

Abstract
Cyclin E, a regulatory subunit of cyclin-dependent kinase 2 (CDK2), is central to the initiation of DNA replication at the G1/S-checkpoint. Tight temporal control of cyclin E is essential to the coordination of cell cycle processes and the maintenance of genome integrity. Overexpression of cyclin E in human tumors was first observed in the 1990s and led to the identification of oncogenic roles for deregulated cyclin E in experimental models. A decade later, low molecular weight cyclin E isoforms (LMW-E) were observed in aggressive tumor subtypes. Compared with full-length cyclin E, LMW-E hyperactivates CDK2 through increased complex stability and resistance to the endogenous inhibitors p21CIP1 and p27KIP1. LMW-E is predominantly generated by neutrophil elastase-mediated proteolytic cleavage, which eliminates the N-terminal cyclin E nuclear localization signal and promotes cyclin E’s accumulation in the cytoplasm. Compared with full-length cyclin E, the aberrant localization and unique stereochemistry of LMW-E dramatically alters the substrate specificity and selectivity of CDK2, increasing tumorigenicity in experimental models. Cytoplasmic LMW-E, which can be assessed by immunohistochemistry, is prognostic of poor survival and predicts resistance to standard therapies in cancer patients. These patients may benefit from therapeutic modalities targeting the altered biochemistry of LMW-E or its associated vulnerabilities.
Keywords: cyclin E, low molecular weight cyclin E (LMW-E), cell cycle, neutrophil elastase
G1-S Checkpoint Control
Fundamentally, cell division requires a cell to first replicate its genomic DNA and then segregate the replicated chromosomes into separate daughter cells. At the cellular level, the coordination of these processes is complex and requires precise temporal control to maintain genome integrity and cell viability. Tissue-level control of cell division is coordinated by a vast array of signals, including soluble factors, cell-cell interactions, and cell-matrix interactions. Individual cells integrate these social cues with readouts of cellular status (e.g. telomere length, DNA integrity, and metabolic state) to inform cell division processes. As Pardee first reported in 1974 (1), these varying cellular inputs converge on a core regulator governing the decision of a cell to replicate its genome or remain quiescent.
Although Pardee predicted a single R-factor underlying the G1-S–phase restriction point, the point at which commitment to cell division is complete (2), subsequent research has revealed a more complicated regulatory network. At its core, cell cycle progression is orchestrated by the orderly expression of cyclins (regulatory subunits), which sequentially activate cyclin-dependent kinases (CDKs; catalytic subunits). Each phase of the cell cycle has a unique configuration of cyclins and CDKs that govern the cell division machinery. G1 cyclin/CDK activity represents the major integration point of tissue-level controls on cell division (3). Sustained deviation in the activity of these CDKs, independent of tissue-level inputs, promotes tumor initiation and progression (4,5).
Cyclin E and Cell Cycle Control
In prevailing models of the mammalian cell cycle, cyclin D–CDK4/6 activity, stimulated by an abundance of mitogenic over inhibitory inputs, induces cells to enter G1-phase from quiescence (G0) by partially phosphorylating pRb (6). The E2F-family transcription factors are bound and sequestered by hypophosphorylated pRb in G0 but are released by phosphorylated pRb during G1 progression and directly enhance cyclin E transcription (7). Cyclin D–CDK4/6 activity tapers off at the end of G1, signaling a shift in cell cycle regulation via cyclins from D-type to E-type. The cyclin E–CDK2 complex further phosphorylates pRb, enhancing the E2F-mediated upregulation of S-phase–specific genes (8). Cyclin E-CDK2 activity is subject to both intrinsic and extrinsic control throughout G1-phase, in part through the binding of endogenous CDK inhibitors (CKi), including p21CIP1 and p27Kip1 (9). Cyclin E–CDK2 reaches peak activity in late G1-phase through positive feedback mechanisms, including the enhanced E2F-mediated transcription of cyclin E and phosphorylation-induced inactivation of key inhibitors (e.g. p27, p21 and SMAD3) (10–12). Eventually, cyclin E–CDK2 becomes insensitive to intrinsic and extrinsic inputs mediating irreversible commitment to cell division (the restriction point) (2). As cells progress through S-phase, cyclin E undergoes rapid ubiquitin proteasome–mediated degradation (13,14). CDK2 activity is maintained by cyclin A throughout S-phase and into G2-phase (14).
At the G1-S–phase transition, cyclin E participates in the initiation of DNA replication through several mechanisms beyond the E2F-mediated activation of S-phase gene transcription. [1] Cyclin E–CDK2 phosphorylates p220NPAT, thereby stimulating histone biosynthesis (15,16); [2] Cyclin E triggers centrosome duplication through a CDK2-dependant mechanism mediated by the phosphorylation of centrosomal proteins (e.g. nucleophosmin B23 and CP110) (17,18) and through a CDK2-independent mechanism dependent on a centrosome localization signal within cyclin E (19); [3] Cyclin E–CDK2 regulates DNA replication licensing and the origin of replication firing through interactions with the origin replication complex subunits (e.g. CDC6, CDT1, and the minichromosome maintenance protein complex), in a manner that is predominantly kinase independent (20–22).
Evidence from studies with knockout mice demonstrates considerable redundancy among different cyclin/CDK complexes. Cyclin E/CDK2 is largely dispensable for mammalian cell cycles. Cyclin E1/E2 double-knockout mice are viable, although the failure of endoreduplication requires placental rescue during gestation (23–26). Cells derived from these animals demonstrate specific defects in their ability to enter G1-phase from G0-phase, which are due in part to defective DNA replication licensing. CDK2 knockout mice are also viable, but they are infertile (27). Critically, cyclin E–null mouse embryonic fibroblasts are resistant to oncogenic transformation, indicating that cyclin E may play a critical role in tumor development (26). In established cancer cell lines, dominant-negative CDK2 dramatically inhibits cell cycle progression (28). Collectively, experimental evidence demonstrating that cyclin E is largely dispensable for adult cell cycle progression but critical to cancer cell cycle progression suggests that specifically targeting cyclin E–CDK2 could have a favorable therapeutic index.
Cyclin E Overexpression in Human Cancers
Cyclin E expression is commonly deregulated in many cancer types, including breast, lung, colorectal, gastric, lymphoma, leukemia, and osteosarcoma (29–38). Cyclin E overexpression has several underlying mechanisms including gene amplification, transcriptional upregulation, and disruption of cyclin E degradation. [1] Cyclin E gene amplification is observed in many common tumor types, particularly ovarian (22%) (39), esophageal/gastric (18%) (40,41), endometrial (14%) (42), bladder (7%) (43), pancreatic (6%) (44), and non-small cell lung cancer (5%) (45) (Supplementary Figure 1A). [2] Transcriptional upregulation of cyclin E occurs in tumors with inactivated pRb, due to an increase in E2F-mediated transcription. Inactivation of pRb is a frequent event during tumorigenesis, as a consequence of excessive CDK4/6 activity (due to the activation of certain oncogenes, such as cyclin D1 and RAS), the genetic inactivation of the pRb gene, or the introduction of viral oncoproteins (e.g. HPV16-E7) mediating degradation or sequestration of pRb (46). The transcriptional upregulation of cyclin E can also result from c-MYC overexpression or amplification, which directly activates the cyclin E promoter (47,48). [3] Disruption of cyclin E degradation is frequently observed following genetic inactivation of FBW7, a critical subunit of the ubiquitin protein ligase complex responsible for directing cyclin E degradation (49–51). Inactivation of FBW7 in several tumor types is commonly observed in Cancer Genome Atlas (TCGA) databases (52), including endometrial (20%), colorectal (17%), cervical, (12%), bladder (9%), and head and neck cancer (8%) (Supplementary Figure 1B).
Cyclin E overexpression leads to enhanced CDK2 activity and cell cycle progression, thereby reducing the ability of cells to regulate the G1-S transition. Owing to functional redundancy, cyclin E–CDK2 overexpression has the ability to phosphorylate pRb, even if cyclin D–CDK4/6 complexes have been rendered inactive by the overexpression of p16 (53). In experimental models, cyclin E overexpression is sufficient to induce tumorigenesis in vivo. Approximately 10% of transgenic mice with overexpression of human cyclin E in the mammary gland develop mammary carcinomas (54). Cyclin E overexpression also dramatically enhances the transformation by oncogenes such as HRAS (55). A likely key mechanism of cyclin E–mediated oncogenesis is the induction of genomic instability (56–58), which was not observed following the overexpression of cyclin D1 or cyclin A (57). However, recent studies suggest that cyclin D1 overexpression can also induce genomic instability in a CDK-independent fashion by promoting the transcriptional induction of a specific set of genomic instability–inducing genes (59–62). This suggests that deregulation of cyclins and CDKs may act synergistically to cause genomic instability. Mechanistically, cyclin E overexpression increases DNA replication beyond the capacity of cellular nucleotide pools, causing replication stress and replication-induced DNA damage (63). In p53-deficient cells, cyclin E overexpression causes centrosome amplification, leading to polyploidy (64). Cyclin E mutants incapable of activating CDK2 or initiating the G1-S-transition are nonetheless able to transform rat embryonic fibroblasts in cooperation with HRAS as long as the centrosome localization signal is intact (65).
Tumor-Specific Low-Molecular-Weight Cyclin E isoforms
A 1993 study characterizing deregulated cyclin expression in breast cancer cell lines revealed that the cells accumulate several truncated forms of the cyclin E protein of unknown biological consequence (29). These truncated isoforms, collectively termed low-molecular-weight cyclin E (LMW-E), are restricted to tumor cell lines and do not occur in human mammary epithelial cells (HMECs) obtained from disease-free donors (29). In both normal and tumor cells and tissues full length cyclin E (which, hereafter is referred to as FL-cyclin E) is expressed at approximately 50 kDa, as determined by western blot and mass spectrometry (66). Five tumor-specific LMW-E isoforms with molecular weights ranging from 45 to 33 kDa have been identified (66,67). Digestion of cyclin E with neutrophil elastase (NE) recapitulates the characteristic LMW-E pattern seen in cancer cells (66,68) (Figure 1a). In addition, at least two other proteases, calcium-dependent calpain (69) and calpain-2 (70), have been linked to the generation of LMW-E in tumor cells.
Figure 1: The unique biochemical activities of LMW-E versus FL cyclin E and their consequences.
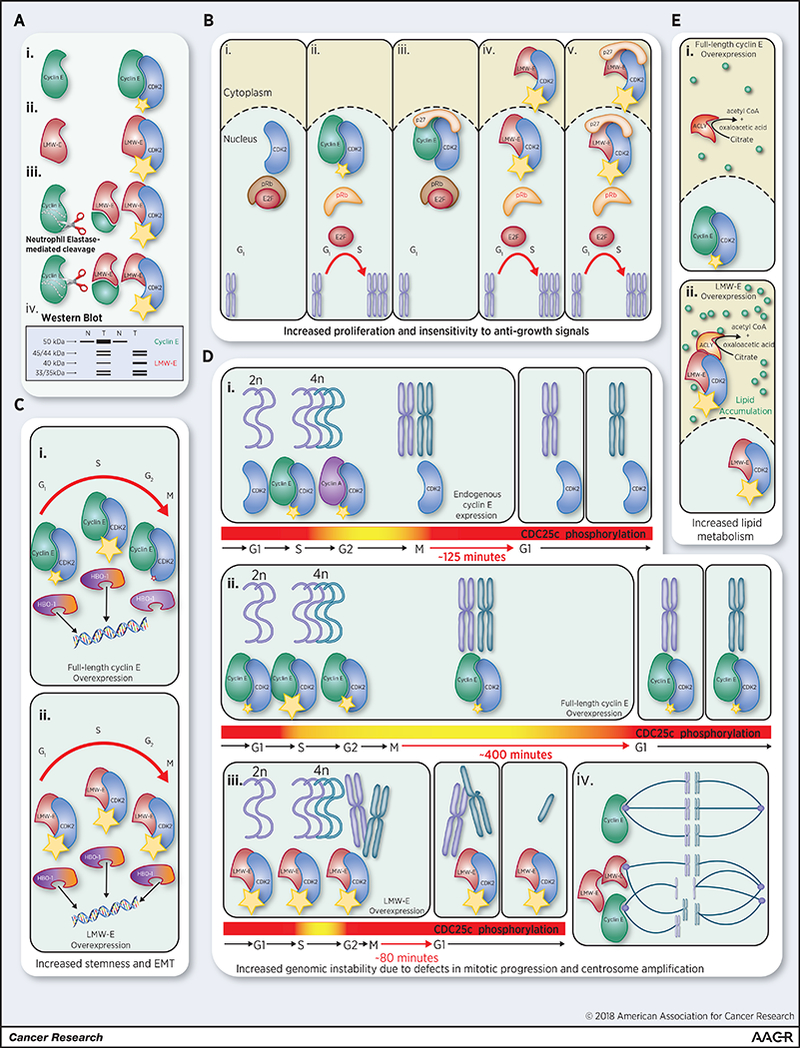
(A) (i) Cyclin E (FL 50 kDa) is an activating subunit of CDK2 that promotes kinase activity (small yellow star) during the G1-S-phase transition. Tumor-specific LMW-E isoforms are generated by (ii) alternative translation from methionine 46 (40 kDa) and (iii) NE-mediated cleavage of full-length cyclin E at two N-terminal sites (45/44 kDa and 35/33 kDa; doublets due to phosphorylation events) (66,105). LMW-E isoforms demonstrate higher binding affinity for CDK2, promoting hyperactivation of the kinase (large yellow star). (iv) In western blot analysis of normal (N) and tumor (T) tissue (using a C-terminally-directed antibody), LMW-E isoforms characteristically resolve as five distinct bands beneath FL cyclin E. In half of LMW-E-expressing breast tumors, cyclin E is also overexpressed; in the other half, however, LMW-E is expressed in the absence of full-length cyclin E (B). (i) When the pRb pathway is unaltered by oncogenic events, hypophosphorylated pRb binds to and sequesters E2F family transcription factors in G0-phase. (ii) FL cyclin E activates CDK2, leading to the hyperphosphorylation and inactivation of pRb, thereby releasing E2Fs to activate S-phase gene expression and progression. (iii) However, CDK inhibitors (e.g. p27) can inhibit the FL cyclin E-CDK2 complex (even if FL cyclin E is overexpressed) and prevent S-phase progression. (iv) Hyperactive LMW-E-CDK2 complexes can localize to the nucleus and hyperphosphorylate pRB; (v) even in the presence of CDK inhibitors, thereby promoting insensitivity to negative growth signals. (C) Hyperactive LMW-E-CDK2 complexes have other consequences. (i) FL Cyclin E-CDK2 can phosphorylate the substrate HBO-1; however, (ii) only constitutive hyperphosphorylation by LMW-E-CDK2 can promote HBO-1-dependant EMT and stemness properties, suggesting that cell cycle context-independent phosphorylation of HBO-1 alters its histone acetyltransferase activity in a pro-tumorigenic manner. (D) The proper timing of DNA replication and mitosis is essential to genome integrity. (i) One mechanism to ensure the fidelity of this process is feedback control through CDC25c, which promotes the proper timing of mitotic entry and exit through the activation of cyclin B-CDK1 and PLK1. (ii) Overexpression of FL cyclin E results in the improper phosphorylation of CDC25c and premature mitotic entry, but maintains the phosphorylation of CDC25c, delaying mitotic progression to cytokinesis and thereby largely preventing genomic instability. (iii) LMW-E overexpression also initiates premature mitotic entry; however unlike FL cyclin E, LMW-E cannot sustain CDC25c phosphorylation, resulting in faster mitotic exit and genomic instability. (iv) Genomic instability is further promoted by centrosome amplification induced by both FL cyclin E and LMW-E overexpression. (E) (i) FL cyclin E is largely restricted to the nucleus and therefore has limited opportunities to interact with cytoplasmic proteins. (ii) In contrast, LMW-E lacks an N-terminal nuclear localization signal promoting its accumulation in the cytoplasm where it can interact with novel binding partners including ACLY. LMW-E-CDK2 enhances ACLY activity (independent of phosphorylation), thereby promoting intracellular lipid accumulation and pro-tumorigenic phenotypes, including migration and invasion.
Clinically, LMW-E isoforms (71,72) have been observed in multiple tumor types including breast cancer (29), ovarian cancer (73,74), melanoma (75), colorectal cancer (76–79), lung cancer (80), bladder cancer, and renal cell carcinomas (81). However these isoforms have not been detected in matched adjacent histologically normal tissues. (82). One analysis of total cyclin E (FL-cyclin E + LMW-E) by western blot in 395 breast cancer patients revealed a statistically significant association with distant metastases and reduced overall survival (83). At a median follow-up of 6.4 years, 91.7% of patients with total cyclin E overexpression developed either local and/or distant metastases compared with 7% of patients without total cyclin E overexpression (p < 0.001). In multivariable analysis, total cyclin E expression was an independent prognostic variable and a better predictor of disease-specific and overall survival compared with nodal status, estrogen receptor (ER) status, or even IIIB-IV stage. Before this study, the prognostic value of cyclin E expression had been evaluated only on the basis of nuclear staining in immunohistochemistry studies. These studies revealed no consistent association between cyclin E overexpression and survival (84). These results led us to study the features distinguishing LMW-E from FL-cyclin E, in order to better understand their roles in the development of aggressive cancers.
Generation of LMW-E by intracellular Neutrophil Elastase
The observation that tumor-specific LMW-E could be generated from FL-cyclin E by the serine protease NE was an unexpected finding (Figure 1a). In breast cancer patients, high levels of NE have been shown to be prognostic for poor overall, metastasis-free, and disease-specific survival, and also predictive of resistance to standard therapies (85–88). High levels of NE have also been found in bladder cancer, lung cancer, and pancreatic cancer (86,88–93). One study using the loxP-Stop-loxP K-rasG12D mouse model of lung cancer showed that NE knockout severely limits tumor growth and proliferation. This study also identified another intracellular target of NE in mouse and human cells, the insulin receptor substrate-1 (IRS-1), which increases phosphatidylinositol 3-kinase (PI3K) activity through enhanced association with the platelet-derived growth factor receptor, ultimately promoting Akt activation (94). Our group has evaluated the role of NE in the C3(1)/Tag mouse model of triple-negative breast cancer (TNBC), reproducing the finding in the lung cancer model that NE-knockout reduces tumor growth and proliferation with no obvious effect on metastasis (95). In these models, the relative importance of NE-mediated pro-tumorigenic mechanisms (activation of PI3K, generation of LMW-E, enhanced inflammation etc.) is an active area of investigation.
In vitro models demonstrate that NE can undergo endocytosis following its association with the cancer cell surface receptor neurophilin-1 (96) in a clathrin- and dynamin-dependent manner (97). Uptake of NE from the extracellular environment has been shown to result in generation of LMW-E (68), as well as, degradation of IRS-1 (98). Serine protease inhibitors are essential to the regulation of NE activity. Elafin, an endogenous WAP-domain-containing NE inhibitor, is downregulated in invasive breast cancer compared with breast tissue from disease-free donors (99,100). Downregulation of elafin sensitizes HMECs to exogenous NE-induced proliferation, suggesting that elafin is a counterbalance against the mitogenic effects of NE, including the intracellular generation of LMW-E (95).
Oncogenic Functions of LMW-E
Overexpression of LMW-E and FL-cyclin E are not mutually exclusive, and both have oncogenic properties. However, we posit that LMW-E has several properties that make it a particularly potent mediator of cell cycle deregulation and tumorigenesis when compared with FL-cyclin E. These properties of LMW-E include hyperactivation of CDK2, altered substrate interactions, and novel interactions within the cytoplasmic compartment, as described in more detail below.
[1]. Hyperactivation of CDK2 (Figure 1b):
Compared with FL-cyclin E, LMW-E has a higher binding affinity for CDK2 (101). Furthermore, LMW-E/CDK2 complexes cannot be inhibited by p21CIP1 or p27KIP1 (102). These factors contribute to significantly higher levels of CDK2-associated kinase activity when CDK2 is in complex with LMW-E compared to FL-cyclin E (66,103). Furthermore, LMW-E is less susceptible than FL-cyclin E to nuclear FBW7-mediated ubiquitin-directed proteosomal degradation, and thus is more stable than FL-cyclin E (104,105). Although LMW-E accumulates in the cytoplasm, it can shuttle to the nucleus when bound to CDK2 and phosphorylate conventional targets required for S-phase progression (e.g. pRB). In a three-dimensional mammary epithelial culture system, the overexpression of hyperactive LMW-E disrupted acinar morphogenesis, enhanced proliferation, and increased tumorigenicity as compared with FL-cyclin E. In this system, LMW-E phenotypes were dependent on their ability to hyperactivate CDK2 (106). In HMECs, the sustained phosphorylation of histone acetyltransferase HBO1 by hyperactive LMW-E-CDK2, but not FL-cyclin E-CDK2, promotes the enrichment of cells with epithelial-to-mesenchymal transition (EMT) and cancer-stem-like phenotypes in an HBO-1-dependent manner (Figure 1c) (107).
[2]. Altered Substrate Interactions:
Novel and atypical properties that arise from the altered structure of LMW-E may be similarly consequential in enhancing tumorigenesis. Although CDKs are the catalytic subunit, interactions with substrates are governed in part by the individual cyclins. Given that LMW-E isoforms are missing a significant part of the N-terminus, one would predict that LMW-E and FL-cyclin E would have different substrate interactions when in complex with CDK2. This is illustrated by the interaction between CDK2 and CDC25c (Figure 1c). The inducible overexpression of either LMW-E or FL-cyclin E induces CDC25c phosphorylation, increasing its activity. CDC25c promotes the activation of cyclin-B-CDK1 and PLK1, causing premature mitotic entry in cyclin E overexpressing cells (108,109). LMW-E-overexpressing cells, however, fail to sustain CDC25c phosphorylation/activity and thus exit mitosis much faster than control cells (not overexpressing any form of cyclin E). In stark contrast, cells overexpressing FL-cyclin E maintain high levels of CDC25c phosphorylation and activity, which sustains hyperactivation of CDK1 and PLK1 and thereby substantially delays mitotic exit as compared with control cells (108). The significant shortening of mitosis in LMW-E overexpressing cells prevents proper chromosome segregation, promotes cytokinesis failure, and leads to the generation of multinucleated cells with supernumerary centrosomes (Figure 1d). These defects are not seen at high levels with overexpression of FL-cyclin E (109,110). One study of 331 breast cancer patients revealed a significant correlation between high LMW-E expression, centrosome amplification, and polyploidy (p = 0.0003). Patients with these features had significantly lower rates of disease-specific survival (p = 0.02) (109).
[3]. Novel Interactions of Cytoplasmic LMW-E (Figure 1e):
FL-cyclin E is normally translated in the cytoplasm, where it forms a complex with CDK2, and is then transported to the nucleus via the importin-dependent pathway (71,72,111). The nuclear localization signal sequence is found at the cyclin E gene’s N-terminus, which is cleaved off in the LMW-E isoforms (112). Fractionation and protein complementation assays demonstrate that the LMW-E-CDK2 complexes preferentially accumulate in the cytoplasm (104). Accumulation of LMW-E-CDK2 in the cytoplasm provides this complex the opportunity to interact with novel partners that FL-cyclin E-CDK2 would not encounter in the nucleus. One recent study identified ATP-citrate lyase (ACLY), an enzyme required for the conversion of citrate to oxaloacetic acid and acetyl-CoA, as a novel LMW-E–binding protein in the cytoplasmic compartment (113). Acetyl-CoA is required for a diverse set of cellular processes, including the synthesis of lipids for membrane biogenesis and histone acetylation. ACLY activity is higher in tumors than in disease-free tissues and it plays an important role in tumorigenesis (114–116). Interaction with LMW-E increases ACLY activity, thereby increasing lipid levels in the cells in a CDK2-independent fashion. That LMW-E has CDK2-independent functions is not surprising, as FL-cyclin E has several such roles as well (e.g., in the initiation of DNA replication and centrosomal duplication) (26). Experimentally, purified FL-cyclin E can also interact with ACLY. In cells, however, FL-cyclin E is largely confined to the nucleus and ACLY resides in the cytoplasm, therefore LMW-E has greater opportunity to alter ACLY activity than FL-cyclin E. Significantly, ACLY activity has important roles in tumorigenesis, and its knockdown attenuates several oncogenic features of LMW-E-expressing cells (113).
Cytoplasmic mislocalization may be a common feature of cyclin deregulation in cancer. Cyclin D1, which lacks a canonical nuclear localization signal, is known to shuttle in complex with CDKs between the nucleus and cytoplasm at certain stages of the cell cycle. Cyclin D1 overexpression dramatically increases the cytoplasmic pool of cyclin D1 (117). Evidence suggests that excessive cytoplasmic cyclin D1 has important oncogenic effects, including increased cell migration and invasion (118,119), through direct interactions with several components of the cytoskeleton and cell migration machinery, including paxillin phosphorylation, which increases Rac1 activity (120,121).
Tumorigenic and pro-metastatic activity of LMW-E
Both primary mammary tumor formation and metastasis are markedly enhanced in LMW-E transgenic mice as compared with FL-cyclin E-overexpressing mice. LMW-E overexpression in the mammary gland is sufficient to induce mammary adenocarcinomas in 27% of mice, whereas FL-cyclin E expression in the mammary gland induces mammary adenocarcinomas in only 10.4% of the transgenic mice (54,122). Metastasis was observed in 25% of LMW-E tumor-bearing animals compared with only 8.3% of animals with tumors arising in the FL-cyclin E background. In this model, LMW-E overexpression results in the spontaneous loss of heterozygosity at the p14ARF locus, suggesting that LMW-E cooperates with p53 inactivation to generate an aggressive tumor phenotype. CDK2 wild-type or heterozygous mice succumbed to mammary tumors with mean latencies of 16 and 19.5 months, respectively, whereas CDK2 nullizygous littermates did not display tumors through 24 months of observation (123). Furthermore, continuous administration of two different CDK inhibitors significantly delayed LMW-E-induced mammary tumor formation. Examination of breast cancer specimens using immunohistochemistry reveal that about half the patients whose tumors express LMW-E (i.e., cytoplasmic cyclin E) but no detectable levels of FL-cyclin E (nuclear) (described in more detail in the section to follow) (124–126). Therefore, a critical direction for future research should be the development of novel mouse models of LMW-E expression in the absence of endogenous mouse cyclin E, which may reveal new biology and increase our understanding of the clinical problem (71).
Future Directions: The Translational Potential of LMW-E
The prognostic significance of LMW-E:
Western blot analysis, the primary assay for evaluating LMW-E in experimental systems, is impractical for routine clinical use. Given the cytoplasmic mislocalization of LMW-E, we hypothesized that an immunohistochemical assay designed to score the nuclear and cytoplasmic compartments for expression of cyclin E and phosphorylated CDK2 (p-CDK2) would provide a surrogate measure of LMW-E expression in human tumor specimens with the potential for clinical application. Once developed, this immunohistochemical analysis was applied to tumor samples from 1676 breast carcinoma patients (124). In this study, cytoplasmic cyclin E strongly correlated with cytoplasmic p-CDK2, high tumor grade, estrogen receptor (ER)-negative status, progesterone receptor (PR)-negative status, and human epidermal growth factor receptor 2 (HER2)-positive status (all p < 0.0001) (124). In multivariable analysis, LMW-E and p-CDK2 predicted recurrence-free and overall survival, suggesting that this assay could be used to identify patients with aggressive breast cancers (124). Recently (126), we expanded this analysis to 2,494 breast cancer patients from four different patient cohorts. In multivariable analysis, cytoplasmic cyclin E staining was associated with the greatest risk of recurrence across all breast cancer subtypes. Collectively, these studies suggest that cytoplasmic cyclin E is likely to identify patients with the highest likelihood of recurrence consistently across different patient cohorts and breast cancer subtypes. Standardization and Clinical Laboratory Improvement Amendments (CLIA) certification of this assay will facilitate the stratification of patients for LMW-E or FL-cyclin E-directed therapies.
Cytoplasmic cyclin E as a predictive biomarker:
In experimental models, LMW-E mediates resistance to endocrine therapies such as aromatase inhibitors and fulvestrant (110,127). In one study, breast cancer patients with LMW-E overexpressing tumors (cytoplasmic cyclin E) who received aromatase inhibitors in the neo-adjuvant setting had significantly worse recurrence-free survival than did patients whose tumors did not express cytoplasmic cyclin (128). Recently, three CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) in combination with either letrozole or fulvestrant have been evaluated in clinical trials for the treatment of advanced hormone receptor positive (HR+) breast cancer (129,130). These trials showed that patients who received a CDK4/6 inhibitor in addition to letrozole or fulvestrant had progression-free survival durations that were at least twice those of patients who received letrozole or fulvestrant alone (131–133). The US Food and Drug Administration (FDA) has now approved all three CDK4/6 inhibitors for the treatment of post-menopausal women with HR positive, HER2-negative metastatic breast cancer (134–137). Despite these promising clinical advances, a major limitation in the use of CDK4/6 inhibitors is the lack of reliable biomarkers to identify patients with intrinsic and/or acquired resistance to these agents. Although previous in vitro studies have shown that pRb, cyclin D, and p16 could predict response to palbociclib (138–140), results from phase II/III trials have not shown a significant correlation between response and the expression of p16, pRb (131), and Ki67 expression; CCND1 amplification (141); or (3) PIK3CA and ESR1 (142) mutational status (143). A recent study from our group demonstrated that together pRb and LMW-E status predicts response in HR-positive breast cancer patients treated with palbociclib (144). Specifically, a multivariable Cox proportional hazards model showed that pRb and LMW-E were the only factors significantly associated with the progression-free interval in both patients treated with palbociclib plus letrozole and those treated with palbociclib plus fulvestrant, with hazard ratios of 0.2 and 0.09 (pRb-negative), 3.2 and 5.2 (LMW-E–positive), and 9.2 and 23.8 (both pRb-negative and LMW-E–positive) respectively (144). These studies suggest that an immunohistochemical assay for pRb and LMW-E can be used clinically to identify patients who are likely to have a sustained response to CDK4/6 inhibitors in combination with endocrine therapy.
Targeting LMW-E overexpressing tumors (Figure 2).
Figure 2: The prognostic significance of LMW-E.
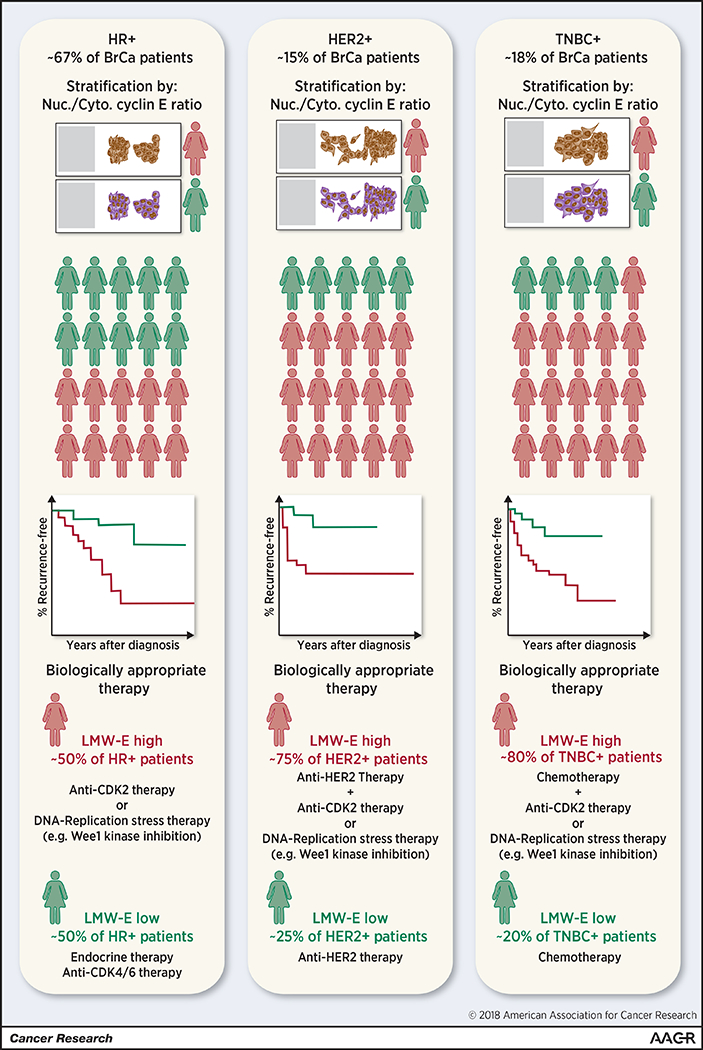
Clinically, breast cancers are stratified into three groups, HR positive breast cancer, HER2 positive breast cancer, and TNBC based on the pathohistological assessment the of ER, PR, and HER2 expression. These breast tumor types are characterized by dramatic differences in clinical course and are treated using tailored therapeutic approaches. Immunohistochemical analysis of cyclin E or phosphorylated CDK2 and scoring according to the nuclear (FL)-to-cytoplasmic (LMW) cyclin E ratio identifies a patient population in each breast cancer subtype expressing high levels of LMW-E relative to FL cyclin E (approximately 50% of HR positive cancers, 75% of HER2 positive cancers, and 80% of TNBC). Within each of these subtypes, patients whose tumors express high LMW-E relative to FL-cyclin E have significantly worse survival outcomes than patients whose tumors predominantly express FL cyclin E. Data reviewed here suggest that LMW-E-expressing tumors are resistant to commonly used targeted therapeutics and may benefit from a combination of current therapeutic approaches with either anti-CDK2 based therapy or therapeutic strategies targeting specific vulnerabilities of LMW-E overexpressing tumors.
Studies in pre-clinical models suggest that tumors that are resistant to CDK4/6 inhibition (e.g. pRb-negative and LMW-E positive tumors) (144) or that become resistant to endocrine therapy (145) are likely to respond to CDK2 inhibitors such as roscovitine or dinaciclib. CDK2 may also be a therapeutic target in other aggressive tumor types. In a model of treatment-naïve aggressive HER2-positive breast cancer cells expressing LMW-E, the combination of trastuzumab (targeting HER2) and roscovitine led to synergistic cell killing (146). The administration of roscovitine or dinaciclib prior to treatment with doxorubicin is also synthetically lethal in both TNBC xenografts (147) and inflammatory breast cancer cell lines (125) expressing high levels of LMW-E. In addition, the specific knockdown of CDK2 significantly inhibits LMW-E driven tumor proliferation and increases apoptosis (123). The development of inhibitors that specifically target CDK2 without inhibiting other CDKs is critical to a comprehensive therapeutic approach targeting the deregulated cell cycle. Such inhibitors could be translated into biomarker (cytoplasmic cyclin E)-driven clinical trials.
A significant problem with the current generation of CDK2 inhibitors is their lack of specificity and their associated toxicity. A combination therapy approached could be used to reduce the toxicity of current CDK inhibitors by maximizing the therapeutic effect and thereby shortening the treatment duration. For example, the combination of the PLK4 inhibitor CFI-400945 and roscovitine can, by generating supernumerary centrosomes and inhibiting centrosome clustering respectively, synergistically cause multipolar anaphase catastrophe and death in lung cancer cells (148). Another possibility is to target the specific vulnerabilities of tumors with high LMW-E and FL-cyclin E expression. Such tumors, for example, have high levels of replication stress and DNA damage. The inhibition of Wee1 kinase, which normally blocks mitosis by specifically phosphorylating CDK1, would force LMW-E and FL-cyclin E-expressing tumor cells with high levels of unrepaired DNA damage into the cell cycle, causing many to undergo mitotic catastrophe and cell death. NE-directed therapies (149), developed for the treatment of inflammatory disease (e.g. chronic obstructive pulmonary disease), could also be repurposed to specifically inhibit the generation of LMW-E and the activation of other pro-tumorigenic pathways in tumor cells. Overall, LMW-E-expressing tumors, which can be identified on the basis of cytoplasmic cyclin E expression, are among the most aggressive tumors, particularly in breast cancer patients. Identifying treatment strategies that are effective for patients with these tumors is a top priority.
Supplementary Material
Acknowledgments
Financial Support
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health through grants to K.K. (CA1522218, CA223772) and MD Anderson’s Cancer Center Support Grant (P30CA016672); by the Cancer Prevention and Research Institute of Texas through a grant to K.K. (RP170079); and by Susan G. Komen for the Cure through a grant to K.K.H. (KG100521) and a post-doctoral fellowship grant to J.P.W.C. (PDF14302675).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A 1974;71:1286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardee AB. G1 events and regulation of cell proliferation. Science 1989;246:603–8. [DOI] [PubMed] [Google Scholar]
- 3.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 2008;9:910–6. [DOI] [PubMed] [Google Scholar]
- 4.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994;266:1821–8. [DOI] [PubMed] [Google Scholar]
- 5.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene 2005;24:2909–15. [DOI] [PubMed] [Google Scholar]
- 6.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001;1:222–31. [DOI] [PubMed] [Google Scholar]
- 7.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009;9:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002;2:910–7. [DOI] [PubMed] [Google Scholar]
- 9.Koff A, Giordano A, Desia D, Yamashita K, Harper JW, Elledge SJ, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992;257:1689–94. [DOI] [PubMed] [Google Scholar]
- 10.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997;11:1464–78. [DOI] [PubMed] [Google Scholar]
- 11.Zhu H, Nie L, Maki CG. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J Biol Chem 2005;280:29282–8. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004;430:226–31. [DOI] [PubMed] [Google Scholar]
- 13.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 2007;26:131–43. [DOI] [PubMed] [Google Scholar]
- 14.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001;294:173–7. [DOI] [PubMed] [Google Scholar]
- 15.Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, et al. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev 2000;14:2283–97. [PMC free article] [PubMed] [Google Scholar]
- 16.Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, et al. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 2000;14:2298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 2002;3:339–50. [DOI] [PubMed] [Google Scholar]
- 18.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000;103:127–40. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto Y, Maller JL. A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science 2004;306:885–8. [DOI] [PubMed] [Google Scholar]
- 20.Rao H, Stillman B. The origin recognition complex interacts with a bipartite DNA binding site within yeast replicators. Proc Natl Acad Sci U S A 1995;92:2224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furstenthal L, Kaiser BK, Swanson C, Jackson PK. Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J Cell Biol 2001;152:1267–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coverley D, Laman H, Laskey RA. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol 2002;4:523–8. [DOI] [PubMed] [Google Scholar]
- 23.Weiss A, Herzig A, Jacobs H, Lehner CF. Continuous Cyclin E expression inhibits progression through endoreduplication cycles in Drosophila. Curr Biol 1998;8:239–42. [DOI] [PubMed] [Google Scholar]
- 24.Follette PJ, Duronio RJ, O’Farrell PH. Fluctuations in cyclin E levels are required for multiple rounds of endocycle S phase in Drosophila. Curr Biol 1998;8:235–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, et al. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. Embo J 2003;22:4794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, et al. Cyclin E ablation in the mouse. Cell 2003;114:431–43. [DOI] [PubMed] [Google Scholar]
- 27.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol 2003;13:1775–85. [DOI] [PubMed] [Google Scholar]
- 28.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science 1993;262:2050–4. [DOI] [PubMed] [Google Scholar]
- 29.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A 1993;90:1112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuse T, Hirata T, Naiki H, Hitomi S, Wada H. Prognostic significance of cyclin E overexpression in resected non-small cell lung cancer. Cancer Res 2000;60:242–4. [PubMed] [Google Scholar]
- 31.Leach FS, Elledge SJ, Sherr CJ, Willson JK, Markowitz S, Kinzler KW, et al. Amplification of cyclin genes in colorectal carcinomas. Cancer Res 1993;53:1986–9. [PubMed] [Google Scholar]
- 32.Muller-Tidow C, Metzger R, Kugler K, Diederichs S, Idos G, Thomas M, et al. Cyclin E is the only cyclin-dependent kinase 2-associated cyclin that predicts metastasis and survival in early stage non-small cell lung cancer. Cancer Res 2001;61:647–53. [PubMed] [Google Scholar]
- 33.Erlanson M, Landberg G. Prognostic implications of p27 and cyclin E protein contents in malignant lymphomas. Leuk Lymphoma 2001;40:461–70. [DOI] [PubMed] [Google Scholar]
- 34.Iida H, Towatari M, Tanimoto M, Morishita Y, Kodera Y, Saito H. Overexpression of cyclin E in acute myelogenous leukemia. Blood 1997;90:3707–13. [PubMed] [Google Scholar]
- 35.Yasui W, Akama Y, Kuniyasu H, Yokozaki H, Semba S, Shimamoto F, et al. Expression of cyclin E in human gastric adenomas and adenocarcinomas: correlation with proliferative activity and p53 status. J Exp Ther Oncol 1996;1:88–94. [PubMed] [Google Scholar]
- 36.Akama Y, Yasui W, Yokozaki H, Kuniyasu H, Kitahara K, Ishikawa T, et al. Frequent amplification of the cyclin E gene in human gastric carcinomas. Jpn J Cancer Res 1995;86:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molendini L, Benassi MS, Magagnoli G, Merli M, Sollazzo MR, Ragazzini P, et al. Prognostic significance of cyclin expression in human osteosarcoma. Int J Oncol 1998;12:1007–11. [PubMed] [Google Scholar]
- 38.Schraml P, Bucher C, Bissig H, Nocito A, Haas P, Wilber K, et al. Cyclin E overexpression and amplification in human tumours. J Pathol 2003;200:375–82. [DOI] [PubMed] [Google Scholar]
- 39.Au-Yeung G, Lang F, Azar WJ, Mitchell C, Jarman KE, Lackovic K, et al. Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition. Clin Cancer Res 2017;23:1862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huber AR, Tan D, Sun J, Dean D, Wu T, Zhou Z. High expression of carbonic anhydrase IX is significantly associated with glandular lesions in gastroesophageal junction and with tumorigenesis markers BMI1, MCM4 and MCM7. BMC Gastroenterol 2015;15:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller CT, Moy JR, Lin L, Schipper M, Normolle D, Brenner DE, et al. Gene amplification in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clin Cancer Res 2003;9:4819–25. [PubMed] [Google Scholar]
- 42.DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S, et al. The genetic landscape of endometrial clear cell carcinomas. J Pathol 2017;243:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu YP, Kohaar I, Moore LE, Lenz P, Figueroa JD, Tang W, et al. The 19q12 bladder cancer GWAS signal: association with cyclin E function and aggressive disease. Cancer Res 2014;74:5808–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandhu V, Wedge DC, Bowitz Lothe IM, Labori KJ, Dentro SC, Buanes T, et al. The Genomic Landscape of Pancreatic and Periampullary Adenocarcinoma. Cancer Res 2016;76:5092–102. [DOI] [PubMed] [Google Scholar]
- 45.Piao J, Sun J, Yang Y, Jin T, Chen L, Lin Z. Target gene screening and evaluation of prognostic values in non-small cell lung cancers by bioinformatics analysis. Gene 2018;647:306–11. [DOI] [PubMed] [Google Scholar]
- 46.Martin LG, Demers GW, Galloway DA. Disruption of the G1/S transition in human papillomavirus type 16 E7-expressing human cells is associated with altered regulation of cyclin E. J Virol 1998;72:975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997;387:422–6. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Roger I, Solomon DL, Sewing A, Land H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene 1997;14:2373–81. [DOI] [PubMed] [Google Scholar]
- 49.Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 2001;413:311–6. [DOI] [PubMed] [Google Scholar]
- 50.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001;413:316–22. [DOI] [PubMed] [Google Scholar]
- 51.Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol 2003;163:1255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ge Z, Leighton JS, Wang Y, Peng X, Chen Z, Chen H, et al. Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Rep 2018;23:213–26 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gray-Bablin J, Zalvide J, Fox MP, Knickerbocker CJ, DeCaprio JA, Keyomarsi K. Cyclin E, a redundant cyclin in breast cancer. Proc Natl Acad Sci U S A 1996;93:15215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bortner DM, Rosenberg MP. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol 1997;17:453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haas K, Johannes C, Geisen C, Schmidt T, Karsunky H, Blass-Kampmann S, et al. Malignant transformation by cyclin E and Ha-Ras correlates with lower sensitivity towards induction of cell death but requires functional Myc and CDK4. Oncogene 1997;15:2615–23. [DOI] [PubMed] [Google Scholar]
- 56.Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 1999;283:851–4. [DOI] [PubMed] [Google Scholar]
- 57.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature 1999;401:297–300. [DOI] [PubMed] [Google Scholar]
- 58.Sutter T, Dansranjavin T, Lubinski J, Debniak T, Giannakudis J, Hoang-Vu C, et al. Overexpression of cyclin E protein is closely related to the mutator phenotype of colorectal carcinoma. Int J Colorectal Dis 2002;17:374–80. [DOI] [PubMed] [Google Scholar]
- 59.Casimiro MC, Crosariol M, Loro E, Ertel A, Yu Z, Dampier W, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. J Clin Invest 2012;122:833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget 2012;3:224–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casimiro MC, Di Sante G, Crosariol M, Loro E, Dampier W, Ertel A, et al. Kinase-independent role of cyclin D1 in chromosomal instability and mammary tumorigenesis. Oncotarget 2015;6:8525–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Casimiro MC, Arnold A, Pestell RG. Kinase independent oncogenic cyclin D1. Aging (Albany NY) 2015;7:455–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011;145:435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, et al. Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 2000;19:1635–46. [DOI] [PubMed] [Google Scholar]
- 65.Geisen C, Moroy T. The oncogenic activity of cyclin E is not confined to Cdk2 activation alone but relies on several other, distinct functions of the protein. J Biol Chem 2002;277:39909–18. [DOI] [PubMed] [Google Scholar]
- 66.Porter DC, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruki S, et al. Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol 2001;21:6254–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harwell RM, Porter DC, Danes C, Keyomarsi K. Processing of cyclin E differs between normal and tumor breast cells. Cancer Res 2000;60:481–9. [PubMed] [Google Scholar]
- 68.Mittendorf EA, Alatrash G, Qiao N, Wu Y, Sukhumalchandra P, St John LS, et al. Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res 2012;72:3153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang XD, Rosales JL, Magliocco A, Gnanakumar R, Lee KY. Cyclin E in breast tumors is cleaved into its low molecular weight forms by calpain. Oncogene 2003;22:769–74. [DOI] [PubMed] [Google Scholar]
- 70.Libertini SJ, Robinson BS, Dhillon NK, Glick D, George M, Dandekar S, et al. Cyclin E both regulates and is regulated by calpain 2, a protease associated with metastatic breast cancer phenotype. Cancer Res 2005;65:10700–8. [DOI] [PubMed] [Google Scholar]
- 71.Loeb KR, Chen X. Too much cleavage of cyclin E promotes breast tumorigenesis. PLoS Genet 2012;8:e1002623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moore JD. In the wrong place at the wrong time: does cyclin mislocalization drive oncogenic transformation? Nature reviews Cancer 2013;13:201–8. [DOI] [PubMed] [Google Scholar]
- 73.Bedrosian I, Lu KH, Verschraegen C, Keyomarsi K. Cyclin E deregulation alters the biologic properties of ovarian cancer cells. Oncogene 2004;23:2648–57. [DOI] [PubMed] [Google Scholar]
- 74.Davidson B, Skrede M, Silins I, Shih Ie M, Trope CG, Florenes VA. Low-molecular weight forms of cyclin E differentiate ovarian carcinoma from cells of mesothelial origin and are associated with poor survival in ovarian carcinoma. Cancer 2007;110:1264–71. [DOI] [PubMed] [Google Scholar]
- 75.Bales E, Mills L, Milam N, McGahren-Murray M, Bandyopadhyay D, Chen D, et al. The low molecular weight cyclin E isoforms augment angiogenesis and metastasis of human melanoma cells in vivo. Cancer Res 2005;65:692–7. [PubMed] [Google Scholar]
- 76.Corin I, Di Giacomo MC, Lastella P, Bagnulo R, Guanti G, Simone C. Tumor-specific hyperactive low-molecular-weight cyclin E isoforms detection and characterization in non-metastatic colorectal tumors. Cancer Biol Ther 2006;5:198–203. [DOI] [PubMed] [Google Scholar]
- 77.Milne AN, Carvalho R, Jansen M, Kranenbarg EK, van de Velde CJ, Morsink FM, et al. Cyclin E low molecular weight isoforms occur commonly in early-onset gastric cancer and independently predict survival. J Clin Pathol 2008;61:311–6. [DOI] [PubMed] [Google Scholar]
- 78.Corin I, Larsson L, Bergstrom J, Gustavsson B, Derwinger K. A study of the expression of Cyclin E and its isoforms in tumor and adjacent mucosa, correlated to patient outcome in early colon cancer. Acta Oncol 2010;49:63–9. [DOI] [PubMed] [Google Scholar]
- 79.Zhou YJ, Xie YT, Gu J, Yan L, Guan GX, Liu X. Overexpression of cyclin E isoforms correlates with poor prognosis in rectal cancer. Eur J Surg Oncol 2011;37:1078–84. [DOI] [PubMed] [Google Scholar]
- 80.Koutsami MK, Tsantoulis PK, Kouloukoussa M, Apostolopoulou K, Pateras IS, Spartinou Z, et al. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol 2006;209:512–21. [DOI] [PubMed] [Google Scholar]
- 81.Nauman A, Turowska O, Poplawski P, Master A, Tanski Z, Puzianowska-Kuznicka M. Elevated cyclin E level in human clear cell renal cell carcinoma: possible causes and consequences. Acta Biochim Pol 2007;54:595–602. [PubMed] [Google Scholar]
- 82.Keyomarsi K, O’Leary N, Molnar G, Lees E, Fingert HJ, Pardee AB. Cyclin E, a potential prognostic marker for breast cancer. Cancer Res 1994;54:380–5. [PubMed] [Google Scholar]
- 83.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med 2002;347:1566–75. [DOI] [PubMed] [Google Scholar]
- 84.Porter PL, Barlow WE, Yeh I-T, Lin MG, Yuan XP, Donato E, et al. p27Kip1 and Cyclin E Expression and Breast Cancer Survival After Treatment with Adjuvant Chemotherapy. J Natl Cancer Inst 2006;98:1723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Foekens JA, Ries C, Look MP, Gippner-Steppert C, Klijn JG, Jochum M. Elevated expression of polymorphonuclear leukocyte elastase in breast cancer tissue is associated with tamoxifen failure in patients with advanced disease. Br J Cancer 2003;88:1084–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foekens JA, Ries C, Look MP, Gippner-Steppert C, Klijn JG, Jochum M. The prognostic value of polymorphonuclear leukocyte elastase in patients with primary breast cancer. Cancer Res 2003;63:337–41. [PubMed] [Google Scholar]
- 87.Yamashita J, Ogawa M, Shirakusa T. Free-form neutrophil elastase is an independent marker predicting recurrence in primary breast cancer. J Leukoc Biol 1995;57:375–8. [DOI] [PubMed] [Google Scholar]
- 88.Akizuki M, Fukutomi T, Takasugi M, Takahashi S, Sato T, Harao M, et al. Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: long-term follow-up results in 313 patients. Neoplasia 2007;9:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grant AJ, Russell PJ, Raghavan D. Elastase activities of human bladder cancer cell lines derived from high grade invasive tumours. Biochem Biophys Res Commun 1989;162:308–15. [DOI] [PubMed] [Google Scholar]
- 90.Kamohara H, Sakamoto K, Mita S, An XY, Ogawa M. Neutrophil elastase inhibitor (ONO-5046.Na) suppresses the proliferation, motility and chemotaxis of a pancreatic carcinoma cell line, Capan-1. Res Commun Mol Pathol Pharmacol 1997;98:103–8. [PubMed] [Google Scholar]
- 91.Taniguchi K, Yang P, Jett J, Bass E, Meyer R, Wang Y, et al. Polymorphisms in the promoter region of the neutrophil elastase gene are associated with lung cancer development. Clin Cancer Res 2002;8:1115–20. [PubMed] [Google Scholar]
- 92.Yamashita JI, Ogawa M, Ikei S, Omachi H, Yamashita SI, Saishoji T, et al. Production of immunoreactive polymorphonuclear leucocyte elastase in human breast cancer cells: possible role of polymorphonuclear leucocyte elastase in the progression of human breast cancer. Br J Cancer 1994;69:72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008;90:227–42. [DOI] [PubMed] [Google Scholar]
- 94.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 2010;16:219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Caruso JA, Akli S, Pageon L, Hunt KK, Keyomarsi K. The serine protease inhibitor elafin maintains normal growth control by opposing the mitogenic effects of neutrophil elastase. Oncogene 2015;34:3556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kerros C, Tripathi SC, Zha D, Mehrens JM, Sergeeva A, Philips AV, et al. Neuropilin-1 mediates neutrophil elastase uptake and cross-presentation in breast cancer cells. J Biol Chem 2017;292:10295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gregory AD, Hale P, Perlmutter DH, Houghton AM. Clathrin Pit-mediated Endocytosis of Neutrophil Elastase and Cathepsin G by Cancer Cells. J Biol Chem 2012;287:35341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang M, Zou Z, Maass N, Sager R. Differential expression of elafin in human normal mammary epithelial cells and carcinomas is regulated at the transcriptional level. Cancer Res 1995;55:2537–41. [PubMed] [Google Scholar]
- 100.Caruso JA, Karakas C, Zhang J, Yi M, Albarracin C, Sahin A, et al. Elafin is downregulated during breast and ovarian tumorigenesis but its residual expression predicts recurrence. Breast Cancer Res 2014;16:3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wingate H, Puskas A, Duong M, Bui T, Richardson D, Liu Y, et al. Low molecular weight cyclin E is specific in breast cancer and is associated with mechanisms of tumor progression. Cell Cycle 2009;8:1062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wingate H, Zhang N, McGarhen MJ, Bedrosian I, Harper JW, Keyomarsi K. The tumor-specific hyperactive forms of cyclin E are resistant to inhibition by p21 and p27. J Biol Chem 2005;280:15148–57. [DOI] [PubMed] [Google Scholar]
- 103.Wingate H, Bedrosian I, Akli S, Keyomarsi K. The low molecular weight (LMW) isoforms of cyclin E deregulate the cell cycle of mammary epithelial cells. Cell Cycle 2003;2:461–6. [PubMed] [Google Scholar]
- 104.Delk NA, Hunt KK, Keyomarsi K. Altered subcellular localization of tumor-specific cyclin E isoforms affects cyclin-dependent kinase 2 complex formation and proteasomal regulation. Cancer Res 2009;69:2817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mull BB, Cox J, Bui T, Keyomarsi K. Post-translational modification and stability of low molecular weight cyclin E. Oncogene 2009;28:3167–76. [DOI] [PubMed] [Google Scholar]
- 106.Duong MT, Akli S, Wei C, Wingate HF, Liu W, Lu Y, et al. LMW-E/CDK2 deregulates acinar morphogenesis, induces tumorigenesis, and associates with the activated b-Raf-ERK1/2-mTOR pathway in breast cancer patients. PLoS Genet 2012;8:e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duong MT, Akli S, Macalou S, Biernacka A, Debeb BG, Yi M, et al. Hbo1 is a cyclin E/CDK2 substrate that enriches breast cancer stem-like cells. Cancer Res 2013;73:5556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bagheri-Yarmand R, Nanos-Webb A, Biernacka A, Bui T, Keyomarsi K. Cyclin E deregulation impairs mitotic progression through premature activation of Cdc25C. Cancer Res 2010;70:5085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bagheri-Yarmand R, Biernacka A, Hunt KK, Keyomarsi K. Low molecular weight cyclin E overexpression shortens mitosis, leading to chromosome missegregation and centrosome amplification. Cancer Res 2010;70:5074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, et al. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res 2004;64:3198–208. [DOI] [PubMed] [Google Scholar]
- 111.Moore JD, Yang J, Truant R, Kornbluth S. Nuclear import of Cdk/cyclin complexes: identification of distinct mechanisms for import of Cdk2/cyclin E and Cdc2/cyclin B1. J Cell Biol 1999;144:213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moore JD, Kornbluth S, Hunt T. Identification of the nuclear localization signal in Xenopus cyclin E and analysis of its role in replication and mitosis. Mol Biol Cell 2002;13:4388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lucenay KS, Doostan I, Karakas C, Bui T, Ding Z, Mills GB, et al. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res 2016;76:2406–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311–21. [DOI] [PubMed] [Google Scholar]
- 115.Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 2012;72:3709–14. [DOI] [PubMed] [Google Scholar]
- 116.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009;324:1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alao JP, Gamble SC, Stavropoulou AV, Pomeranz KM, Lam EW, Coombes RC, et al. The cyclin D1 proto-oncogene is sequestered in the cytoplasm of mammalian cancer cell lines. Mol Cancer 2006;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Body S, Esteve-Arenys A, Miloudi H, Recasens-Zorzo C, Tchakarska G, Moros A, et al. Cytoplasmic cyclin D1 controls the migration and invasiveness of mantle lymphoma cells. Sci Rep 2017;7:13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fuste NP, Ferrezuelo F, Gari E. Cyclin D1 promotes tumor cell invasion and metastasis by cytoplasmic mechanisms. Mol Cell Oncol 2016;3:e1203471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fuste NP, Fernandez-Hernandez R, Cemeli T, Mirantes C, Pedraza N, Rafel M, et al. Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat Commun 2016;7:11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meng H, Tian L, Zhou J, Li Z, Jiao X, Li WW, et al. PACSIN 2 represses cellular migration through direct association with cyclin D1 but not its alternate splice form cyclin D1b. Cell Cycle 2011;10:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Akli S, Van Pelt CS, Bui T, Multani AS, Chang S, Johnson D, et al. Overexpression of the low molecular weight cyclin E in transgenic mice induces metastatic mammary carcinomas through the disruption of the ARF-p53 pathway. Cancer Res 2007;67:7212–22. [DOI] [PubMed] [Google Scholar]
- 123.Akli S, Van Pelt CS, Bui T, Meijer L, Keyomarsi K. Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res 2011;71:3377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Karakas C, Biernacka A, Bui T, Sahin AA, Yi M, Akli S, et al. Cytoplasmic Cyclin E and Phospho-Cyclin-Dependent Kinase 2 Are Biomarkers of Aggressive Breast Cancer. Am J Pathol 2016;186:1900–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alexander A, Karakas C, Chen X, Carey JP, Yi M, Bondy M, et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget 2017;8:14897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hunt KK, Karakas C, Ha MJ, Biernacka A, Yi M, Sahin AA, et al. Cytoplasmic Cyclin E Predicts Recurrence in Patients with Breast Cancer. Clin Cancer Res 2017;23:2991–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Akli S, Bui T, Wingate H, Biernacka A, Moulder S, Tucker SL, et al. Low-molecular-weight cyclin E can bypass letrozole-induced G1 arrest in human breast cancer cells and tumors. Clin Cancer Res 2010;16:1179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Doostan I, Karakas C, Kohansal M, Low KH, Ellis MJ, Olson JA Jr., et al. Cytoplasmic Cyclin E Mediates Resistance to Aromatase Inhibitors in Breast Cancer. Clin Cancer Res 2017;23:7288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 2016;6:353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med 2016;375:1738–48. [DOI] [PubMed] [Google Scholar]
- 131.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25–35. [DOI] [PubMed] [Google Scholar]
- 132.Finn RS, Martin M, Rugo HS, Jones SE, Im S-A, Gelmon KA, et al. PALOMA-2: Primary results from a phase III trial of palbociclib (P) with letrozole (L) compared with letrozole alone in postmenopausal women with ER+/HER2– advanced breast cancer J Clin Oncol 2016;34:suppl; abstr 507. [Google Scholar]
- 133.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med 2015;373:209–19. [DOI] [PubMed] [Google Scholar]
- 134.Veronesi U, Salvadori B, Luini A, Greco M, Saccozzi R, del Vecchio M, et al. Breast conservation is a safe method in patients with small cancer of the breast. Long term results of three randomised trials on 1973 patients. Eur J Cancer 1995;31A:1574–9. [DOI] [PubMed] [Google Scholar]
- 135.Fisher B, Anderson S. Conservative surgery for the management of invasive and noninvasive carcinoma of the breast: NSABP trials. National Surgical Adjuvant Breast and Bowel Project. World J Surg 1994;18:63–9. [DOI] [PubMed] [Google Scholar]
- 136.FDA Breakthrough Therapy Designation to Abemaciclib for Breast Cancer. Oncology Times 2015;37:21. [Google Scholar]
- 137.Santella L The role of calcium in the cell cycle: facts and hypotheses. Biochemical & Biophysical Research Communications 1998;244:317. [DOI] [PubMed] [Google Scholar]
- 138.Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 2011;17:1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, Dunn GP, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A 2010;107:11501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro-oncology 2012;14:870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Clark AS, Karasic TB, DeMichele A, Vaughn DJ, O’Hara M, Perini R, et al. Palbociclib (PD0332991)-a Selective and Potent Cyclin-Dependent Kinase Inhibitor: A Review of Pharmacodynamics and Clinical Development. JAMA Oncol 2016;2:253–60. [DOI] [PubMed] [Google Scholar]
- 142.Turner NC, Jiang Y, O’Leary B, Hrebien S, Cristofanilli M, Andre F, et al. Efficacy of palbociclib plus fulvestrant (P+F) in patients (pts) with metastatic breast cancer (MBC) and ESR1 mutations (mus) in circulating tumor DNA (ctDNA). J Clin Oncol 2016;34:suppl; abstr 512. [Google Scholar]
- 143.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17:425–39. [DOI] [PubMed] [Google Scholar]
- 144.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Doostan I, Karakas C, Kohansal M, Low KH, Ellis MJ, Olson JA, et al. Cytoplasmic Cyclin E Mediates Resistance to Aromatase Inhibitors in Breast Cancer. Clin Cancer Res 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mittendorf EA, Liu Y, Tucker SL, McKenzie T, Qiao N, Akli S, et al. A novel interaction between HER2/neu and cyclin E in breast cancer. Oncogene 2010;29:3896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jabbour-Leung NA, Chen X, Bui T, Jiang Y, Yang D, Vijayaraghavan S, et al. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol Cancer Ther 2016;15:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kawakami M, Mustachio LM, Zheng L, Chen Y, Rodriguez-Canales J, Mino B, et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc Natl Acad Sci U S A 2018;115:1913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Groutas WC, Dou D, Alliston KR. Neutrophil elastase inhibitors. Expert Opin Ther Pat 2011;21:339–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.