reduces hepatic steatosis and oxidative stress induced by ethanol and low-protein diet in mice. Chemico-biological interactions. 2014;217:19–27. doi: 10.1016/j.cbi.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 19.Wu W, Zhu B, Peng X, et al. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochemical and biophysical research communications. 2014;443:68–73. doi: 10.1016/j.bbrc.2013.11.057. [DOI] [PubMed] [Google Scholar]
- 20.Bertola A, Mathews S, Ki SH, et al. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nature protocols. 2013;8:627–37. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3920–5. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Liu Y, Sidhu A, et al. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. American journal of physiology Gastrointestinal and liver physiology. 2012;303:G32–41. doi: 10.1152/ajpgi.00024.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. The American journal of physiology. 1998;275:G605–11. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 24.Hartmann P, Hochrath K, Horvath A, et al. Modulation of the intestinal bile acid-FXR-FGF15 axis improves alcoholic liver disease in mice. Hepatology. 2017 doi: 10.1002/hep.29676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.