

Abstract
The term amyloid has historically been used to describe fibrillar aggregates formed as the result of protein misfolding and that are associated with a range of diseases broadly termed amyloidoses. The discovery of ‘functional amyloids’ expanded the amyloid umbrella to encompass aggregates structurally similar to disease-associated amyloids but that engage in a variety of biologically useful tasks without incurring toxicity. The mechanisms by which functional amyloid systems ensure nontoxic assembly has provided insights into potential therapeutic strategies for treating amyloidoses. Some of the most-studied functional amyloids are ones produced by bacteria. Curli amyloids are extracellular fibers made by enteric bacteria that function to encase and protect bacterial communities during biofilm formation. Here we review recent studies highlighting microbial functional amyloid assembly systems that are tailored to enable the assembly of non-toxic amyloid aggregates.
Keywords: functional amyloid, protein misfolding, microbial amyloids, nature-inspired therapeutics, toxic oligomer
Introduction
Amyloid fibers are insoluble protein aggregates that share specific biophysical and chemical properties [1]. While the primary sequences of amyloid-forming proteins lack unifying signatures, most amyloid aggregates possess β-rich secondary structure where the β-strands align perpendicular to the long axis of the unbranched fibril [2]. Therefore, amyloids are best described structurally as ordered fibrillar aggregates with a repeating cross-strand structure. Amyloid formation is well known for its association with protein misfolding and neurodegenerative disease [2]. The amyloid-linked diseases include Alzheimer’s, Parkinson’s, type II diabetes and prion diseases, among many others [1,2]. In each case, the conversion of non-amyloid proteins to thermodynamically stable amyloids occurs as the result of protein misfolding [2].
However, amyloid formation is not always the product of protein misfolding and can sometimes be a positive contributor to cellular biology. ‘Functional’ amyloids capitalize on the intrinsic properties of the amyloid fold, including its thermodynamic stability, to facilitate various cellular tasks [3–9]. Functional amyloids form via dedicated pathways and the diversity of functional amyloids across cellular life suggest the amyloid fold represents an essential protein folding state. Recent studies have elucidated mechanisms of regulation for numerous functional amyloid and amyloid-like systems, including the discovery of spatially controlled nucleation mechanisms, biological amyloid inhibitors, reversibility of aggregation and protease cleavages which precipitate functional amyloid formation, controlling the precise cellular moment at which amyloid formation can occur [6,10–14, 107, 108]. A better understanding of the mechanisms employed by functional amyloid systems to mitigate the inherent toxicity of amyloid formation should inspire the development of novel therapeutics. This review will highlight two key features of functional amyloid assembly systems that differentiate them from disease-associated amyloids: 1) the rapid and efficient conversion of unstructured peptides into functional amyloid polymers, and 2) the structural integrity of functional amyloid fibers.
The path to amyloid can be toxic
Mature amyloid fibers are recognized for their stable, highly-repetitive, β-rich quaternary structure [2]. However, amyloidogenic peptides often start as intrinsically disordered structures that assemble into transient oligomeric states before reaching the amyloid state [1]. In fact, it is the oligomeric intermediates, rather than full-length fibers, that are the toxic species in several disease-associated amyloidoses [15–23]. The kinetics of amyloid formation is routinely measured in vitro using the amyloid-binding dye thioflavin-T (ThT) due to the ability of ThT to fluoresce upon intercalation between β-strands along the axis of growing fibers [24]. Monitoring the emission of ThT fluorescence typically reveals three distinct phases of amyloid polymerization: a lag phase, a growth phase, and a plateau phase (Figure 1a). The lag phase reports on a critical rate-limiting step during which fibril formation is undetectably low (Figure 1a). Early in the lag phase, primary nucleation dominates the process [25]. During primary nucleation, protein monomers aggregate to precipitate formation of a minimal ‘nucleus’ which is then poised to template the rapid conversion of additional subunits into the growing amyloid fiber. The elongation phase is characterized by sequential addition of non-fibrillar monomers or oligomers to the tip of the growing fiber [25]. Various kinetic models have been proposed to describe the major molecular events occurring during each phase [25]. The simplest of these models is the nucleated polymerization (NP) model, which was adapted from the model proposed for the formation of scaffolding proteins like actin, although more complex two-state kinetic models have also been proposed [26–30]. An overview of these mechanisms are shown in Figure 1b. For several disease-associated amyloids, the formation of amyloid can proceed via a pathway generating non-fibrillar, pre-amyloid oligomers which are cytotoxic [15–23].
Figure 1.
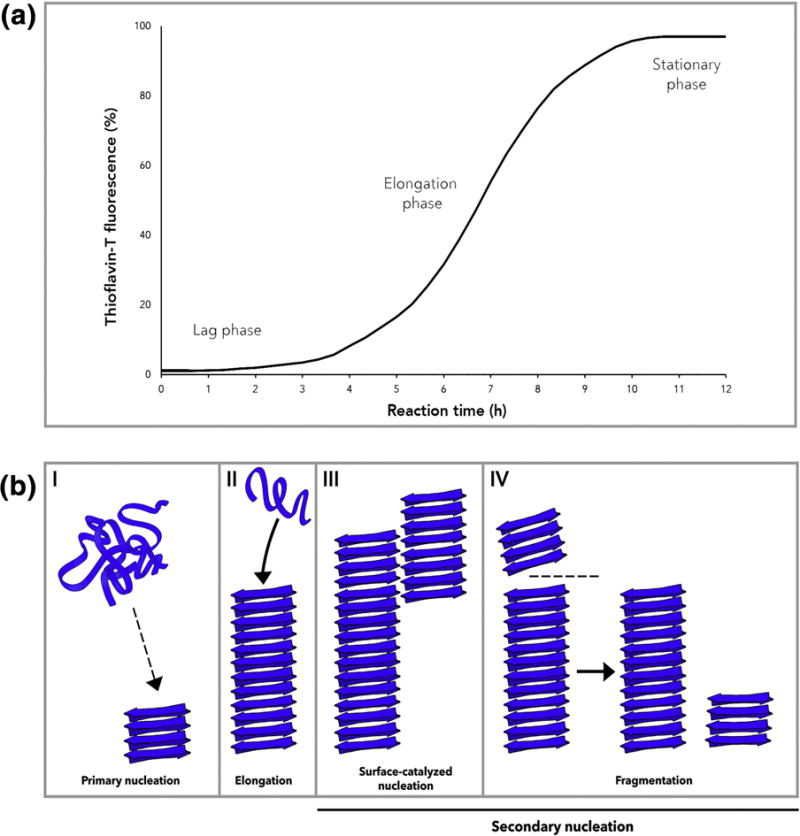
(A) Kinetics of amyloid polymerization can be monitored with ThT fluorescence [24]. Amyloid polymerization occurs via a rate-limiting lag phase, growth or elongation phase and stationary phase. (B) Mechanisms of amyloid polymerization. In (I) primary nucleation, protein monomers form a minimal nucleus competent for elongation [25]. (II) Elongation occurs when monomers add on to an existing fibril. (III) Surface-catalyzed nucleation and (IV) fragmentation describe two modes of secondary nucleation [26–28]. In surface-catalyzed nucleation, new fibrils are nucleated on the surface of an existing fibril in a monomer-dependent process [26,27]. Fragmentation occurs when an existing fibril breaks to produce two new independent fibrillar units capable of undergoing elongation [28].
The observation of structural polymorphisms, or variations in fibril structure, is an emerging characteristic of multiple disease-associated amyloids [31,32]. Structural polymorphisms have been observed for several disease-associated amyloids, including amyloid-β, α-synuclein, PrP and tau [33–40]. For functional amyloids that rely on predictable fibril architecture to participate in structural roles and ligand-binding activities, polymorphisms might produce unwanted or non-functional polymers that might liberate toxic species. The following sections will focus on recent studies revealing intrinsic mechanisms by which functional amyloid systems may circumvent the toxic pathways followed by disease-associated amyloids.
Rapid fibrillization by functional amyloids can limit cytotoxicity: Lessons from the curli system
If the production of toxic intermediates is a hallmark of disease-associated amyloid fibrillization, do functional amyloids limit the formation of amyloid intermediates? The assembly of curli amyloid fibers by enteric bacteria represents a finely tuned system for controlled amyloid polymerization [41]. Curli are extracellular functional amyloids that mediate host cell adhesion and provide structural integrity to bacterial communities called biofilms [3]. E. coli bacteria that express curli and form biofilms are resistant to many environmental stressors [42–51]. Curli biogenesis relies on tightly regulated transcriptional, secretory and assembly systems. Curli production is encoded by the csg operons, csgBAC and csgDEFG [52] (Figure 2). CsgD is the ‘master regulator’ of curli biogenesis, as it activates transcription of the csgBAC operon [53–55]. CsgA and CsgB are the major and minor curlin subunits, respectively [3,10,11,56]. CsgA and CsgB are both amyloid forming proteins, but are secreted across the bacterial outer membrane via the CsgG secretion complex as unstructured, monomeric subunits [57–59]. On the cell surface, CsgB is proposed to fold efficiently into a β-rich ‘nucleator’ due to electrostatic interactions with the outer membrane, assuming a conformation competent to template the rapid polymerization of CsgA [11,56,60,61]. The growth of curli amyloid fibers continues as additional CsgA molecules are secreted through the CsgG complex and interact with the growing fiber tip (Figure 2). CsgE and CsgF are accessory proteins that promote efficient secretion of CsgA across the outer membrane and nucleation into amyloid on the cell surface [62,63]. In the periplasmic space, the highly aggregation prone CsgA protein is maintained in a soluble and unstructured state by CsgC, a specific and potent periplasmic inhibitor of curli formation [13,14]. While this coordinated machinery is necessary to ensure that amyloid formation occurs at the right place and time, biophysical characteristics of curli fibers may also play a role in protecting the cell from the potentially detrimental effects associated with amyloid formation.
Figure 2.
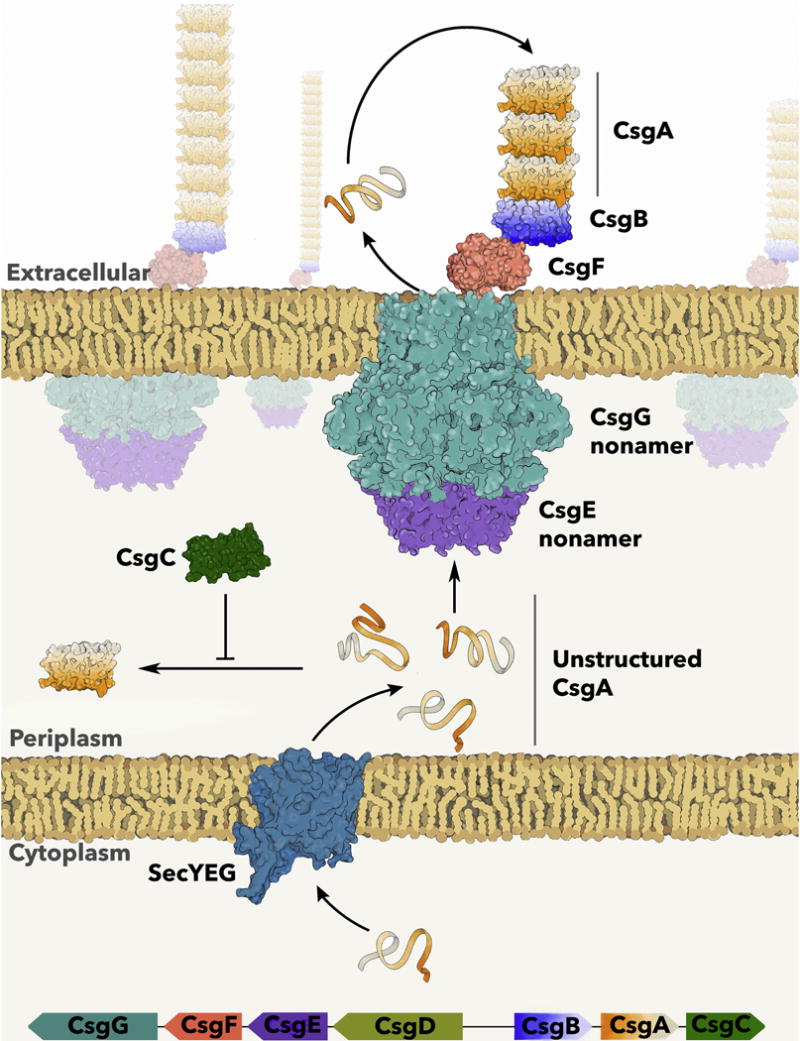
The csg operons encode the curli-specific genes [52]. CsgA is the major curlin subunit and is secreted into the periplasmic space in an unstructured conformation via SecYEG [3]. CsgA is maintained as unstructured in the periplasmic space by the amyloid inhibitor, CsgC [13]. Soluble CsgA is secreted to the outer membrane via the nonameric CsgG pore and nonameric CsgE adaptor complex [58,59,63]. CsgA assumes a β-sheet rich conformation upon interaction with the minor curlin subunit and nucleator protein, CsgB, which is tethered to CsgG via CsgF [11,56,60,62].
CsgA is secreted as an unstructured protein of roughly 13 kDa composed of five imperfect repeats, R1–R5. When incorporated in an amyloid fiber, it has been predicted that each repeat includes a strand-loop-strand motif which stacks between neighboring repeats, stabilized by hydrogen bonds in a β-helix [3,12,64,65,79,106] (Figure 3a). In this conformation, each CsgA monomer would contribute five consecutive, covalently linked β-strands to the fibril spine. Terminal repeats R1 and R5 mediate heteronucleation with the nucleator protein, CsgB, and self-association while internal repeats R2, R3 and R4 contain critical ‘gatekeeper residues’ which modulate amyloid formation [12,66]. The architecture predicted for each monomer in the fibrillar state is precisely organized to facilitate efficient, regulated nucleation. Each outer membrane-tethered CsgB monomer exposes a template for recognition by an R1 or R5 repeat on a recently secreted, unstructured CsgA monomer, potentially precipitating rapid association and adoption of β-sheet rich structure [12]. The exposure of R1 or R5 on the growing tip of a curli fiber would similarly contribute to efficient elongation, readily providing a recognition site for subsequently secreted CsgA monomers. While the precise mechanism of surface-catalyzed nucleation has not been elucidated, it is tempting to postulate that the necessity of an exposed lateral face of an R1 or R5 repeat to permit for the correct folding of a CsgA monomer into an amyloid-competent component potentially restricts the ability of CsgA to form surface-catalyzed fibrils with structural integrity comparable to that of fibrils nucleated from the cell surface.
Figure 3.
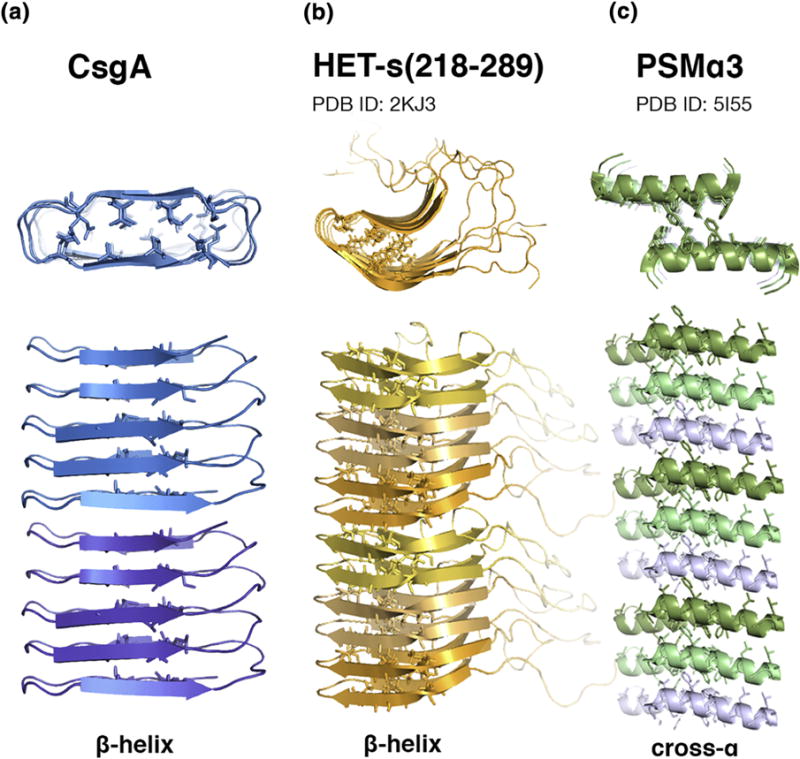
(A) The proposed structure of CsgA in the amyloid form assumes a β-helical conformation with a rectangular core [3,12,64,65,79,106]. (B) HET-s(218–289) forms a β-helical structure by stacking sets of ‘pseudo-repeats’ [81,89,90]. (C) The amyloid formed from PSMα3 peptide assumes a ‘cross-α’ structure [100].
Recent in vitro kinetic studies of CsgA polymerization have demonstrated the efficiency of unseeded nucleation [67,68]. CsgA polymerization monitored by ThT fluorescence fits a polymerization model consistent with primary homonucleation and elongation, notably excluding the presence of surface-catalyzed nucleation or fragmentation, which have been described as responsible for liberating toxic oligomeric species for some disease-associated amyloids [16,21,22,67]. By complementing kinetic modelling with dynamic imaging using atomic force microscopy (AFM), Sleutel et al. have recently been able to synthesize a comprehensive picture of CsgA polymerization in isolation [68]. While CsgA polymerization occurs via a characteristic lag time during which a significant population of fibers are undetectable monitoring ThT fluorescence, transmission electron microscopy (TEM) demonstrates individual fibers begin to appear almost immediately after CsgA is exchanged out of its denaturing purification buffer [68]. Results indicate that these fibers polymerize directly from unstructured CsgA monomers, lacking transition through a non-fibrillar, oligomeric intermediate [68,69]. Dynamic imaging of growing fibers using AFM demonstrates the formation of curled fibers capable of self-terminating with single growth poles which do not show evidence of surface-catalyzed nucleation [68]. Dynamic imaging further demonstrates the absence of a ‘minimal stable fragment size,’ or minimal nucleus size, for polymerizing CsgA, below which monomer assembly is unstable and results in nucleus breakdown, observable with the 10 nm resolution of the imaging technique [68]. For scale, their predicted width of folded CsgA is 3.2nm [68]. Together, these results compose a distinct picture of amyloid polymerization kinetics. CsgA polymerization occurs rapidly and does not occur via a non-fibrillar intermediate. The authors further postulate the nucleus size may approach the size of an individual monomer, as has been suggested previously [68,70]. This mechanism would dramatically lower the kinetic barrier for amyloid formation while permitting for rapid fibrillization without the generation of structurally divergent, toxic oligomers.
Curli Fibers Polymerize via Specific Nucleation and Adopt a Highly Stable Amyloid-like Fold
In contrast to the disease-associated amyloids, CsgA has not been shown to adopt polymorphic structures and can form ordered fibrils under a wide range of conditions [67]. Nucleation specificity and the overall stability of the mature curli amyloid fiber may be responsible for the structural integrity. CsgA nucleation by the dedicated nucleator protein, CsgB, provides a mechanism for curli fibers to be templated predictably and homogeneously [11]. CsgA and CsgB share approximately 30% sequence similarity and the first four repeats of CsgB share a motif similar to the β-strand-loop-β-strand motif repeated in the R1–R5 repeats of CsgA [11,60]. The first four repeats of CsgB lacking the fifth repeat which does not contain the CsgA-like motif, CsgBtrunc, is itself amyloidogenic and is able to effectively nucleate the polymerization of CsgA by reducing the lag time in vitro [11]. Notably, islet amyloid polypeptide protein (IAPP) fibers are unable to seed CsgA when incubated with CsgA at similar w/w% ratios, suggesting that nucleation of CsgA is at least somewhat specific [11]. The specific nucleation process by which CsgA rapidly polymerizes on the surface of E. coli facilitates homogenous fiber morphology and helps ensure precise temporal and spatial control of fiber growth.
In addition to polymerizing via a specific nucleation process, CsgA contains critical gatekeeper residues that modulate amyloid activity within the medial repeats R2, R3 and R4 [66]. While the terminal repeats R1 and R5 mediate CsgA-CsgA interactions and are interchangeable in vivo, R2, R3 and R4 contain residues that temper the amyloidogenicity of CsgA [66]. A CsgA mutant lacking its gatekeeper residues, called CsgA*, polymerizes more rapidly than CsgA and its overexpression in E. coli significantly reduces viability compared to CsgA [66]. Wang et al. propose that this decrease in viability could be due to increased propensity of CsgA* to aggregate relative to CsgA, suggesting that the gatekeeper residues may play a role in quenching fiber formation kinetics so that amyloid aggregates do not form at the wrong time or place [66].
Perhaps the most striking distinguishing structural feature of CsgA is its adoption of a fold distinct from the discontinuous in-register parallel β-sheet structure observed for many prion strains and disease-associated amyloids, including Aβ, amylin, tau, and α-synuclein [65,71–78]. Solid-state NMR and electron microscopy data reveal that CsgA does not adopt an in-register parallel β-sheet structure, but likely adopts a structure in which individual monomers adopt a β-helix or β-solenoid-like fold [65]. A β-helical structure for CsgA was constructed computationally and compared to experimental data [79]. In this model, CsgA monomers may form a left- or right-handed β-helix when integrated into a fiber with each of the five repeats wrapping around the sides of the enclosed hydrophobic ‘rectangular’ core while neighboring repeats participate in intermolecular stacking along the fibril axis within each subunit [79].
β-solenoids and β-helices are formed by coiling of β-strands and feature a characteristic hydrophobic core [80]. β-solenoids often function as viral or bacterial adhesins, exposing elongated lateral faces which may participate in binding activities [80]. While this fold is common in bacterial proteins, it has been suggested as a variant of an amyloid fold only fairly recently [65,79,81,82]. The adoption of this fold by functional amyloid systems could represent a means to mimic the efficiency and functionality of amyloid-like superstructure without the potentially risky adoption of an amyloid fold composed of discontinuous, mated β-sheets. If CsgA adopts a β-helical structure in curli fibers, the presence of five repeating units with sequence similarity forming the windings of the helix would support this proposed evolutionary mechanism [80]. Kajava et al. note that, as repetitive sequences experience deletions and duplications more rapidly than sequences without repetition, β-helical proteins composed of repetitive sequences may be common at the cell surface due to their ability to respond sensitively to environmental changes [80,83,84]. Evidence for the adoption of a β-helical fold by CsgA monomers within curli fibers in vivo would provide insight into the mechanism by which this functional amyloid may have evolved and remained nontoxic to its host organism.
The Prion-Forming Domain of HET-s Mimics a β-solenoid-like Fold and Auto-Inhibitory Mechanism
The β-helix-like amyloid fold also appears in the fibrillar form of the HET-s prion of Podospora anserina, which is required for a process known as heterokaryon incompatibility (Figure 3b) [78,85,86]. The het-s locus is one of several loci checked before colony fusion occurs, which is dependent on a critical number of loci in the genome being identical [78]. Incompatibility between the het-s and het-S loci is dependent on two distinct states of the gene products, the amyloid form, HET-s, and the non-amyloid form, HET-S, which vary by 13 residues [87]. Both HET-s and HET-S possess a C-terminal prion-forming domain from residues 218–289, which is amyloid-competent by itself, and an N-terminal globular domain (1–227) which overlap [88].
The solid-state NMR spectra of HET-s(218–289) fibrils reveals a β-solenoid structure, in which each molecule contributes two complete windings to the solenoid [81]. This structure has since been confirmed for the prion forming domain within the full-length HET-s prion [89,90]. Again, the β-solenoid HET-s structure differs from the in-register parallel β-sheet structure observed for many disease-associated amyloids [65,71–78,81]. The β-strands that comprise the HET-s β-helix have been termed ‘pseudo-repeats,’ as they share ~30% sequence identity with each other [81,82]. The presence of these pseudo-repeats in the β-strands of HET-s(218-289) is reminiscent of the repeats present in CsgA and CsgB and, like CsgA, the HET-s structure is stabilized by favorable intramolecular charge interactions between stacked residues [80,81]. Notably, structural polymorphisms have not been reported for HET-s fibrils.
The interaction between HET-s and HET-S may be directly responsible for mediating heterokaryon incompatibility [5,88]. Greenwald et al. suggest that HET-S can adopt a conformation competent to inhibit prion formation of HET-s in trans which is toxic or which promotes the formation of a toxic oligomer-like species with HET-s [88]. The inhibitory conformation adopted by HET-S reportedly ‘render[s] the seed or fibril sterile for further growth’ [88,90]. This mechanism is reminiscent of a broader class of solenoid-capping activities routinely observed to prevent polymerization of soluble proteins with solenoidal windings [80,91–96]. In summary, the structural integrity and fibril homogeneity of CsgA and HET-s(218–289) is maintained by intramolecular contacts that stabilize the fibril spine composed of stacked β-helices [65,81,89,90]. The ability of functional amyloids to assume a single, stable fibril architecture minimizes the risk of potentially toxic structural polymorphisms.
A Functional Amyloid Peptide of S. aureus PSMα3 Assumes a Unique Fibril Architecture
Phenol-soluble modulin α3 (PSMα3) belongs to a class of peptides that are involved in virulence and biofilm structuring in Staphylococcus aureus [97–99]. Biofilms enriched with PSMs show increased resistance to dispersal and decreased vulnerability to enzymatic degradation [97]. PSMα3 can also be highly cytotoxic [99]. Like other disease-associated and functional amyloids characterized to date, PSMα3 binds ThT, demonstrating time-dependent fibrillization, and binds the amyloid indicator dye Congo red [97]. In contrast to other amyloids for which structures have been determined, an amyloidogenic peptide derived from PSMα3 forms long, unbranched amyloid fibrils composed of stacked α-helices in a ‘cross-α’ spine (Figure 3c) [100]. In this fibril architecture, the helices are stacked in-register with a tightly associated, hydrophobic interface between paired sheets, strongly suggesting similarity to in-register cross-β amyloid spines [100]. Furthermore, the cytotoxicity of the PSMα3 peptide to HEK293 and human T cells is dependent upon fibrillization; α-helical secondary structure alone is not sufficient to confer cytotoxicity [100]. The uncommon ‘cross-α’ structure of PSMα3 peptide fibrils illuminates another example of a functional amyloid system building an amyloid-like scaffold to perform necessary biological activities via a specifically tailored and controlled structural mechanism circumventing autotoxicity [97,98,100].
Summary and Perspectives
Structural and kinetic insights into functional amyloid systems has revealed the blueprint for safely and efficiently assembling amyloid fibrils [65,79,81,100]. These fibrils exploit the functionality of amyloids by adopting an amyloid-like superstructure while potentially minimizing the risk of generating polymorphic structures and liberating toxic intermediates. A detailed understanding of the mechanisms by which functional amyloids circumvent cellular toxicity has applications for the rational design of therapeutics aimed at mitigating the toxicity of disease-associated amyloids. The two key intrinsic safety mechanisms employed by the functional amyloid systems described in this review are rapid fibrillization kinetics that bypass the formation of toxic oligomers, and the assumption of a single, stable fibril conformation without structural polymorphisms [67–69,79,81,89,90]. These risk-minimizing mechanisms prevent the formation of the proposed key determinants of toxicity conferred by disease-associated amyloids, outlining a strategy to therapeutically target amyloid-related toxicity by structurally stabilizing fibrillary aggregates or by expediting the fibrillization of amyloidogenic proteins [16–23,101–105].
Highlights.
Structural studies reveal unique fibril structures for microbial functional amyloids.
The unique and atypical fibril structures may help ensure structural integrity.
Curli fibers capitalize on intrinsic amyloid properties to fulfill biological functions.
Functional amyloid assembly is optimized to promote rapid and efficient polymer formation.
An understanding of risk-minimizing mechanisms used by functional amyloid systems can guide therapeutic strategies targeting disease-associated amyloids.
Acknowledgments
We thank current and past members of the Chapman for helpful discussions. Our lab gratefully acknowledges the support of National Institutes of Health grant R01GM118651 and R21 AI137535.
Abbreviations
- ThT
thioflavin-T
- csg
curli-specific gene
- PSMα3
phenol-soluble modulin α3
- NP
nucleated polymerization
- TEM
transmission electron microscopy
- AFM
atomic force microscopy
- NMR
nuclear magnetic resonance
- PrP
prion protein
- Aβ
amyloid-β
- IAPP
islet amyloid polypeptide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–5. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, Dijkhuizen L, Wosten HA. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–26. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773–8. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Kelly J, Balch WE. Amyloid as a natural product. J Cell Biol. 2003;161:461–2. doi: 10.1083/jcb.200304074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 10.Hammar M, Bian Z, Normark S. Nucleator-dependent intercellular assembly of adhesive curli organelles in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:6562–6. doi: 10.1073/pnas.93.13.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci USA. 2007;104:12494–9. doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Hammer ND, Chapman MR. The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem. 2008;283:21530–9. doi: 10.1074/jbc.M800466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans ML, Chorell E, Taylor JD, Aden J, Gotheson A, Li F, Koch M, Sefer L, Matthews SJ, Wittung-Stafshede P, Almqvist F, Chapman MR. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell. 2015;57:445–55. doi: 10.1016/j.molcel.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans ML, Schmidt JC, Ilbert M, Doyle SM, Quan S, Bardwell JC, Jakob U, Wickner S, Chapman MR. E. coli chaperones DnaK, Hsp33 and Spy inhibit bacterial functional amyloid assembly. Prion. 2011;5:323–34. doi: 10.4161/pri.5.4.18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SJ, Nam E, Lee HJ, Savelieff MG, Lim MH. Towards an understanding of amyloid-beta oligomers: characterization, toxicity mechanisms, and inhibitors. Chem Soc Rev. 2017;46:310–323. doi: 10.1039/c6cs00731g. [DOI] [PubMed] [Google Scholar]
- 16.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A. 2013;110:9758–63. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong HR, An SSA. Causative factors for formation of toxic islet amyloid polypeptide oligomer in type 2 diabetes mellitus. Clin Interv Aging. 2015;10:1873–1879. doi: 10.2147/CIA.S95297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 19.Fodera V, Librizzi F, Groenning M, van de Weert M, Leone M. Secondary nucleation and accessible surface in insulin amyloid fibril formation. J Phys Chem B. 2008;112:3853–8. doi: 10.1021/jp710131u. [DOI] [PubMed] [Google Scholar]
- 20.Padrick SB, Miranker AD. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry (Mosc) 2002;41:4694–703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 21.Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, Sparr E, Linse S. Secondary nucleation of monomers on fibril surface dominates alpha-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. 2017;50:e6. doi: 10.1017/S0033583516000172. [DOI] [PubMed] [Google Scholar]
- 22.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci USA. 2011;108 doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils, Biochim. Biophys Acta. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arosio P, Knowles TP, Linse S. On the lag phase in amyloid fibril formation. Phys Chem Chem Phys. 2015;17:7606–18. doi: 10.1039/c4cp05563b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen SIA, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM, Knowles TPJ. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys. 2011;135:065105. doi: 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee CT, Terentjev EM. Mechanisms and rates of nucleation of amyloid fibrils. J Chem Phys. 2017;147:105103. doi: 10.1063/1.4995255. [DOI] [PubMed] [Google Scholar]
- 28.Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–7. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 29.Michaels TC, Knowles TP. Role of filament annealing in the kinetics and thermodynamics of nucleated polymerization. J Chem Phys. 2014;140:214904. doi: 10.1063/1.4880121. [DOI] [PubMed] [Google Scholar]
- 30.Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33:S67–78. doi: 10.3233/JAD-2012-129001. [DOI] [PubMed] [Google Scholar]
- 31.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 32.Tycko R. Physical and structural basis for polymorphism in amyloid fibrils: Amyloid Polymorphism. Protein Sci. 2014;23:1528–1539. doi: 10.1002/pro.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heise H, Celej MS, Becker S, Riedel D, Pelah A, Kumar A, Jovin TM, Baldus M. Solid-state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant alpha-synuclein. J Mol Biol. 2008;380:444–450. doi: 10.1016/j.jmb.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 36.Gath J, Bousset L, Habenstein B, Melki R, Böckmann A, Meier BH. Unlike Twins: An NMR Comparison of Two α-Synuclein Polymorphs Featuring Different Toxicity. PLoS ONE. 2014;9:e90659. doi: 10.1371/journal.pone.0090659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andronesi OC, von Bergen M, Biernat J, Seidel K, Griesinger C, Mandelkow E, Baldus M. Characterization of Alzheimer’s-like Paired Helical Filaments from the Core Domain of Tau Protein Using Solid-State NMR Spectroscopy. J Am Chem Soc. 2008;130:5922–5928. doi: 10.1021/ja7100517. [DOI] [PubMed] [Google Scholar]
- 38.Daebel V, Chinnathambi S, Biernat J, Schwalbe M, Habenstein B, Loquet A, Akoury E, Tepper K, Müller H, Baldus M, Griesinger C, Zweckstetter M, Mandelkow E, Vijayan V, Lange A. β-Sheet Core of Tau Paired Helical Filaments Revealed by Solid-State NMR. J Am Chem Soc. 2012;134:13982–13989. doi: 10.1021/ja305470p. [DOI] [PubMed] [Google Scholar]
- 39.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cobb NJ, Apostol MI, Chen S, Smirnovas V, Surewicz WK. Conformational Stability of Mammalian Prion Protein Amyloid Fibrils Is Dictated by a Packing Polymorphism within the Core Region. J Biol Chem. 2014;289:2643–2650. doi: 10.1074/jbc.M113.520718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans ML, Chapman MR. Curli biogenesis: Order out of disorder. Biochim Biophys Acta BBA - Mol Cell Res. 2014;1843:1551–1558. doi: 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen A, Jonsson A, Normark S. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature. 1989;338:652–5. doi: 10.1038/338652a0. [DOI] [PubMed] [Google Scholar]
- 43.Nasr A, Olsén A, Sjöbring U, Müller-Esterl W, Björck L. Assembly of human contact phase proteins and release of bradykinin at the surface of curli-expressing Escherichia coli. Mol Microbiol. 1996;20:927–935. doi: 10.1111/j.1365-2958.1996.tb02534.x. [DOI] [PubMed] [Google Scholar]
- 44.Arnqvist A, Olsén A, Pfeifer J, Russell DG, Normark S. The Crl protein activates cryptic genes for curli formation and fibronectin binding in Escherichia coli HB101. Mol Microbiol. 1992;6:2443–2452. doi: 10.1111/j.1365-2958.1992.tb01420.x. [DOI] [PubMed] [Google Scholar]
- 45.Sjöbring U, Pohl G, Olsén A. Plasminogen, absorbed by Escherichia coli expressing curli or by Salmonella enteritidis expressing thin aggregative fimbriae, can be activated by simultaneously captured tissue-type plasminogen activator (t-PA) Mol Microbiol. 1994;14:443–452. doi: 10.1111/j.1365-2958.1994.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 46.Uhlich GA, Cooke PH, Solomon EB. Analyses of the Red-Dry-Rough Phenotype of an Escherichia coli O157:H7 Strain and Its Role in Biofilm Formation and Resistance to Antibacterial Agents. Appl Environ Microbiol. 2006;72:2564–2572. doi: 10.1128/AEM.72.4.2564-2572.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryu JH, Beuchat LR. Biofilm Formation by Escherichia coli O157:H7 on Stainless Steel: Effect of Exopolysaccharide and Curli Production on Its Resistance to Chlorine. Appl Environ Microbiol. 2005;71:247–254. doi: 10.1128/AEM.71.1.247-254.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeter C, Matthysse AG. Characterization of the binding of diarrheagenic strains of E. coli to plant surfaces and the role of curli in the interaction of the bacteria with alfalfa sprouts. Mol Plant-Microbe Interact MPMI. 2005;18:1235–1242. doi: 10.1094/MPMI-18-1235. [DOI] [PubMed] [Google Scholar]
- 49.Vidal O, Longin R, Prigent-Combaret C, Dorel C, Hooreman M, Lejeune P. Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J Bacteriol. 1998;180:2442–2449. doi: 10.1128/jb.180.9.2442-2449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DePas WH, Syed AK, Sifuentes M, Lee JS, Warshaw D, Saggar V, Csankovszki G, Boles BR, Chapman MR. Biofilm formation protects Escherichia coli against killing by Caenorhabditis elegans and Myxococcus xanthus. Appl Environ Microbiol. 2014;80:7079–7087. doi: 10.1128/AEM.02464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Rochon M, Lamprokostopoulou A, Lünsdorf H, Nimtz M, Römling U. Impact of biofilm matrix components on interaction of commensal Escherichia coli with the gastrointestinal cell line HT-29. Cell Mol Life Sci CMLS. 2006;63:2352–2363. doi: 10.1007/s00018-006-6222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammar MR, Arnqvist A, Bian Z, Olsen A, Normark S. Expression of two csg operons is required for production of fibronectin- and Congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol. 1995;18:661–670. doi: 10.1111/j.1365-2958.1995.mmi_18040661.x.. [DOI] [PubMed] [Google Scholar]
- 53.Ogasawara H, Yamamoto K, Ishihama A. Role of the Biofilm Master Regulator CsgD in Cross-Regulation between Biofilm Formation and Flagellar Synthesis. J Bacteriol. 2011;193:2587–2597. doi: 10.1128/JB.01468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zogaj X, Bokranz W, Nimtz M, Romling U. Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect Immun. 2003;71:4151–8. doi: 10.1128/IAI.71.7.4151-4158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dudin O, Geiselmann J, Ogasawara H, Ishihama A, Lacour S. Repression of flagellar genes in exponential phase by CsgD and CpxR, two crucial modulators of Escherichia coli biofilm formation. J Bacteriol. 2014;196:707–15. doi: 10.1128/JB.00938-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bian Z, Normark S. Nucleator function of CsgB for the assembly of adhesive surface organelles in Escherichia coli. EMBO J. 1997;16:5827–5836. doi: 10.1093/emboj/16.19.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor JD, Zhou Y, Salgado PS, Structure PA. Atomic resolution insights into curli fiber biogenesis. Structure. 2011 doi: 10.1016/j.str.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loferer H, Hammar M, Normark S. Availability of the fibre subunit CsgA and the nucleator protein CsgB during assembly of fibronectin-bindingcurliis limited by the intracellular concentration of the novel lipoprotein CsgG. Mol Microbiol. 1997;26:11–23. doi: 10.1046/j.1365-2958.1997.5231883.x. [DOI] [PubMed] [Google Scholar]
- 59.Goyal P, Krasteva PV, Gerven VN, Nature GF. Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG. Nature. 2014 doi: 10.1038/nature13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammer ND, McGuffie BA, Zhou Y, Badtke MP, Reinke AA, Brannstrom K, Gestwicki JE, Olofsson A, Almqvist F, Chapman MR. The C-terminal repeating units of CsgB direct bacterial functional amyloid nucleation. J Mol Biol. 2012;422:376–89. doi: 10.1016/j.jmb.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swasthi HM, Mukhopadhyay S. Electrostatic lipid–protein interactions sequester the curli amyloid fold on the lipopolysaccharide membrane surface. J Biol Chem. 2017;292:19861–72. doi: 10.1074/jbc.M117.815522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nenninger AA, Robinson LS, Hultgren SJ. Localized and efficient curli nucleation requires the chaperone-like amyloid assembly protein CsgF. Proc Natl Acad Sci U A. 2009;106:900–5. doi: 10.1073/pnas.0812143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nenninger AA, Robinson LS, Hammer ND, Epstein EA, Badtke MP, Hultgren SJ, Chapman MR. CsgE is a curli secretion specificity factor that prevents amyloid fibre aggregation. Mol Microbiol. 2011;81:486–99. doi: 10.1111/j.1365-2958.2011.07706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeBenedictis EP, Ma D, Keten S. Structural predictions for curli amyloid fibril subunits CsgA and CsgB. RSC Adv. 2017;7:48102–48112. [Google Scholar]
- 65.Shewmaker F, McGlinchey RP, Thurber KR, McPhie P, Dyda F, Tycko R, Wickner RB. The functional curli amyloid is not based on in-register parallel beta-sheet structure. J Biol Chem. 2009;284:25065–76. doi: 10.1074/jbc.M109.007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Zhou Y, Ren JJ, Hammer ND, Chapman MR. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proc Natl Acad Sci U S A. 2010;107:163–8. doi: 10.1073/pnas.0908714107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor JD, Hawthorne WJ, Lo J, Dear A, Jain N, Meisl G, Andreasen M, Fletcher C, Koch M, Darvill N, Scull N, Escalera-Maurer A, Sefer L, Wenman R, Lambert S, Jean J, Xu Y, Turner B, Kazarian SG, Chapman MR, Bubeck D, de Simone A, Knowles TP, Matthews SJ. Electrostatically-guided inhibition of Curli amyloid nucleation by the CsgC-like family of chaperones. Sci Rep. 2016;6:24656. doi: 10.1038/srep24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sleutel M, Van den Broeck I, Van Gerven N, Feuillie C, Jonckheere W, Valotteau C, Dufrene YF, Remaut H. Nucleation and growth of a bacterial functional amyloid at single-fiber resolution. Nat Chem Biol. 2017;13:902–908. doi: 10.1038/nchembio.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dueholm MS, Nielsen SB, Hein KL, Nissen P, Chapman M, Christiansen G, Nielsen PH, Otzen DE. Fibrillation of the major curli subunit CsgA under a wide range of conditions implies a robust design of aggregation. Biochemistry. 2011;50:8281–90. doi: 10.1021/bi200967c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Smith DR, Jones JW, Chapman MR. In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem. 2007;282:3713–9. doi: 10.1074/jbc.M609228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s -amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jayasinghe SA, Langen R. Identifying Structural Features of Fibrillar Islet Amyloid Polypeptide Using Site-directed Spin Labeling. J Biol Chem. 2004;279:48420–48425. doi: 10.1074/jbc.M406853200. [DOI] [PubMed] [Google Scholar]
- 73.Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural Organization of α-Synuclein Fibrils Studied by Site-directed Spin Labeling. J Biol Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 75.Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci. 2004;101:10278–10283. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry (Mosc) 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q Rev Biophys. 2008;41:265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 78.Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. Protein inheritance (prions) based on parallel in-register beta-sheet amyloid structures. Bioessays. 2008;30:955–64. doi: 10.1002/bies.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tian P, Boomsma W, Wang Y, Otzen DE, Jensen MH, Lindorff-Larsen K. Structure of a functional amyloid protein subunit computed using sequence variation. J Am Chem Soc. 2015;137:22–5. doi: 10.1021/ja5093634. [DOI] [PubMed] [Google Scholar]
- 80.Kajava AV, Squire JM, Parry DA. Beta-structures in fibrous proteins. Adv Protein Chem. 2006;73:1–15. doi: 10.1016/S0065-3233(06)73001-7. [DOI] [PubMed] [Google Scholar]
- 81.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–6. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 82.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–8. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buard J, Vergnaud G. Complex recombination events at the hypermutable minisatellite CEB1 (D2S90) EMBO J. 1994;13:3203–3210. doi: 10.1002/j.1460-2075.1994.tb06619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. A census of protein repeats. J Mol Biol. 1999;293:151–160. doi: 10.1006/jmbi.1999.3136. [DOI] [PubMed] [Google Scholar]
- 85.Glass NL, Kaneko I. Fatal attraction: nonself recognition and heterokaryon incompatibility in filamentous fungi. Eukaryot Cell. 2003;2:1–8. doi: 10.1128/EC.2.1.1-8.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saupe SJ. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev. 2000;64:489–502. doi: 10.1128/mmbr.64.3.489-502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turcq B, Deleu C, Denayrolles M, Bégueret J. Two allelic genes responsible for vegetative incompatibility in the fungus Podospora anserina are not essential for cell viability. Mol Gen Genet MGG. 1991;228:265–269. doi: 10.1007/BF00282475. [DOI] [PubMed] [Google Scholar]
- 88.Greenwald J, Buhtz C, Ritter C, Kwiatkowski W, Choe S, Maddelein ML, Ness F, Cescau S, Soragni A, Leitz D, Saupe SJ, Riek R. The mechanism of prion inhibition by HET-S. Mol Cell. 2010;38:889–99. doi: 10.1016/j.molcel.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Melckebeke H, Wasmer C, Lange A, Ab E, Loquet A, Bockmann A, Meier BH. Atomic-resolution three-dimensional structure of HET-s(218–289) amyloid fibrils by solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:13765–75. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- 90.Wasmer C, Schutz A, Loquet A, Buhtz C, Greenwald J, Riek R, Bockmann A, Meier BH. The molecular organization of the fungal prion HET-s in its amyloid form. J Mol Biol. 2009;394:119–27. doi: 10.1016/j.jmb.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 91.Emsley P, Charles IG, Fairweather NF, Isaacs NW. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature. 1996;381:90–2. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- 92.Richardson JS, Richardson DC. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leinala EK, Davies PL, Doucet D, Tyshenko MG, Walker VK, Jia Z. A beta-helical antifreeze protein isoform with increased activity. Structural and functional insights. J Biol Chem. 2002;277:33349–52. doi: 10.1074/jbc.M205575200. [DOI] [PubMed] [Google Scholar]
- 94.Liou YC, Tocilj A, Davies PL, Jia Z. Mimicry of ice structure by surface hydroxyls and water of a beta-helix antifreeze protein. Nature. 2000;406:322–4. doi: 10.1038/35018604. [DOI] [PubMed] [Google Scholar]
- 95.Cordell SC, Anderson RE, Lowe J. Crystal structure of the bacterial cell division inhibitor MinC. EMBO J. 2001;20:2454–61. doi: 10.1093/emboj/20.10.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Clantin B, Hodak H, Willery E, Locht C, Jacob-Dubuisson F, Villeret V. The crystal structure of filamentous hemagglutinin secretion domain and its implications for the two-partner secretion pathway. Proc Natl Acad Sci U S A. 2004;101:6194–9. doi: 10.1073/pnas.0400291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8:e1002744. doi: 10.1371/journal.ppat.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schwartz K, Ganesan M, Payne DE, Solomon MJ, Boles BR. Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol Microbiol. 2016;99:123–34. doi: 10.1111/mmi.13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laabei M, Jamieson WD, Yang Y, van den Elsen J, Jenkins ATA. Investigating the lytic activity and structural properties of Staphylococcus aureus phenol soluble modulin (PSM) peptide toxins. Biochim Biophys Acta. 2014;1838:3153–3161. doi: 10.1016/j.bbamem.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 100.Tayeb-Fligelman E, Tabachnikov O, Moshe A, Goldshmidt-Tran O, Sawaya MR, Coquelle N, Colletier JP, Landau M. The cytotoxic Staphylococcus aureus PSMalpha3 reveals a cross-alpha amyloid-like fibril. Science. 2017;355:831–833. doi: 10.1126/science.aaf4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schütz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Güntert P, Böckmann A, Glockshuber R, Meier BH. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew Chem Int Ed Engl. 2015;54:331–335. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc Natl Acad Sci USA. 2016;113:E4976–E4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J Am Chem Soc. 2016;138:9663–74. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fändrich M, Grigorieff N. Peptide dimer structure in an Aβ(1–42) fibril visualized with cryo-EM. Proc Natl Acad Sci USA. 2015;112:11858–11863. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tycko R. Amyloid polymorphism: structural basis and neurobiological relevance. Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guenther EL, Cao Q, Trinh H, Lu J, Sawaya MR, Cascio D, Boyer DR, Rodriguez JA, Hughes MP, Eisenberg DS. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat Struct Mol Biol. 2018;6:463–471. doi: 10.1038/s41594-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luo F, Gui X, Zhou H, Li Y, Liu X, Zhao M, Li D, Li X, Liu C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat Struct Mol Biol. 2018;25:341–346. doi: 10.1038/s41594-018-0050-8. [DOI] [PubMed] [Google Scholar]