

Abstract
Selenocysteine-containing proteins (selenoproteins) have been implicated in the regulation of various cell signaling pathways, many of which are linked to colorectal malignancies. In this in-depth excurse into the selenoprotein literature, we review possible roles for human selenoproteins in colorectal cancer, focusing on the typical hallmarks of cancer cells and their tumor-enabling characteristics. Human genome studies of single nucleotide polymorphisms in various genes coding for selenoproteins have revealed potential involvement of glutathione peroxidases, thioredoxin reductases, and other proteins. Cell culture studies with targeted down-regulation of selenoproteins and studies utilizing knockout/transgenic animal models have helped elucidate the potential roles of individual selenoproteins in this malignancy. Those selenoproteins, for which strong links to development or progression of colorectal cancer have been described, may be potential future targets for clinical interventions.
Keywords: colorectal cancer, glutathione peroxidases, inflammation, selenium, selenoproteins, single nucleotide polymorphisms, thioredoxin reductases
Graphical abstract
Introduction
Colorectal cancer (CRC) is a common malignancy, affecting millions of people worldwide. In the USA, an estimated 95,520 new colon cancer cases and 39,910 new rectal cancer cases, and an estimated 50,260 deaths due to CRC were expected for 2017 [1,2]. Similarly, in Germany, 61,000 new CRC cases were expected for 2016 [3], and CRC has been the second leading type of cancer in terms of incidence across Europe [4]. Worldwide, well over one million new cases are estimated to be diagnosed annually [5]. Selenium (Se) is an essential trace mineral that occurs naturally in the environment as both inorganic and organic chemical species. Soil Se concentrations vary geographically with the erosion of rocks containing selenites and selenides. A lower Se status, usually as a result of a lower Se intake, has been found to be associated with increased risk of colorectal malignancies. In contrast, an increased Se intake was associated with lower risk of colonic adenoma recurrence [6–8]. Therefore, for populations living in areas with lower soil Se, such as in some areas of China, Europe, and Africa, Se supplementation may be recommended. As with most other nutrients, the intake-benefit/risk correlation of dietary Se follows a U-shaped curve, where both inadequate and excess Se have been shown to result in deleterious effects in humans [9].
Many in vitro, animal, and clinical studies have demonstrated the importance of Se in (human) health, including its role in CRC. Estimated from food consumption patterns in 27 countries, an early ecological study found significant inverse correlations between dietary Se intake and cancers of the large intestine and rectum, among others [10]. Subsequently, two major clinical trials evaluated the role of dietary Se supplementation in cancer prevention: the Nutritional Prevention of Cancer (NPC) trial and the Se and Vitamin E Cancer Prevention Trial (SELECT). The results of the NPC trial, where participants received a daily oral dose of 200 µg Se in brewer’s yeast, showed no effect on the recurrence of the primary outcome, skin cancers; however, risks of total cancer mortality and cancers of the prostate, colon, and rectum with Se-supplementation compared to placebo controls were reduced [11]. In SELECT, participants received 200 µg L-selenomethionine per day and/or Vitamin E to assess mitigating effects on prostate cancer risk. After a 5.5 year median follow up, no decrease in prostate cancer incidence was found, and the trial was halted [12,13]. Subsequent analyses showed that the relative risk for colorectal adenoma occurrence was not significantly reduced in participants of the Se intervention arm compared to placebo. Serum levels in the subjects used in SELECT indicated that participants were Se replete at baseline [14]. Thus, as has been argued repeatedly, the type of Se supplementation might be important, as the mechanisms of intestinal absorption of Se differ depending on the chemical form of the element [15], as well as host factors such as sex, age, nutritional status [16], and the composition and activity of the intestinal microbiome. Furthermore, it is possible that Se supplementation in terms of cancer preventive effects, may only benefit those individuals with low-to-adequate baseline Se [14,17,18].
For the chemopreventive effects of Se observed in in vitro and animal studies, as well as in earlier human clinical trials (with lower participants’ serum Se levels), several mechanisms and pathways have been proposed, including decreased oxidative stress, altered metabolism of carcinogens, regulation of immune and apoptotic pathways, and DNA repair [19]. The exact mechanism of how Se modifies these many pathways remains to be elucidated. Importantly, it has become increasingly recognized that, very often, the biological effects of Se appear to be mediated through various selenoproteins. Selenoproteins contain at least one selenocysteine (Sec), which is the 21st amino acid in the genetic code. It is co-translationally inserted into selenoproteins by recoding of the UGA stop codon (see reviews in [20–22]). The human selenoproteome is encoded by 25 genes [23]. While the function of some selenoproteins is still unknown, their significant role in human health is increasingly uncovered. Among those selenoproteins that have been characterized, many have been shown to play a role in redox regulation, thyroid hormone metabolism, protein folding, or disulfide isomerization [21].
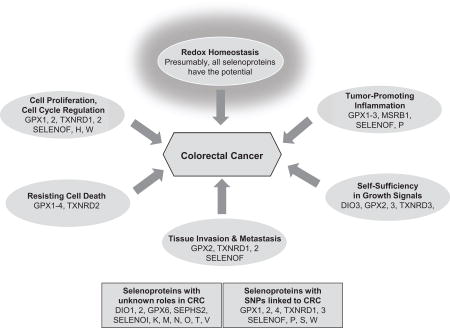
Whereas other reviews have addressed aspects of Se and selenoproteins in cancers in the general sense [20,24–26], our review will concentrate on those selenoproteins with suspected roles in CRC, grouping them, as possible, by their known or suspected functions according to the ‘hallmarks of cancer’ and enabling characteristics, as described by Hanahan and Weinberg [27,28]. It should be noted that these cancer hallmarks and other characteristics are interconnected [29], and that especially redox signaling and its regulation in homeostasis are important overarching themes, networking the individual contributors in pathways that continue to be investigated.
Hallmark: Redox Homeostasis/Deregulated Redox Signaling
Organisms rely on effective cellular redox homeostasis, balancing production and elimination of oxidants from both exogenous and endogenous sources. Reactive oxygen species (ROS), whether byproducts of metabolism or purposefully made species, are important signaling molecules, utilized by cells to affect signaling programs, activation and inactivation of transcription factors and other target proteins. A substantial amount of endogenous ROS is generated by members of the NADPH oxidase (NOX) family of enzymes during redox-mediated kinase signaling [30]. To maintain ROS below toxic levels, and to mediate ROS signal duration, organisms evolved defense enzyme systems and mediators of redox signaling pathways (reviewed in [31,32]), which includes catalases, peroxiredoxins, and importantly, selenoproteins such as members of the glutathione peroxidase and thioredoxin reductase families. Redox signaling is involved in the regulation of many proteins and signaling cascades that are connected to various hallmarks and enabling characteristics of cancer [31]. Consequently, redox homeostasis is often observed to be deregulated in cancer cells, leading to increased ROS levels, but also increased activities of enzymes involved in cell maintenance. Most selenoproteins are oxidoreductases or possess a thioredoxin-like fold, suggesting potential involvement in redox functions. Redox homeostasis is maintained in various cell compartments, most notably in cytosol and mitochondria. Selenoproteins with expression in colorectal tissues and well-established functions in redox control, and are therefore described in this section, include glutathione peroxidases and thioredoxin reductases, SELENOP, and SELENOW. The endoplasmic reticulum (ER) is another critical compartment for redox control that exists in an oxidative environment necessary for efficient disulfide bond formation and control of the unfolded protein response (UPR) (reviewed in [33]). The UPR, a consequence of disrupting the ER homeostasis as a result of accumulation of unfolded proteins in the ER, leads to degradation of misfolded proteins and an increase in the production of molecular chaperones involved in protein folding. It may also trigger intracellular oxidative stress, and initiation of pro-apoptotic signals. The UPR may also directly interact with ROS-mediated ER calcium efflux [34]. Thus, for ER-resident selenoproteins in general, molecular crosstalk to ER- and oxidative stress-mediated signaling pathways, and intrinsic apoptotic pathways should be considered. Six mammalian selenoproteins (SELENOF, K, M, N, S, T) had originally been identified as ER-residents [35]. Recently, SELENOI has been added as an additional ER-resident selenoprotein [36]. For some of these, a role in the UPR (SELENOF, SELENOK, SELENOS) or regulation of calcium homeostasis (SELENOK, M, N, T) has been suggested (reviewed in [37]). Thus far, it appears as if evidence among ER-resident selenoproteins for a role in cancer is limited to SELENOF and SELENOS, which are described in this section.
There are several isozymes of glutathione peroxidase (GPX) in mammals. In humans, among eight known GPXs, five are selenoproteins (GPX 1,2,3,4,6). GPXs reduce hydrogen peroxide, organic hydroperoxides and/or phospholipid hydroperoxides via their substrate, glutathione (GSH). GPXs are present in the cytosol of all cell types, including epithelial cells of the gastrointestinal tract [21], and are discussed below primarily with relevance to CRC. Extensive in-depth reviews on GPXs in cancer in general, their functions as they relate to metabolism of hydroperoxides, and their likely functions in cancer promotion and progression have been published recently [38,39]. Among the glutathione peroxidases, GPX1 was the first identified, and is likely the most in-depth investigated enzyme. It is also one of the most studied selenoenzymes in general [40–42]. Its cytosolic expression is regulated by the Se status of the organism, and is particularly abundant during erythropoiesis [43]. GPX1 expression has been implicated in various human diseases, including risk and development of cancers. GPX1 has been shown to be altered at both protein and mRNA levels in tissues of patients with CRC [44–47]. Much of the contributions and functions of GPX1 has been elucidated using in vitro and in vivo knockout models. The major role of GPX1 has always been thought to relate to amelioration of peroxide-mediated deleterious effects. In healthy tissues, this would be considered anti-tumorigenic through prevention of cytotoxicity and inflammation. In transformed cells, GPX1 activity could be considered pro-tumorigenic, as it would prevent oxidation-mediated initiation of apoptosis [38,39]. However, in several types of cancers, GPX1 is reported to be down-regulated [38], hinting at a potential interesting dichotomy or tissue-specificity in its function. Therefore, while very limited evidence has been presented, it is likely that the ubiquitously expressed GPX1 may be involved in colorectal tumorigenesis. It should be emphasized, however, that especially in colorectal tissues, another GPX isoform, GPX2, is considered highly relevant [48], and is thought to possibly functionally compensate for reduced levels of GPX1 [49].
GPX2 appears to be involved in various human malignancies, such as squamous cell carcinoma and, in particular, CRC. Expression and mRNA stability of GPX2 are maintained under lower Se availability, indicating that GPX2 ranks high in the hierarchy of GPX selenoproteins [50,51]. Based on its basal expression profile initially identified in rodent gastrointestinal tissues, GPX2 has often been referred to as the gastrointestinal GPX, although it is found to be expressed in many other tissues, especially of epithelial origin [48,50,52–54], and even in embryonic stem cells [55]. Within the gastrointestinal tract, it is found in the intestinal crypt epithelium, specifically in the rapidly proliferating crypt bases [52,56]. GPX2 has been thought originally to function as a defense mechanism against dietary hydroperoxides, to which the gastrointestinal mucosa is exposed regularly [51]. More importantly, GPX2 functions to mediate signal duration of endogenously created ROS by forming a disulfide with glutathione [30]. This may be especially important as a response to ROS generated by NOX, whose expression is often dysregulated during carcinogenesis [38]. Thus, increased expression of GPX2, which is regulated by NF-E2-related factor-2 [57,58], has been suggested as a mechanism against severe oxidative stress [59], including conditions where increased cell proliferation is likely stimulated as a response to stress.
Three mammalian thioredoxin reductases (TXNRD) are structurally similar and contain a C-terminal extension that harbors Sec [60]. These enzymes were suggested to mediate nutritional Se intake-mediated effects on health and disease in human populations [61]. TXNRDs function in NADPH-dependent reduction of thioredoxins, which in turn maintain cysteines in cellular proteins in the reduced state. TXNRDs can reduce other substrates as well, including ascorbic acid and hydrogen peroxide, and also certain inorganic and organic forms of Se (e.g., selenite and selenodiglutathione). Thus, TXNRDs play a key role in redox homeostasis and Se metabolism, and therefore may affect human health and disease directly through their redox function, and indirectly through other Se-dependent functions (reviewed in [21,61–64]).
TXNRD1 is a gene for which several splice variants are known, which are thought to be pivotal components of diverse cellular signaling pathways and redox-related mechanisms [61]. These encode a number of protein variants, which are thought to either possess glutaredoxin and thioredoxin reductase activity, induce polymerization of cytoskeletal proteins, or modulate the transcriptional activity of estrogen receptors α and/or β. These isoform-specific functions are likely tissue-type specific [61,63,65–67]. Importantly for tumorigenesis, TXNRD1 has been shown to promote functions of known tumor suppressors, including p53, protein kinase C, and the phosphatase and tensin homolog PTEN, as well as functions of transcription factors, such as NFκB, for which both pro-tumorigenic and tumor suppressing roles have been described [68]. It is important to note that both p53, often referred to as the ‘guardian of the genome’, and PTEN play significant roles in cell cycle regulation, but also in the cellular response to oxidative stress, thus mitigating potential ROS-induced DNA damage [69,70]. However, in an earlier study using a human colon cancer cell line, p53 repressed TXNRD1 expression, indicating a p53-dependent regulation of TXNRD1 [71]. Interestingly, TXNRD1 also appears to show a stabilizing effect on p53 [72]. TXNRD1 can also inhibit Src tyrosine kinase, and thus limit hydrogen peroxide-mediated induction of growth signals and impact vascular endothelial growth factor expression (reviewed in [62]). The roles that ROS and proteins with anti- and/or pro-oxidative functions play in the initiation, progression, or prevention of tumors are highly complex [73,74], and, as the extensive literature on this topic shows, this is demonstrated for several selenoproteins in general, and TXNRD1 in particular.
Thus, it is not entirely unexpected that TXNRD1 appears to have roles in preventing as well as promoting and sustaining cancer (see reviews in [20,61,75–78]). On the one hand, TXNRD1 has been found to suppress tumor initiation, growth, invasion and metastasis. Much of this ability appears to be through support of redox homeostasis, and this is also thought to be the main aspect in the prevention of malignancies such as CRC. The activity of TXNRD1 was detected in biopsy specimens from normal mucosa of colorectal tissues, but not in samples from the ileum [50], and increased expression levels of TXNRD1 were detected in colorectal carcinoma specimens [47]. Targeted down-regulation of TXNRD1 in human colon cancer cells made the cells more susceptible to apoptosis mediated by mitomycin C, a CRC therapeutic drug [79], and sensitized colon cancer cells to cytotoxicity and ER stress by methylseleninic acid, resulting in increased autophagy and apoptosis [80]. On the other hand, increased levels of thioredoxin [81] and TXNRD1 have been found in many cancer cells, suggesting a tumor protective function, thus supporting tumor promotion and progression. Both oxidative stress and hypoxia are common features in malignancies, pointing to the importance of redox systems in tumorigenesis. Interestingly, hypoxia has been shown to down-regulate TXNRD1 expression, and, whereas TXNRD1 deficiency increases hypoxia-induced ROS levels, overexpression of TXNRD1 attenuates the hypoxia-induced increase of ROS [82].
SELENOP, which was previously known as SeP, SELP, SEPP, or commonly SEPP1, is different from other selenoproteins in that it contains several Sec residues. The actual Sec number varies among species; in humans, this gene contains 10 Sec codons, and thus the protein includes multiple Sec. SELENOP is predominantly expressed in the liver and secreted into the plasma, where its main function is to transport Se to peripheral tissues. At present, plasma SELENOP expression is frequently used as a biomarker for the organism’s Se status [83]. SELENOP is also abundant in healthy tissues of the intestinal tract as well as in human colon cancer cell lines [45,84,85]. In target tissues, SELENOP is endocytosed via apoER2 receptors in tissues such as brain and testis, or the lipoprotein receptor megalin in the proximal tubule of the kidney (see review in [86]). SELENOP is then degraded intracellularly to free Se for synthesis of other selenoproteins. In addition to the nine Sec residues in the C-terminal domain, which are used to transport Se, the single Sec residue in the N-terminal domain is located within a redox motif. It likely functions in a redox capacity [21,87], as findings from a mouse study with a non-functional serine in place of Sec at residue 40 in the N-terminal domain supported [88].
SELENOW, formerly known as Selw or Sepw1, appears highly expressed in tissues from skeletal muscle, heart, and brain [89]. A possible antioxidant function was suggested in studies using human cancer cell lines overexpressing SELENOW [90]. The presence of the thioredoxin-like fold, and evidence from studies in primary cultured cells derived from embryonic cerebral cortex demonstrating SELENOW’s role in protection of neurons from oxidative stress [91], all point towards a redox function for this gene. mRNA expression analyses of mice on various Se diets suggested that SELENOW expression in colorectal tissues is sensitive to Se intake [92]. As a matter of fact, studies regarding SELENOW levels in response to dietary Se, especially in poultry, are abundant, and suggest that SELENOW may be useful as a biomarker for Se status of the organism [93]. Unfortunately, much less information is available regarding a possible role of SELENOW in human malignancies, especially CRC.
The 15 kDa selenoprotein ‘SELENOF’, formerly known as SEP15, is in the category of stress-related selenoproteins, indicating its expression is dependent on available Se. Expression of SELENOF in humans and mice is high in organs that have been shown to be protected from malignancy via Se, including normal liver, kidney, brain, and testis, with the highest levels detected in normal thyroid and prostate tissues [94,95]. The exact molecular function of this ER-resident protein is still under investigation. Mechanistically, Labunskyy et al. [96] showed upregulation of SELENOF in response to adaptive UPR, whereas acute ER stress led to rapid degradation of SELENOF by proteasomes. However, SELENOF deficiency in mouse fibroblasts did not result in detectable ER stress, and the in vivo knockout model resulted in only mild oxidative stress in liver [97]. Recent evidence points to the function relating to its role as a gatekeeper in the regulation of folding of proteins such as immunoglobulins that exit the ER, sorting, and transport from the ER to the Golgi network thereby supporting redox quality control of disulfide-rich glycoproteins [21,98]. Supporting evidence includes the findings that SELENOF forms a complex with UDP-glucose:glycoprotein glucosyltransferase, an enzyme involved in the quality control of glycoprotein folding in the ER, and that mice lacking systemic SELENOF expression developed cataracts compared to littermate controls, presumably through accumulation or clumping of misfolded proteins [97]. How this particular function relates to CRC remains to be investigated; however, given the changes of other cancer-enabling characteristics observed in CRC cells and animals with severely reduced or ablated SELENOF expression, a role for SELENOF in redox-mediated colorectal tumorigenesis is likely.
SELENOS (previously known as SelS, SepS1, Tanis, or VIMP) is predominantly found in the ER membrane, although cell surface expression and detection of a secreted form in human serum have also been described [99]. Whereas the function of the secreted form remains unclear, the ER-resident form of SELENOS is a transmembrane protein, and it has been implicated with ER-associated protein degradation and protein unfolding response [100]. Given that a human genotyping study linked SELENOS to estrogen-linked CRC risk [101], an indirect role for SELENOS in CRC is plausible.
Hallmark: Resisting Cell Death
High cellular levels of ROS, redox stress, and disruption of ER homeostasis may lead cells to activate signaling pathways initiating apoptosis [33]. The intrinsic apoptotic pathway is mitochondria-mediated, and both cytosolic and mitochondrial subcellular localization has been described for members of GPX and TXNRD families.
GPX1 is primarily thought of as a cytosolic selenoprotein. However, its mitochondrial matrix expression in various tissue types has also been described [102]. In a colon cancer cell model, p53 induced GPX1 mRNA expression [71], which may suggest a potential role for GPX1 in p53-mediated apoptosis.
Various tissue types express the cytosolic housekeeping selenoprotein GPX4, which is the only enzyme capable of reducing phospholipid hydroperoxides within cell membranes and lipoproteins [21,103]. Phospholipid hydroperoxides may regulate different redox-signaling pathways than hydrogen peroxide, but cross-talk likely exists [102]. Down-regulation of GPX4 via RNAi in the human intestinal epithelial cell line Caco2 showed that lack of GPX4 results in increased levels of mitochondrial ROS and decreased mitochondrial ATP levels. These in vitro studies suggest that GPX4 plays a major role in oxidative phosphorylation and mitochondrial dysfunction pathways, thus protecting mitochondria from oxidative damage [104]. Interestingly, GPX4 has recently been shown to be the key component in controlling ferroptosis-induced cancer cell death [105], a feature distinct from the better-known apoptotic cell death. Whereas its specific impact in CRC remains to be elucidated, given that GPX4-mediated regulation of ferroptosis is essential for hepatocyte function and survival [106], an important role for this selenoprotein is plausible.
The cytosolic GPX2 has also been suggested to play a role in apoptosis. This was demonstrated, for example, in a mouse model of inflammation-mediated carcinogenesis [56,107], where GPX2 deficiency correlated with increased rate of apoptosis in crypt bases of the intestinal epithelium [107]. Furthermore, in an artificial system of GPX2-overexpressing colonospheres derived from patients harboring colorectal liver metastases [108], these cells were found to inhibit hypoxia-induced effects on caspase processing, likely pointing to GPX2’s role in redox regulation and apoptosis. Mechanistically, its ability to regulate apoptosis is thought to be, at least in part, mediated by NOX1. This member of the NADPH oxidase family is expressed in various epithelial tissues, including in the apical membrane of enterocytes in the gastrointestinal tract, and is often induced in patients with inflammatory bowel disease [109]. This was demonstrated in mice deficient in GPX1/GPX2 [110,111], where increased NOX1 expression correlated with an increase in apoptosis in crypt epithelium. As a result of increased apoptosis, leakage of bacterial products into circulation may occur, which likely contributes to inflammation [110], a tumor enabling characteristic further described below [28].
Intriguingly, targeted downregulation of GPX3 in the human colon cancer Caco2 cells led to increased apoptosis. This is in contrast to findings in vivo, where GPX3 deficiency in a knockout mouse model of chemically-induced inflammatory carcinogenesis did not impact apoptotic markers [112]. The mechanism and GPX3’s potential relevance to CRC in vivo continues to be elucidated.
TXNRD2 encodes a mitochondrial TXNRD form, believed to be of importance in redox regulation in this cellular compartment [113]. Dysregulation of redox status is frequently observed in cancers, and TXNRD2 expression has been shown to be upregulated in some malignancies [114,115]. Because down-regulation of TXNRD2 in lung carcinoma cells induced apoptosis [115], and because TXNRD2 is a putative target of the WNT-signaling pathway [116], which is often deregulated in CRC, TXNRD2 may be acting as an oncogene in the context of tumor progression by modulating apoptosis signaling.
Hallmark: Cell Proliferation, Cell Cycle Regulation
Increased or uncontrolled cell proliferation is probably the best known hallmark of cancer cells. Whereas cell cycle regulators, including cyclins, cyclin-dependent kinases and their up- and down-stream regulators often play a role, it should be noted that redox signaling also modulates cell cycle entry and transition (reviewed in [29]). Thus, extensive crosstalk exists between redox homeostasis described above and cell proliferation described here. Accordingly, selenoproteins that are involved in redox homeostasis are often also involved in regulation of cell proliferation.
GPX1 appears to be involved in a number of cell signaling pathways, and much has been learned from targeted downregulation of the gene in cells, or systemic knockout or overexpression in animals. However, the results are conflicting and thus far only provide limited information for colorectal malignancies. Overexpression of GPX1 in non-CRC cell lines resulted in reduced proliferation [117], decreased rate of UV-induced DNA damage [118], and aberrant phosphorylation of extracellular-regulated kinases [119], among other effects, suggesting at least a potential role in regulation of cell proliferation in CRC. Using GPX1/GPX2 knockout mice, NOX1 has been implicated as a contributor to proliferation in intestinal epithelia [111], further suggesting an indirect role of GPXs in cell proliferation. However, it is difficult to assess, whether this is due to lack of GPX1, GPX2, or both, though it should be noted that, unlike GPX1/GPX2 double-knockout mice, animals deficient solely in GPX1 have not been reported to spontaneously develop tumors [120]. GPX2 is highly expressed in mucosal epithelia of the intestinal tract, specifically in the crypt base of colorectal epithelia. In addition to the evidence of an increase of proliferating cells in GPX1/GPX2 knockout mice [120], it appears that GPX2 is regulated via the WNT/β-catenin pathway, an important pathway in CRC, which is particularly active in proliferative crypt compartments of colorectal tissues [116].
Among the TXNRDs, evidence suggests that TXNRD1 affects cell proliferation. A mouse colon carcinoma cell line with Txnrd1 ablation via RNAi showed decreased cell proliferation with concomitant strong increase in Cyclin B1 Interacting-Protein-1 mRNA expression [78], as well as decreased anchorage-independent growth. It should be noted that, in colorectal tissues specifically, there appears to be some discrepancy between in vitro and in vivo findings, as Lechner et al. described using reverse transcription-PCR and in situ hybridization techniques [121]. Lack of TXNRD1 expression was apparent in neoplastic areas of colonic cancer tissue, and in epithelial cells in colonic mucosa. Interestingly, TXNRD1 expression was detectable in human colon cancer HT29 cells grown in monolayers, but not when grown as spheroids or as tumors in SCID mice, which may complicate comparative analyses across studies [121]. Mitochondrial TXNRD2, in addition to its potential role in other cancer-enabling characteristics, appears to promote cell proliferation, cell invasion and migration [115], which was shown primarily in mouse embryonic fibroblasts, where activation of hypoxia-inducible factor-1α signaling failed in absence of TXNRD2 [122]. TXNRD2 activity appears to be positively regulated by p53R2 [123]. Thus, evidence from other malignancies suggests that TXNRD2 might contribute to CRC as well. Both TXNRD2 and TXNRD3, along with GPX2 as described above, had been identified as novel WNT targets [116] suggesting involvement of additional pathways that may be linked to regulation of cell proliferation.
In vitro and in vivo experiments uncovered colon tissue-specific roles for SELENOF. Murine [124] and human [125] colon cancer cell lines, in which SELENOF was selectively down-regulated via RNAi, exhibited a reversed cancer phenotype, as demonstrated by decreased cell proliferation and altered cell cycle progression [124–126]. This suggests a role for SELENOF in cell cycle regulation. Similarly, in an in vivo model, Selenof knockout mice developed fewer chemically-induced pre-neoplastic lesions than littermate controls [127], indicating that low expression of SELENOF may be protective during the early stages of colon tumorigenesis. This further suggests a role of SELENOF expression in promoting cell proliferation and colorectal malignancies, which is supported by gene expression analyses in both cell lines and mouse colon tissues.
SELENOH (SELH, C11orf31, C17orf10), which resides in the nucleoli [89], appears to have expression in colorectal tissues that is sensitive to Se intake [92]. In a recent publication [128], SELENOH expression was found to be increased in tumor tissue and in colorectal cancer lines. Using targeted downregulation of SELENOH in CRC, the authors observed a cell cycle shift from G1- to S-phase in cells lacking SELENOH, likely mediated by down-regulation of the cyclin-dependent kinase inhibitor 1a, p21. These observations suggest SELENOH’s involvement in cell cycle progression as well as in cellular differentiation in CRC cells. Furthermore, loss of SELENOH appeared to promote cell migration in cell culture, as well as tumor formation in mice subcutaneously injected with SELENOH knockdown cells or controls. These observations clearly indicate an important role for SELENOH in regulating cell proliferation and tumorigenesis in CRC, which would benefit from further investigations.
It has been suggested that SELENOW, at least in breast and prostate epithelial cells, may facilitate the G1 to S-phase transition via modulating expression of a cyclin-dependent kinase inhibitor, and thus enable cell cycle progression [129]. This further supports the hypothesis of SELENOW contributing to proliferation and possibly tumorigenesis. However, direct evidence supporting a role for SELENOW in CRC thus far has not been found.
Hallmark: Metastatic Ability (Activating invasion and metastasis)
The ability of primary tumors to invade adjacent tissues and metastasize involves complex processes. Often, this includes downregulated or inactivated expression of cell-adhesion molecules and upregulation of matrix-degrading proteases [28], which results in the acquisition of anchorage-independent growth abilities. Three selenoproteins, GPX2, SELENOF, and TXNRD1, appear to function similarly in this case. Human colon cancer cells with targeted down-regulation of GPX2 expression demonstrated that GPX2 supported anchorage-independent growth, as well as in vivo tumor growth, but inhibited the potential for cell migration [130]. Mouse [124] and human colorectal cancer cells [125] in which SELENOF was down-regulated via RNAi demonstrated not only decreased cell proliferation, but also dramatically decreased anchorage-independent growth. Similar observations were noted in the same mouse colon carcinoma cell line with decreased Txnrd1 expression: anchorage-independent growth, and metastatic ability were severely limited. Surprisingly, a combined targeted down-regulation of both SELENOF and TXNRD1 did not reverse cancer phenotypes, hinting to a more complicated interplay between selenoproteins in CRC [78]. Unfortunately, other information regarding TXNRD1’s role in metastasis is limited to tissues outside the intestinal tract (reviewed in [37]), including targeted downregulation of TXNRD1 in mouse lung carcinoma cells that had similarly resulted in the reversal in the morphology and anchorage-independent growth properties [131]. The mitochondrial TXNRD2 also appears to promote cell invasion and migration [115], which was shown in mouse embryonic fibroblasts, where activation of hypoxia-inducible factor-1α signaling failed in the absence of TXNRD2 [122]. TXNRD2 activity appears to be positively regulated by p53R2 [123]. Thus, evidence from other malignancies suggests that TXNRD2 might contribute to CRC as well.
Enabling Characteristic: Tumor-Promoting Inflammation
The mammalian immune system often responds to various tumor types with an inflammatory response. Because inflammation can result in increased production of ROS and growth factors that promote proliferative signaling, an unanticipated side effect of inflammation is often an enhanced tumor promotion [28]. The emerging hallmark ‘Evading immune destruction’, in which Hanahan and Weinberg addressed avoiding both immune surveillance and concomitant restriction of tumor growth, is tightly linked to the enabling characteristic of tumor-promoting inflammation. Both the innate and adaptive arms of the immune system, which includes B cells, T cells, natural killer cells, macrophages, etc., contribute to immune surveillance and thus potentially restricting tumor growth. However, this appears to be mostly related to virus-induced cancers [28]. Of course, colon tissues and tumors may be infiltrated by immune cells, which may express genes active in immune surveillance and growth restriction, and such gene expression may be mediated by selenoproteins [132]. Furthermore, many immune cells are implicated in inflammation, which also may be modulated by selenoproteins [133], and this may result in increased production of ROS and growth factors in colorectal tissues, promoting proliferative signaling as described in the next section. Presented here are a few selenoproteins that have been implicated, mostly indirectly, in the regulation of colorectal inflammation.
A transgenic mouse model expressing the human GPX1 gene developed a greater number of chemically-induced skin tumors [134], potentially mediated through suppressed inflammation and decreased apoptosis, and mice overexpressing GPX1 have also been shown to develop insulin resistance and elevated body fat accretion [135,136]. The latter might suggest some connection to intestinal cancers through obesity-related inflammation. Using transiently transfected Caco2 cells with a ~60% diminished GPX1 expression compared to controls, GPX1 knockdown cells responded with a Se depletion-mediated effect on NFκB signaling and a reduced cytokine response, suggesting that expression of GPX1 may be required for tumor necrosis factor α (TNFα)-mediated activation of NFκB signaling [137]. Gpx1 knockout mice appear to have a normal phenotype, but are highly sensitive to oxidative stress-inducing agents (reviewed in [38]). This could predispose Gpx1-knockout mice to inflammation-induced CRC, but such studies are very limited. Transgenic mice, where selenoprotein expression, including expression of GPX1, was reduced via mutation in Sec-tRNA, developed a larger number of chemically-induced pre-neoplastic colon lesions, suggesting that GPX1 and expression/activity of other selenoproteins is needed to guard against colorectal tumorigenesis [138]. This is in contrast to a study where Gpx1 knockout mice were regenerating jejunal epithelium more efficiently after γ-irradiation damage compared to controls [49]. The authors suggested that lack of GPX1, which is known to inhibit cyclooxygenase activity, allowed for increase in pro-inflammatory prostaglandin E2 (PGE2) levels. In turn, this supports regeneration of intestinal crypts [139], which is also supported by non-statistically significant observations in jejunal tissues in the previous study [49].
Not surprisingly, intestinal GPX2 is also expected to play a role in the control of colorectal inflammation and tumorigenesis [57]. Evidence for the latter is the preferential expression of GPX2 in the intestinal crypts, where intestinal stem cells reside, and in the Paneth cells of healthy ileum epithelia, which are known to play a major role in mucosal immunity. In contrast, a more diffuse GPX2 distribution has been reported in various stages of malignancy [52]. Earlier studies found a significant increase in transcripts of GPX2 in colorectal adenomas, carcinomas, and during epithelial cell neoplastic transformation. This not only suggested a vital role of GPX2 in the gastrointestinal tract, but pointed to a possible pro-tumorigenic function [45,53,140,141], positively correlating with proliferation and differentiation of tumors, as well as associating with early tumor recurrence [142]. In animal knockout models, lack of GPX2 resulted in fewer chemically-induced pre-neoplastic colon lesions [107], further suggesting that this protein contributes to the formation of colorectal malignancies. GPX2 was also found to inhibit inflammation-mediated tumorigenesis [143], suggesting a dual role for this selenoprotein.
The intestinal microbiome is an important modulator of colon health, reflects an organism’s health, and appears tightly linked to inflammation status of the host. The regulation of the intestinal microflora by dietary Se has been investigated in mice, which demonstrated an increased microbial diversity in response to increased Se in the diet in addition to an increase in the host’s Se status and selenoproteome expression [144]. Amongst the in vivo studies utilizing transgenic mice with knockouts of selenoproteins, the gut microbiome has been investigated thus far only in Gpx1/Gpx2 double-knockout mice. In contrast to animals deficient in only Gpx1, the mice, in which both Gpx1 and Gpx2 were deleted, spontaneously developed mucosal inflammation of the ileum and colon, symptoms consistent with inflammatory bowel disease [145]. The inflammatory response is likely induced through luminal contents, including bacteria, entering the lamina propria upon lack of proper barrier function, usually provided by GPXs. This was shown in Gpx1/Gpx2 double-knockout mice that exhibited pathology and inflammation in the ileal and colonic epithelia when bacterial colonization of the intestinal tract with commensal microflora without any known pathogens occurred at birth. In contrast, Gpx1/Gpx2 double-knockout mice raised under germ-free conditions did not develop pathologies or malignancies [120]. When barrier function is not restored, persistent inflammation in the colon may drive tumorigenesis, which is, in turn, likely modulated by the presence of certain microbes. Interestingly, the severity of the inflammatory response also depends on the host’s genetic background and associated differences in intestinal microbiota, as has been shown with two different mouse strains, each lacking both Gpx1 and Gpx2 [146]. The majority of these studies suggest that GPX2, much like GPX1, functions in maintenance of colonic epithelial barrier integrity, protecting against oxidative stress during bacteria-associated inflammation. Its role in suppressing apoptosis may also explain why GPX2 transcripts are elevated in CRC cells, which would benefit tumor cells in their efforts to further proliferate. Further studies are required to help explain this dual role, which appears to depend on the cancer stage and the involvement of inflammation.
Unlike the aforementioned GPX1 and GPX2, the GPX3 isozyme is a secreted form, and thus abundantly found in plasma, where it reduces hydroperoxides. However, GPX3 mRNA is also found expressed in tissues, including in the gastrointestinal tract. Furthermore, extracellular GPX3 is transported via systemic circulation and may subsequently bind to basement membranes of colorectal epithelial cells [112]. Two transcript variants have been described, primarily in reference to promoter hypermethylation and subsequent depressed gene expression observed in various cancers. GPX3 appears to play a role in thyroid, liver and prostate cancers, and correlates with head and neck cancer chemoresistance [147,148]. Studies with knockout mice in a chemically-induced inflammatory colon carcinogenesis model demonstrated the importance of systemic GPX3 expression, as these Gpx3-knockout mice developed increased inflammation and a larger number of colon tumors compared to controls. Subsequent in vitro experiments with the human colon cancer cell line Caco2 suggested that GPX3 played an immunoregulatory role through modulating ROS production and thus ameliorating inflammatory colitis-associated colon tumors [112].
Methionine sulfoxide reductases (MSR) catalyze conversion of methionine sulfoxide to methionine. Among the three MSR families known, MSRBs are responsible for the reduction of methionine-R-sulfoxide residues in proteins, and as such assist in protecting cellular proteins from oxidative stress. MSRB1 (formerly known as SELR, SEPR, or SELX) is found in the cytosol and nucleus of mammalian cells, and it evolved to utilize catalytic Sec [149–151]. Expression of MSRB1 decreases under conditions of Se deficiency [151,152]. Interestingly, studies with macrophages from Msrb1 knockout mice suggest that MSRB1 promotes anti-inflammatory cytokine expression, and as such may limit inflammatory responses [153]. Inflammation is a very important contributor to colorectal malignancies. However, if and how MSRB1 expression may modulate colorectal inflammation or CRC directly has yet to be elucidated.
SELENOP has been linked to inflammatory CRC in an interesting haploinsufficient manner. In comparison to both wild type controls and systemic SELENOP knockout mice, animals that were heterozygous for SELENOP demonstrated an increase in number of tumors, degree of DNA lesions, and dysplasia [86]. However, this finding is complicated by SELENOP’s important function as the hepatocyte-secreted plasma selenium transport protein, which impacts selenoprotein production within macrophages and other tissues, which in turn may regulate colorectal inflammation. Furthermore, dietary Se affects not only SELENOP expression, but also the composition of the intestinal microbiome [144,154], which makes it difficult to discern the direct mechanism by which SELENOP chiefly impacts inflammation.
Hallmark: Self-sufficiency in growth signals
Most CRC cases are sporadic rather than inherited, developing from a benign adenoma, progressing to cancer as a result of accumulated genetic and epigenetic changes [155]. In terms of molecular mechanisms in the development of colorectal tumors, there are three major signaling pathways in the intestinal epithelium that drive cell proliferation and differentiation (i.e., WNT, NOTCH, and BMP [156]). The genes of the WNT/β-catenin signaling pathway are especially known to be functionally altered in tumor cells, thus providing self-sufficiency in cellular growth, which is another hallmark of cancer [27,28]. The WNT/β-catenin-signaling pathway, which may be regulated by NOX1-derived hydrogen peroxide [156], is an integral part of the normal cell proliferation regulation of intestinal epithelial cells, and important for maintenance of epithelial stem cells. β-catenin expression is often up- or de-regulated in colorectal tumors, frequently as a result of mutations in the adenomatous polyposis coli (APC) gene, although non-canonical WNT-signaling, such as the WNT/Ca2+-signaling pathway, may also be involved [157,158]. NOX1, whose expression is increased in malignant colorectal tissues [156], has been suggested to also play a role in the regulation of NOTCH1 and WNT/β-catenin-signaling pathways. As a result, cell proliferation and differentiation are affected, which not only may impact the development of colon epithelia, but also contribute to the proliferative potential of malignant cells [159]. Furthermore, extensive crosstalk between WNT/β-catenin-signaling pathway and other signaling pathways have been described [160,161], including those that Hanahan and Weinberg referred to as ‘motility circuits’ [28]. Some of the proteins involved in such pathways may directly or indirectly involve selenoproteins.
GPX2 was found to inhibit inflammation-mediated tumorigenesis [143]. In order to further investigate the molecular mechanisms, Kipp et al. examined aspects of the canonical WNT/β-catenin cell signaling pathway, and demonstrated that the GPX2 promoter contains several putative β-catenin binding sites. Furthermore, overexpression of β-catenin in cancer cell lines resulted in activation of the GPX2 promoter [162]. This was further substantiated with subsequent in vitro and in vivo studies, where regulation of GPX2 expression by the WNT pathway was demonstrated [116]. Altogether, these findings not only suggest a function of GPX2 in the maintenance and renewal of the intestinal epithelium, but also further supports a function in colorectal tumorigenesis. Similarly, in experiments with GPX3-deficient mice, and paralleled with in vitro experiments using a colon cancer cell line, an increase in proliferation, along with increased nuclear and total β-catenin suggesting a hyperactive WNT signaling, was observed [112].
The gene for TXNRD3 contains an additional N-terminal glutaredoxin domain. Unlike TXNRD1 or 2, this domain allows this isozyme, to participate in both thioredoxin and glutathione systems [23,163]. Using cell culture models, Kipp et al. were able to demonstrate that TXNRD3 may also be a target for WNT, implying a role for TXNRD3 in proliferation, apoptosis and tumorigenesis [116]. Interestingly, mRNA expression of the type 3 deiodinase DIO3, a selenoprotein whose role is to inactivate the thyroid hormone T3, which regulates the bioavailability of thyroid hormones in tissues, was higher in human intestinal adenomas and carcinomas than in healthy intestinal tissue as quantitated in 24 CRC and matched normal tissue from the same patients. Similarly, mRNA and protein levels of DIO3 were highly expressed in six investigated colon cancer cell lines. In contrast, expression was found to be reduced in the most aggressive lesions [164]. The functional meaning of this remains to be elucidated, as expression of thyroid hormone deiodinases varies with development, cell type, and pathology [165]. Surprisingly, it appeared that DIO3 may be a β-catenin target, thus providing a cross-talk between the WNT/β-catenin and the thyroid hormone signaling pathways [164]. With the canonical WNT/β-catenin signaling pathway’s importance in colorectal tumors as described earlier, a significant role for DIO3 in CRCs is likely.
Other contributors: Single nucleotide polymorphisms suggest involvement of selenoproteins in CRC risk
Though not directly attributable to any specifics hallmark of cancer, it is important to note that various epidemiological studies have suggested associations between single nucleotide polymorphisms (SNPs) in at least seven different selenoprotein genes and risk of CRC. SNPs in GPX genes include those in GPX1 at loci 75 (proline → arginine), 192 (alanine → threonine), and 198 (proline → leucine). Of functional importance are especially those at GPX1 locus 198 [166], and allelic loss at GPX1 locus 198 has been correlated with various cancers [167]. A study based on randomly selected samples from a Gastrointestinal Cancers Tissue Bank (Chicago, IL) from tumor and normal adjacent tissue from the same individuals identified loss of GPX1 heterozygosity in CRC cases at a frequency of 42% [168]. Interestingly, subsequent epidemiological analyses did not find genetic variants in the GPX1 gene to be associated with colorectal adenomas [169], nor was loss of heterozygosity associated with benign colorectal adenomas in a population of patients in Tucson (AZ) [170]. While this suggests that loss of GPX1 heterozygosity may occur at a later stage in colorectal tumor development, earlier studies in both a Norwegian population and in a Danish population did not show an association between SNPs in the GPX1 locus 198 and CRC [171,172]. Instead, the latter study showed a significant interaction between alcohol consumption and smoking, which was associated with increased CRC risk in homozygous GPX1 (198) Leucine carriers. Although there is plenty of evidence linking GPX1 to inflammation and other diseases, there doesn’t appear to be a strong consensus regarding an association of SNPs in the GPX1 gene and CRC risk, because overall nutrition, as well as environmental and genetic factors are thought to influence the GPX1 expression, its catalytic activity, and thus its biological effects [173].
Human polymorphisms in the GPX2 gene have been described [169,174,175]. Human polymorphisms in the GPX2 gene initially appeared to be associated with a lower risk for rectal cancers with TP53 mutations; however, this did not remain statistically significant after adjustment for multiple comparisons [175], and no association with GPX2 SNPs were found with colorectal adenoma risk [169]. Thus, it remains to be elucidated, whether genetic variants in GPX2 result in modifications to CRC risk. In a population of CRC patients and controls from the Czech Republic, as well as in a study population from the United Kingdom, a GPX4 SNP in the 3’ UTR at position 718 (rs713041) was associated with the risk of CRC and increased ulcerative colitis. Intriguingly, the variant carried in these two studies was not the same, and in a study in a Korean population, this association was not replicated [176–178]. Using a reporter construct to assess SECIS function, Bermano et al. showed increased reporter activity with the C variant compared to the T variant, independent of Se concentration [176]; however, its relevance in vivo remains unknown. Interestingly, a significant two-loci interaction was observed with this GPX4 SNP and rs960531 in TXNRD2, but if this results in a functional interaction at the protein structure level remains to be elucidated [178]. Further in vivo analyses of GPX4 SNP functions are needed to assess whether the variant with greater activity is beneficial to malignant cells, possibly by combating oxidative stress, which may alter the inflammatory response, and thus aid tumorigenesis.
Evidence for TXNRD1’s involvement in CRC from human studies, using a multiethnic panel of 102 subjects, suggested an overall association with advanced colorectal adenoma risk, resulting in a highly significant 80% risk reduction for carriers of the variant allele at TXNRD1 IVS1-181 C→G [169]. However, because this SNP is located on the small EID3 gene (EP300 Interacting Inhibitor of Differentiation), which is nested within the intron between TXNRD1 splice variants 2 and 3 [179], it remains to be elucidated if this SNP’s association with colorectal adenoma risk is really due to TXNRD1 or speaks more to the function of EID3 [169]. A subsequent analysis of two population-based case-control studies of colon and rectal cancers showed that SNPs in TXNRD1 (rs17202060) correlated with colon cancer, but the association was not statistically significant after adjustment for multiple comparisons. However, interactions between TXNRD1 rs4964778 and non-steroidal anti-inflammatory drug use remained statistically significant for colon cancer [101]. Thus, results from epidemiological analyses suggest that TXNRD1 expression may impact colorectal malignancies, and high expression of TXNRD1 appears to negatively correlate with patient outcome in CRC [180]. However, given the many functions of the various splice variants, and the ability for TXNRD1 to function in both cancer prevention and promotion, it remains to be elucidated just how much of a role these SNPs play in human colorectal malignancies. SNPs in TXNRD2, which is usually found in mitochondria, and SNPs in TXNRD3, which is primarily found expressed in the testis, have also been associated with CRC [101,178]. Additional experiments will be required to directly link TXNRD2 and TXNRD3 with CRC in humans.
Two SNPs in the SELENOF gene have been identified, at positions 811 (C/T) and 1125 (G/A), both located in the 3’ UTR, including one within a SECIS element. Using reporter constructs, Hu et al. found that the T at position 811 and A at position 1125, individually, influenced SECIS function in a Se-dependent manner [181]. In a later study analyzing sequence data obtained from healthy controls and patients with CRC in a Korean population, these SNPs were found to be associated with cancer risk. The minor alleles for T (rs5845) and A (rs5859) were associated with an increased risk of rectal cancer in males (odds ratios of and 2.51, respectively), suggesting links between SELENOF and rectal cancer risk [182]. Furthermore, in vitro data show that a T at position 811 or an A at position 1125 decreases UGA read-through, resulting in decreased expression of full length SELENOF, presumably decreasing CRC risk. It seems that, if expression of SELENOF is Se-dependent and Se levels are lower in individuals with colorectal malignancies, a decreased SELENOF expression in these CRC patients could be beneficial, depending on the stage of tumorigenesis.
In randomly selected samples from controls and patients with advanced distal colon adenomas that were collected as part of a larger cancer screening trial, four SNPs within the SELENOP gene were significantly associated with advanced colorectal adenoma risk. One rare SNP located in the promotor region (−4166 C→G) was shown to be associated with increased risk. Among the other three SNPs, which are located in the 3’ UTR, two SNPs were shown to increase adenoma risk (rs12055266, rs3797310 G→A), and one correlated with decreased risk (rs2972994 C→T) of CRC [169]. In a population of CRC patients from the Czech Republic, the SELENOP (rs7579) SNP was found to be associated with malignancy, where individuals with at least one A allele correlated with increased CRC risk. Furthermore, a significant two-loci interaction was observed between SELENOP and either SELENOF or GPX4, suggesting that SNPs in these selenoproteins may influence risk of CRC [178]. Clearly, there is an association between SELENOP polymorphisms and risk of CRC, which is likely due to functional differences in gene expression.
Colorectal cancer risk was found to be modified by SELENOS SNPs in patients from the Czech Republic, and rs34713741 variants, in particular, were suggested to play a role in colon cancer development [25,178]. Similarly, there was a mean odds ratio of 2.25 in female Koreans for the rs34713741 SNP in SELENOS with the T variant being associated with higher risk of rectal cancer [182]. A subsequent study based on two US populations found that another SELENOS SNP (rs9874) interacted with estrogen to modify colon cancer risk, similarly to what has been described for TXNRD2, SELENOW, and SELENOF [101]. Estrogen has been shown to play complex pro- and anti-inflammatory roles [183], potentially providing a protective role in CRC for specific subpopulations [184].
Human genome studies revealed three SELENOW SNPs (rs10412896; rs3786777; rs2042286) that interacted with estrogen to modify colon cancer or rectal cancer risk [101]. Based on findings in other human malignancies, it is unclear whether SELENOW SNPs affect SELENOW expression, and whether increased SELENOW expression contributes to the development of colorectal tumors. It is possible, that SELENOW expression simply correlates with increased Se status, which contributes to increased risk of colorectal tumorigenesis through pathways that include functions of other selenoproteins sensitive to the organism’s Se status.
Selenoproteins with unknown roles in CRC
For several selenoproteins, expression in tissues of the intestinal tract appear to be minimal or haven’t been described (DIO1, DIO2, GPX 6, SELENOI, SELENOV, SELENOO, SELENOT), therefore currently not supporting a role in CRC. For four additional selenoproteins, relationships to CRC are unclear, but appear to be more likely. SELENOK is a trans-membrane protein that appears to be involved in ER regulation of calcium flux and palmitoylation reactions [185,186]. Evidence from in vitro [187], clinical [185,188] and epidemiological studies [189] suggest that SELENOK expression may be impacted by some malignancies. SELENOM, formerly known as SELM, is a structural homolog to SELENOF. Similarly, SELENOM is an ER-resident protein with potential for redox activity [190], and it has been implicated in preventing oxidative stress [191], and to be indirectly involved in the quality control pathways of protein folding [92,190,192]. Whereas expression of SELENOM appears particularly high in the brain [193,194] and liver tissues [192], information on SELENOM expression in normal or malignant colorectal tissues or cells is sparse. mRNA expression analyses of mice on various Se diets suggested that Selenom expression in colorectal tissues is sensitive to Se intake [92], suggesting a potential role in CRC that would benefit from further elucidation. Similarly, another ER-resident selenoprotein, SELENON (formerly known as SelN or SepN1), is thought to be involved in the cell’s protection against oxidative stress, as well as in redox-related calcium homeostasis (reviewed in [186]), and four SNPs of SELENON (rs11247735; rs2072749; rs4659382; rs718391) have been associated with rectal cancer [101]. Mammalian Selenophosphate Synthetase 2 (SEPHS2, also known as SPS2) is an important selenoenzyme involved in generating the Se donor for Sec, and thus an important regulator of selenoprotein synthesis [195]. Lower mRNA SEPHS2 expression levels were observed in breast cancer tissues [196], and SEPHS2 may play a role by regulating Sec, and thus selenoprotein synthesis. This may impact colorectal expression of selenoproteins and thus indirectly affect pathologies and malignancies in the intestinal tract.
Conclusion
Selenium plays an important role in human health and disease, including colorectal inflammation and cancer. A decreased Se status is often correlated with increased CRC risk. Although the exact mechanisms in many cases remain to be elucidated, selenoproteins appear to affect multiple signaling pathways that reflect those properties of cancer cells often referred to as ‘hallmarks of cancer’ [27,28]. Especially those selenoproteins that link directly or indirectly to redox homeostasis via regulation of oxidative stress, apoptosis, or inflammation and immune responses (e.g., GPX1-4, TXNRD1, SELENOF, SELENOP), but also those that have been linked to the canonical WNT/β-catenin signaling pathway (e.g., DIO3, GPX2, TXNRD3, SELENOP), appear to have a direct impact on CRC risk and development. This is likely because signaling pathways in redox homeostasis network with classical signal transduction pathways in every cancer hallmark and enabling characteristic [29]. Imbalances in these pathways may be the driving force of tumor initiation and progression, thus ultimately affecting colorectal tumorigenesis. In recent years, evidence of the contributions of the intestinal microbiome to colorectal pathogenesis has emerged. Because both dietary Se as well as selenoproteins expression appear to regulate intestinal microflora, this further intertwines selenoproteins with redox homeostasis and inflammation. Human genome studies have linked various SNPs in selenoproteins genes with CRC risk, and there is strong evidence that a number of selenoprotein SNPs have functional consequences (e.g., GPX1-2, SELENOP, TXNRD1). Thus, even though the functions for some selenoproteins remain to be elucidated, strong links have been provided for several selenoproteins and the development or progression of CRC by in vitro, in vivo, and evidence from human clinical trials (Table 1). Thus, selenoproteins could provide targets for cancer treatment or prevention strategies, and additional in vitro, animal, and clinical research is necessary to elucidate such potential.
Table 1.
List of selenoproteins, by category, and their potential role in CRC via pre-clinical or clinical data.
| Selenoprotein Category |
Gene | SNP, relevance for CRC |
Role in CRC | Selected References |
|---|---|---|---|---|
| Glutathione Peroxidases | GPX1 | Various, including functional SNPs | Likely, but unclear | [38,44,110,111,167] |
| GPX2 | Yes, but effect unclear | Implicated in prevention & promotion | [38,52,53,110,111,116,169] | |
| GPX3 | Yes, but effect unclear | Potentially, but unclear | [112,147,148] | |
| GPX4 | Yes, but effect unclear | Potentially, but unclear | [105,177,178] | |
| GPX6 | unknown | unknown | [23] | |
| Thioredoxin Reductases | TXNRD1 | Yes, but effect unclear | Implicated in prevention & promotion | [20,47,61,63,71,75,76] |
| TXNRD2 | Yes, but effect unclear | Potentially, but unclear | [101,178] | |
| TXNRD3 | Yes, but effect unclear | Potentially, but unclear | [101,116] | |
| Thyroid Hormone Deiodinases | DIO1 | Yes, but effect unclear | Unknown | [197] |
| DIO2 | Unknown | Unknown | [35] | |
| DIO3 | Yes, but effect unclear | Likely, possibly through WNT-pathway | [164,197] | |
| ER-resident selenoproteins | SELENOF | Yes, but effect unclear | Implicated in prevention/promotion | [20,76,94,96,127,181,198] |
| SELENOI | Unknown | Unknown | [36] | |
| SELENOK | Yes, but unclear | Unknown | [187,189] | |
| SELENOM | unknown | Unknown | [92,190] | |
| SELENON | Yes, associated with rectal cancer | Potentially, but unclear | [101,186,199] | |
| SELENOS | Yes, CRC risk modified | Likely, possibly through immune response | [100,101,178,182,200] | |
| SELENOT | Unknown, no evidence | Unknown | [201,202] | |
| Other selenoproteins | SELENOH | Yes, no association with CRC | Unknown | [92,101] |
| SELENOO | Unknown | Unknown | [203] | |
| SELENOV | Unknown, no evidence | Unknown | [23] | |
| SELENOW | Yes, CRC association via estrogen | Potentially, possibly through cell cycle regulation | [101] | |
| Methionine sulfoxide reductase | MSRB1 | Unknown | Likely, through immune response | [149,151,153] |
| Selenoprotein Synthesis | SEPHS2 | Unknown | Unknown | [195] |
| Selenium Transport | SELENOP | Yes, CRC risk association | Likely, through immune response or WNT-pathway | [21,53,87,169,178,204] |
Highlights.
Many selenoproteins have been implicated to play a direct or indirect role in CRC
Highly likely role in CRC: DIO3, GPX1-2, TXNRD1, DIO3, SELENOF,P,S
Potential role in CRC: GPX3-4, MSRB1, TXNRD2-3, SELENOM,N,W
Selenoproteins are potential targets for CRC treatment or prevention strategies
Acknowledgments
Funding: This work was supported by Towson University and the National Institutes of Health.
Abbreviations
- CRC
Colorectal Cancer
- DIO
Iodothyronine Deiodinases
- ER
Endoplasmic Reticulum
- GPX
Glutathione Peroxidase
- GSH
Glutathione
- NFκB
Nuclear Factor κ-light-chain-enhancer of activated B cells
- NOX
NADPH-oxidase
- NPC
Nutritional Prevention of Cancer
- PLCO
Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial
- ROS
Reactive Oxygen Species
- Se
Selenium
- Sec
Selenocysteine
- SECIS
Sec Insertion Sequence
- SELECT
Selenium and Vitamin E Cancer Prevention Trial
- SNP
Single Nucleotide Polymorphism
- tRNA
Transfer RNA
- TXNRD
Thioredoxin Reductase
- UPR
Unfolded Protein Response
- UTR
Untranslated Region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–193. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Robert Koch Institut. Cancer in Germany 2011/2012. Berlin, Germany: Robert Koch Institute & Association of Population-based Cancer Registries in Germany; 2016. [Google Scholar]
- 4.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 5.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015 doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 6.Russo MW, Murray SC, Wurzelmann JI, Woosley JT, Sandler RS. Plasma selenium levels and the risk of colorectal adenomas. Nutr Cancer. 1997;28:125–129. doi: 10.1080/01635589709514563. [DOI] [PubMed] [Google Scholar]
- 7.Peters U, Takata Y. Selenium and the prevention of prostate and colorectal cancer. Mol Nutr Food Res. 2008;52:1261–1272. doi: 10.1002/mnfr.200800103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rayman MP. Selenium in cancer prevention: a review of the evidence and mechanism of action. Proc Nutr Soc. 2005;64:527–542. doi: 10.1079/pns2005467. [DOI] [PubMed] [Google Scholar]
- 9.Waters DJ, Chiang EC. Five Threads: How U-Shaped Thinking Weaves Together Dogs, Men, Selenium, and Prostate Cancer Risk. Free Radic Biol Med. doi: 10.1016/j.freeradbiomed.2017.12.039. (in press) [DOI] [PubMed] [Google Scholar]
- 10.Schrauzer GN, White DA, Schneider CJ. Cancer mortality correlation studies--III: statistical associations with dietary selenium intakes. Bioinorg Chem. 1977;7:23–31. doi: 10.1016/s0006-3061(00)80126-x. [DOI] [PubMed] [Google Scholar]
- 11.Clark LC, Combs GFJ, Turnbull BW, Slate E, Chalker D, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957–1963. [PubMed] [Google Scholar]
- 12.Lippman SM, Goodman PJ, Klein EA, Parnes HL, Thompson IM., Jr Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT) J Natl Cancer Inst. 2005;97:94–102. doi: 10.1093/jnci/dji009. [DOI] [PubMed] [Google Scholar]
- 13.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;1 [Google Scholar]
- 14.Lance P, Alberts DS, Thompson PA, Fales L, Wang F, et al. Colorectal adenomas in participants of the SELECT randomized trial of selenium and vitamin E for prostate cancer prevention. Cancer Prev Res. 2017;10:45–54. doi: 10.1158/1940-6207.CAPR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehdi Y, Hornick JL, Istasse L, Dufrasne I. Selenium in the environment, metabolism and involvement in body functions. Molecules. 2013;18:3292–3311. doi: 10.3390/molecules18033292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reilly C. Selenium in food and health. New York, NY: Springer US; 2006. [Google Scholar]
- 17.Duffield-Lillico AJ, Dalkin BL, Reid ME, Turnbull BW, Slate EH, et al. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91:608–612. doi: 10.1046/j.1464-410x.2003.04167.x. [DOI] [PubMed] [Google Scholar]
- 18.Duffield-Lillico AJ, Reid ME, Turnbull BW, Combs GF, Slate EH, et al. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: a summary report of the Nutritional Prevention of Cancer Trial. Cancer Epidemiol Biomarkers Prev. 2002;11:630–639. [PubMed] [Google Scholar]
- 19.Connelly-Frost A, Poole C, Satia JA, Kupper LL, Millikan RC, et al. Selenium, folate, and colon cancer. Nutr Cancer. 2009;61:165–178. doi: 10.1080/01635580802404188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends in Biochemical Sciences. 2014;39:112–120. doi: 10.1016/j.tibs.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: Molecular pathways and physiological roles. Physiological Reviews. 2014;94:739–777. doi: 10.1152/physrev.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinkerton MH, Copeland PR. Eukaryotic mechanisms of selenocysteine incorporation and its reconsitution in vitro. In: Hatfield DL, Schweizer U, Tsuji PA, Gladyshev VN, editors. Selenium - Its molecular biology and role in human health. 4. New York, NY: Springer Science+Business Media, LLC; 2016. [Google Scholar]
- 23.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 24.Méplan C. Association of single nucleotide polymorphisms in selenoprotein genes with cancer risk. Methods Mol Biol. 2018 doi: 10.1007/978-1-4939-7258-6_22. 7258-7256_7222. [DOI] [PubMed] [Google Scholar]
- 25.Meplan C, Hesketh J. Selenium and cancer: a story that should not be forgotten-insights from genomics. Cancer Treat Res. 2014;159:145–166. doi: 10.1007/978-3-642-38007-5_9. [DOI] [PubMed] [Google Scholar]
- 26.Squires J, Berry MJ. Selenium, selenoproteins, and cancer. Hawaii Med J. 2006;65:239–240. [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:50–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Hornsveld M, Dansen TB. The hallmarks of cancer from a redox perspective. Antioxid Redox Signal. 2016;25:300–325. doi: 10.1089/ars.2015.6580. [DOI] [PubMed] [Google Scholar]
- 30.Truong TH, Carroll KS. Redox regulation of protein kinases. Crit Rev Biochem Mol Biol. 2013;48:332–356. doi: 10.3109/10409238.2013.790873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:105. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciccarese F, Ciminale V. Escaping Death: Mitochondrial Redox Homeostasis in Cancer Cells. Front Oncol. 2017;7 doi: 10.3389/fonc.2017.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higa A, Chevet E. Redox signaling loops in the unfolded protein response. Cell Signal. 2012;24:1548–1555. doi: 10.1016/j.cellsig.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Eletto D, Chevet E, Argon Y, Appenzeller-Herzog C. Redox controls UPR to control redox. J Cell Sci. 2014;127:3649–3658. doi: 10.1242/jcs.153643. [DOI] [PubMed] [Google Scholar]
- 35.Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12:839–849. doi: 10.1089/ars.2009.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varlamova EG, Goltyaev MV, Novoselov VI, Fesenko EE. Cloning, intracellular localization, and expression of the mammalian selenocysteine-containing protein SELENOI (SelI) in tumor cell lines. Dokl Biochem Biophys. 2017;476:320–322. doi: 10.1134/S160767291705012X. [DOI] [PubMed] [Google Scholar]
- 37.Marciel MP, Hoffmann PR. Selenoproteins and metastasis. Adv Cancer Res. 2017;136:85–108. doi: 10.1016/bs.acr.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brigelius-Flohé R, Kipp A. Glutathione peroxidases in different stages of carcinogenesis. Biochim Biophys Acta. 2009;11:13. doi: 10.1016/j.bbagen.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Kipp AP. Selenium-dependent glutathione peroxidases during tumor development. Adv Cancer Res. 2017;136:109–138. doi: 10.1016/bs.acr.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Flohé L. The impact of thiol peroxidases on redox regulation. Free Radic Res. 2016;50:126–142. doi: 10.3109/10715762.2015.1046858. [DOI] [PubMed] [Google Scholar]
- 41.Flohé L, Brand I. Kinetics of glutathione peroxidase. Biochim Biophys Acta. 1969;191:541–549. doi: 10.1016/0005-2744(69)90347-7. [DOI] [PubMed] [Google Scholar]
- 42.Flohé L, Günzler WA, Schock HH. Glutathione peroxidase: a selenoenzyme. FEBS Lett. 1973;32:132–134. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- 43.Frampton J, Conkie D, Chambers I, McBain W, Dexter M, et al. Changes in minor transcripts from the alpha 1 and beta maj globin and glutathione peroxidase genes during erythropoiesis. Nucleic Acids Res. 1987;15:3671–3688. doi: 10.1093/nar/15.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochimica et Biophysica Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Murawaki Y, Tsuchiya H, Kanbe T, Harada K, Yashima K, et al. Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 2008;259:218–230. doi: 10.1016/j.canlet.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Nalkiran I, Turan S, Arikan S, Kahraman OT, Acar L, et al. Determination of gene expression and serum levels of MnSOD and GPX1 in colorectal cancer. Anticancer Res. 2015;35:255–259. [PubMed] [Google Scholar]
- 47.Yagublu V, Arthur JR, Babayeva SN, Nicol F, Post S, et al. Expression of selenium-containing proteins in human colon carcinoma tissue. Anticancer Res. 2011;31:2693–2698. [PubMed] [Google Scholar]
- 48.Brigelius-Flohé R, Kipp AP. Physiological functions of GPx2 and its role in inflammation-triggered carcinogenesis. Ann N Y Acad Sci. 2012;1259:19–25. doi: 10.1111/j.1749-6632.2012.06574.x. [DOI] [PubMed] [Google Scholar]
- 49.Esworthy RS, Mann JR, Sam M, Chu FF. Low glutathione peroxidase activity in Gpx1 knockout mice protects jejunum crypts from gamma-irradiation damage. Am J Physiol Gastrointest Liver Physiol. 2000;279 doi: 10.1152/ajpgi.2000.279.2.G426. [DOI] [PubMed] [Google Scholar]
- 50.Mörk H, Lex B, Scheurlen M, Dreher I, Schutze N, et al. Expression pattern of gastrointestinal selenoproteins--targets for selenium supplementation. Nutr Cancer. 1998;32:64–70. doi: 10.1080/01635589809514720. [DOI] [PubMed] [Google Scholar]
- 51.Wingler K, Müller C, Schmehl K, Florian S, Brigelius-Flohé R. Gastrointestinal glutathione peroxidase prevents transport of lipid hydroperoxides in CaCo-2 cells. Gastroenterology. 2000;119:420–430. doi: 10.1053/gast.2000.9521. [DOI] [PubMed] [Google Scholar]
- 52.Florian S, Wingler K, Schmehl K, Jacobasch G, Kreuzer OJ, et al. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic Res. 2001;35:655–663. doi: 10.1080/10715760100301181. [DOI] [PubMed] [Google Scholar]
- 53.Mörk H, al-Taie OH, Bähr K, Zierer A, Beck C, et al. Inverse mRNA expression of the selenocysteine-containing proteins GI-GPx and SeP in colorectal adenomas compared with adjacent normal mucosa. Nutr Cancer. 2000;37:108–116. doi: 10.1207/S15327914NC3701_14. [DOI] [PubMed] [Google Scholar]
- 54.Esworthy RS, Yang L, Frankel PH, Chu FF. Epithelium-specific glutathione peroxidase, Gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J Nutr. 2005;135:740–745. doi: 10.1093/jn/135.4.740. [DOI] [PubMed] [Google Scholar]
- 55.Saretzki G, Armstrong L, Leake A, Lako M, von Zglinicki T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells. 2004;22:962–971. doi: 10.1634/stemcells.22-6-962. [DOI] [PubMed] [Google Scholar]
- 56.Florian S, Krehl S, Loewinger M, Kipp A, Banning A, et al. Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic Biol Med. 2010;49:1694–1702. doi: 10.1016/j.freeradbiomed.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohé R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brigelius-Flohé R, Müller M, Lippmann D, Kipp AP. The yin and yang of nrf2-regulated selenoproteins in carcinogenesis. Int J Cell Biol. 2012;2012:486147. doi: 10.1155/2012/486147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Te Velde AA, Pronk I, de Kort F, Stokkers PC. Glutathione peroxidase 2 and aquaporin 8 as new markers for colonic inflammation in experimental colitis and inflammatory bowel diseases: an important role for H2O2? Eur J Gastroenterol Hepatol. 2008;20:555–560. doi: 10.1097/MEG.0b013e3282f45751. [DOI] [PubMed] [Google Scholar]
- 60.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc Natl Acad Sci U S A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arnér ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;6:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 62.Short SP, Williams CS. Selenoproteins in Tumorigenesis and Cancer Progression. Adv Cancer Res. 2017;136:49–83. doi: 10.1016/bs.acr.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 64.Arnér ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16:420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 65.Björkhem-Bergman L, Johansson M, Morgenstern R, Rane A, Ekström L. Prenatal expression of thioredoxin reductase 1 (TRXR1) and microsomal glutathione transferase 1 (MGST1) in humans. FEBS Open Bio. 2014;4:886–891. doi: 10.1016/j.fob.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cebula M, Moolla N, Capovilla A, Arner ES. The rare TXNRD1_v3 ("v3") splice variant of human thioredoxin reductase 1 protein is targeted to membrane rafts by N-acylation and induces filopodia independently of its redox active site integrity. J Biol Chem. 2013;288:10002–10011. doi: 10.1074/jbc.M112.445932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Damdimopoulou PE, Miranda-Vizuete A, Arner ES, Gustafsson JA, Damdimopoulos AE. The human thioredoxin reductase-1 splice variant TXNRD1_v3 is an atypical inducer of cytoplasmic filaments and cell membrane filopodia. Biochim Biophys Acta. 2009;10:3. doi: 10.1016/j.bbamcr.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 68.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 69.Liu D, Xu Y. p53, oxidative stress, and aging. Antioxid Redox Signal. 2011;15:1669–1678. doi: 10.1089/ars.2010.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kitagishi Y, Matsuda S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review) Int J Mol Med. 2013;31:511–515. doi: 10.3892/ijmm.2013.1235. [DOI] [PubMed] [Google Scholar]
- 71.Gladyshev VN, Factor VM, Housseau F, Hatfield DL. Contrasting patterns of regulation of the antioxidant selenoproteins, thioredoxin reductase, and glutathione peroxidase, in cancer cells. Biochem Biophys Res Commun. 1998;251:488–493. doi: 10.1006/bbrc.1998.9495. [DOI] [PubMed] [Google Scholar]
- 72.Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, et al. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- 73.Harris IS, Brugge JS. Cancer: The enemy of my enemy is my friend. Nature. 2015;527:170–171. doi: 10.1038/nature15644. [DOI] [PubMed] [Google Scholar]
- 74.Schumacker PT. Reactive oxygen species in cancer: a dance with the devil. Cancer Cell. 2015;27:156–157. doi: 10.1016/j.ccell.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Arnér ESJ. Targeting the Selenoprotein Thioredoxin Reductase 1 for Anticancer Therapy. Adv Cancer Res. 2017;136:139–151. doi: 10.1016/bs.acr.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 76.Hatfield DL, Yoo MH, Carlson BA, Gladyshev VN. Selenoproteins that function in cancer prevention and promotion. Biochim Biophys Acta. 2009;11:9. doi: 10.1016/j.bbagen.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hatfield DL, Carlson BA, Tsuji PA, Tobe R, Gladyshev VN. Selenium and Cancer. In: Collins JF, editor. Molecular, Genetic, and Nutritional Aspects of Major and Trace Minerals. New York: Elsevier; 2016. pp. 463–473. [Google Scholar]
- 78.Tsuji PA, Carlson BA, Lee BJ, Gladyshev VN, Hatfield DL. Interplay of selenoproteins and different antioxidant systems in various cancers. In: Hatfield DL, Schweizer U, Tsuji PA, Gladyshev VN, editors. Selenium - Its molecular biology and role in human health. 4. New York: Springer Science+Business Media, LLC; 2016. [Google Scholar]
- 79.Koedrith P, Seo YR. Enhancement of the efficacy of mitomycin C-mediated apoptosis in human colon cancer cells with RNAi-based thioredoxin reductase 1 deficiency. Exp Ther Med. 2011;2:873–878. doi: 10.3892/etm.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Honeggar M, Beck R, Moos PJ. Thioredoxin reductase 1 ablation sensitizes colon cancer cells to methylseleninate-mediated cytotoxicity. Toxicol Appl Pharmacol. 2009;241:348–355. doi: 10.1016/j.taap.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karlenius TC, Tonissen KF. Thioredoxin and cancer: a role for thioredoxin in all states of tumor oxygenation. Cancers. 2010;2:209–232. doi: 10.3390/cancers2020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Naranjo-Suarez S, Carlson BA, Tsuji PA, Yoo MH, Gladyshev VN, et al. HIF-independent regulation of thioredoxin reductase 1 contributes to the high levels of reactive oxygen species induced by hypoxia. PLoS One. 2012;7:13. doi: 10.1371/journal.pone.0030470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Combs GF, Jr, Watts JC, Jackson MI, Johnson LK, Zeng H, et al. Determinants of selenium status in healthy adults. Nutr J. 2011;10:1475–2891. doi: 10.1186/1475-2891-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Al-Taie OH, Uceyler N, Eubner U, Jakob F, Mörk H, et al. Expression profiling and genetic alterations of the selenoproteins GI-GPx and SePP in colorectal carcinogenesis. Nutr Cancer. 2004;48:6–14. doi: 10.1207/s15327914nc4801_2. [DOI] [PubMed] [Google Scholar]
- 85.Speckmann B, Bidmon HJ, Borchardt A, Sies H, Steinbrenner H. Intestinal selenoprotein P in epithelial cells and in plasma cells. Arch Biochem Biophys. 2014;541:30–36. doi: 10.1016/j.abb.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 86.Barrett CW, Short SP, Williams CS. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell Mol Life Sci. 2017;74:607–616. doi: 10.1007/s00018-016-2339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burk RF, Hill KE. Selenoprotein P. an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 88.Kurokawa S, Eriksson S, Rose KL, Wu S, Motley AK, et al. Sepp1(UF) forms are N-terminal selenoprotein P truncations that have peroxidase activity when coupled with thioredoxin reductase-1. Free Radic Biol Med. 2014;69:67–76. doi: 10.1016/j.freeradbiomed.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dikiy A, Novoselov SV, Fomenko DE, Sengupta A, Carlson BA, et al. SelT, SelW, SelH, and Rdx12: genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry. 2007;46:6871–6882. doi: 10.1021/bi602462q. [DOI] [PubMed] [Google Scholar]
- 90.Jeong DW, Kim TS, Chung YW, Lee BJ, Kim IY. Selenoprotein W is a glutathione-dependent antioxidant in vivo. FEBS Letters. 2002;517:225–228. doi: 10.1016/s0014-5793(02)02628-5. [DOI] [PubMed] [Google Scholar]
- 91.Chung YW, Jeong D, Noh OJ, Park YH, Kang SI, et al. Antioxidative role of selenoprotein W in oxidant-induced mouse embryonic neuronal cell death. Mol Cells. 2009;27:609–613. doi: 10.1007/s10059-009-0074-3. [DOI] [PubMed] [Google Scholar]
- 92.Kipp A, Banning A, van Schothorst EM, Méplan C, Schomburg L, et al. Four selenoproteins, protein synthesis, and Wnt signaling are particularly sensitive to limited selenium intake in mouse colon. Mol Nutr Food Res. 2009;53:1561–1572. doi: 10.1002/mnfr.200900105. [DOI] [PubMed] [Google Scholar]
- 93.Kipp AP, Frombach J, Deubel S, Brigelius-Flohé R. Selenoprotein W as biomarker for the efficacy of selenium compounds to act as source for selenoprotein biosynthesis. Methods Enzymol. 2013;527:87–112. doi: 10.1016/B978-0-12-405882-8.00005-2. [DOI] [PubMed] [Google Scholar]
- 94.Gladyshev VN, Jeang KT, Wootton JC, Hatfield DL. A new human selenium-containing protein. Purification, characterization, and cDNA sequence. J Biol Chem. 1998;273:8910–8915. doi: 10.1074/jbc.273.15.8910. [DOI] [PubMed] [Google Scholar]
- 95.Kumaraswamy E, Malykh A, Korotkov KV, Kozyavkin S, Hu Y, et al. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem. 2000;275:35540–35547. doi: 10.1074/jbc.M004014200. [DOI] [PubMed] [Google Scholar]
- 96.Labunskyy VM, Yoo MH, Hatfield DL, Gladyshev VN. Sep15, a thioredoxin-like selenoprotein, is involved in the unfolded protein response and differentially regulated by adaptive and acute ER stresses. Biochemistry. 2009;48:8458–8465. doi: 10.1021/bi900717p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kasaikina MV, Fomenko DE, Labunskyy VM, Lachke SA, Qiu W, et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J Biol Chem. 2011;286:33203–33212. doi: 10.1074/jbc.M111.259218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yim SH, Everley RA, Schildberg FA, Lee SG, Orsi A, et al. Role of Selenof as a gatekeeper of secreted disulfide-rich glycoproteins. Cell Rep. 2018;23:1387–1398. doi: 10.1016/j.celrep.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gao Y, Pagnon J, Feng HC, Konstantopolous N, Jowett JB, et al. Secretion of the glucose-regulated selenoprotein SEPS1 from hepatoma cells. Biochem Biophys Res Commun. 2007;356:636–641. doi: 10.1016/j.bbrc.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 100.Turanov AA, Shchedrina VA, Everley RA, Lobanov AV, Yim SH, et al. Selenoprotein S is involved in maintenance and transport of multiprotein complexes. Biochem J. 2014;462:555–565. doi: 10.1042/BJ20140076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Slattery ML, Lundgreen A, Welbourn B, Corcoran C, Wolff RK. Genetic variation in selenoprotein genes, lifestyle, and risk of colon and rectal cancer. PLoS One. 2012;7:e37312. doi: 10.1371/journal.pone.0037312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Handy DE, Lubos E, Yang Y, Galbraith JD, Kelly N, et al. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J Biol Chem. 2009;284:11913–11921. doi: 10.1074/jbc.M900392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, et al. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med. 2017;112:1–11. doi: 10.1016/j.freeradbiomed.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 104.Cole-Ezea P, Swan D, Shanley D, Hesketh J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free Radic Biol Med. 2012;53:488–497. doi: 10.1016/j.freeradbiomed.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 105.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2017;16:31438–31431. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 106.Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31. doi: 10.1016/j.redox.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Müller MF, Florian S, Pommer S, Osterhoff M, Esworthy RS, et al. Deletion of glutathione peroxidase-2 inhibits azoxymethane-induced colon cancer development. PLoS One. 2013;8:e72055. doi: 10.1371/journal.pone.0072055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jongen JMJ, van der Waals LM, Trumpi K, Laoukili J, Peters NA, et al. Downregulation of DNA repair proteins and increased DNA damage in hypoxic colon cancer cells is a therapeutically exploitable vulnerability. Oncotarget. 2017;8:86296–86311. doi: 10.18632/oncotarget.21145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.El Hassani RA, Benfares N, Caillou B, Talbot M, Sabourin JC, et al. Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol. 2005;288:9. doi: 10.1152/ajpgi.00198.2004. [DOI] [PubMed] [Google Scholar]
- 110.Chu FF, Esworthy RS, Doroshow JH, Grasberger H, Donko A, et al. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 2017;11:144–156. doi: 10.1016/j.redox.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Esworthy RS, Kim BW, Chow J, Shen B, Doroshow JH, et al. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and-2. Free Radic Biol Med. 2014;68:315–325. doi: 10.1016/j.freeradbiomed.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barrett CW, Ning W, Chen X, Smith JJ, Washington MK, et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013;73:1245–1255. doi: 10.1158/0008-5472.CAN-12-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fink EE, Mannava S, Bagati A, Bianchi-Smiraglia A, Nair JR, et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia. 2016;30:104–111. doi: 10.1038/leu.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Choi JH, Kim TN, Kim S, Baek SH, Kim JH, et al. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002;22:3331–3335. [PubMed] [Google Scholar]
- 115.Bu L, Li W, Ming Z, Shi J, Fang P, et al. Inhibition of TrxR2 suppressed NSCLC cell proliferation, metabolism and induced cell apoptosis through decreasing antioxidant activity. Life Sci. 2017;178:35–41. doi: 10.1016/j.lfs.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 116.Kipp AP, Müller MF, Göken EM, Deubel S, Brigelius-Flohé R. The selenoproteins GPx2, TrxR2 and TrxR3 are regulated by Wnt signalling in the intestinal epithelium. Biochim Biophys Acta. 2012;10:7. doi: 10.1016/j.bbagen.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 117.Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, et al. Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther. 2004;15:239–250. doi: 10.1089/104303404322886093. [DOI] [PubMed] [Google Scholar]
- 118.Baliga MS, Wang H, Zhuo P, Schwartz JL, Diamond AM. Selenium and GPx-1 overexpression protect mammalian cells against UV-induced DNA damage. Biol Trace Elem Res. 2007;115:227–242. doi: 10.1007/BF02685998. [DOI] [PubMed] [Google Scholar]
- 119.Autheman D, Sheldon RA, Chaudhuri N, von Arx S, Siegenthaler C, et al. Glutathione peroxidase overexpression causes aberrant ERK activation in neonatal mouse cortex after hypoxic preconditioning. Pediatr Res. 2012;72:568–575. doi: 10.1038/pr.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chu FF, Esworthy RS, Chu PG, Longmate JA, Huycke MM, et al. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004;64:962–968. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 121.Lechner S, Müller-Ladner U, Neumann E, Spöttl T, Schlottmann K, et al. Thioredoxin reductase 1 expression in colon cancer: discrepancy between in vitro and in vivo findings. Lab Invest. 2003;83:1321–1331. doi: 10.1097/01.lab.0000085189.47968.f8. [DOI] [PubMed] [Google Scholar]
- 122.Hellfritsch J, Kirsch J, Schneider M, Fluege T, Wortmann M, et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid Redox Signal. 2015;22:938–950. doi: 10.1089/ars.2014.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Park SJ, Kim HB, Piao C, Kang MY, Park SG, et al. p53R2 regulates thioredoxin reductase activity through interaction with TrxR2. Biochem Biophys Res Commun. 2017;482:706–712. doi: 10.1016/j.bbrc.2016.11.099. [DOI] [PubMed] [Google Scholar]
- 124.Irons R, Tsuji PA, Carlson BA, Ouyang P, Yoo MH, et al. Deficiency in the 15 kDa selenoprotein inhibits tumorigenicity and metastasis of colon cancer cells. Cancer Prev Res. 2010;3:630–639. doi: 10.1158/1940-6207.CAPR-10-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tsuji PA, Naranjo-Suarez S, Carlson BA, Tobe R, Yoo MH, et al. Deficiency in the 15 kDa selenoprotein inhibits colon cancer cell growth. Nutrients. 2011;3:805–817. doi: 10.3390/nu3090805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsuji PA, Carlson BA, Yoo MH, Naranjo-Suarez S, Xu XM, et al. The 15kDa selenoprotein and thioredoxin reductase 1 promote colon cancer by different pathways. PLoS One. 2015;10 doi: 10.1371/journal.pone.0124487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tsuji PA, Carlson BA, Naranjo-Suarez S, Yoo MH, Xu X, et al. Knockout of the 15 kDa selenoprotein protects against chemically-induced aberrant crypt formation in mice. PLoS One. 2012;7:e50574. doi: 10.1371/journal.pone.0050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bertz M, Kühn K, Koeberle SC, Müller MF, Hoelzer D, et al. Selenoprotein H controls cell cycle progression and proliferation of human colorectal cancer cells. Free Radic Biol Med. 2018;9:30020–30020. doi: 10.1016/j.freeradbiomed.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 129.Hawkes WC, Alkan Z. Delayed cell cycle progression from SEPW1 depletion is p53- and p21-dependent in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2011;413:36–40. doi: 10.1016/j.bbrc.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 130.Banning A, Kipp A, Schmitmeier S, Lowinger M, Florian S, et al. Glutathione peroxidase 2 inhibits cyclooxygenase-2-mediated migration and invasion of HT-29 adenocarcinoma cells but supports their growth as tumors in nude mice. Cancer Res. 2008;68:9746–9753. doi: 10.1158/0008-5472.CAN-08-1321. [DOI] [PubMed] [Google Scholar]
- 131.Yoo MH, Xu XM, Carlson BA, Gladyshev VN, Hatfield DL. Thioredoxin reductase 1 deficiency reverses tumor phenotypes and tumorigenicity of lung carcinoma cells. J Biol Chem. 2006;281:13005–13008. doi: 10.1074/jbc.C600012200. [DOI] [PubMed] [Google Scholar]
- 132.Bartolini D, Sancineto L, Fabro de Bem A, Tew KD, Santi C, et al. Selenocompounds in Cancer Therapy: An Overview. Adv Cancer Res. 2017;136:259–302. doi: 10.1016/bs.acr.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 133.Nettleford SK, Prabhu KS. Selenium and selenoproteins in gut inflammation-a review. Antioxidants. 2018;7 doi: 10.3390/antiox7030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lu YP, Lou YR, Yen P, Newmark HL, Mirochnitchenko OI, et al. Enhanced skin carcinogenesis in transgenic mice with high expression of glutathione peroxidase or both glutathione peroxidase and superoxide dismutase. Cancer Res. 1997;57:1468–1474. [PubMed] [Google Scholar]
- 135.Lei XG, Cheng WH. New roles for an old selenoenzyme: evidence from glutathione peroxidase-1 null and overexpressing mice. J Nutr. 2005;135:2295–2298. doi: 10.1093/jn/135.10.2295. [DOI] [PubMed] [Google Scholar]
- 136.McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, et al. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci U S A. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gong G, Méplan C, Gautrey H, Hall J, Hesketh JE. Differential effects of selenium and knockdown of glutathione peroxidases on TNFalpha and flagellin inflammatory responses in gut epithelial cells. Genes Nutr. 2012;7:167–178. doi: 10.1007/s12263-011-0256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Irons R, Carlson BA, Hatfield DL, Davis CD. Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J Nutr. 2006;135:1311–1317. doi: 10.1093/jn/136.5.1311. [DOI] [PubMed] [Google Scholar]
- 139.Kulmacz RJ. Regulation of cyclooxygenase catalysis by hydroperoxides. Biochem Biophys Res Commun. 2005;338:25–33. doi: 10.1016/j.bbrc.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 140.Lin YM, Furukawa Y, Tsunoda T, Yue CT, Yang KC, et al. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene. 2002;21:4120–4128. doi: 10.1038/sj.onc.1205518. [DOI] [PubMed] [Google Scholar]
- 141.Serewko MM, Popa C, Dahler AL, Smith L, Strutton GM, et al. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res. 2002;62:3759–3765. [PubMed] [Google Scholar]
- 142.Emmink BL, Laoukili J, Kipp AP, Koster J, Govaert KM, et al. GPx2 suppression of H2O2 stress links the formation of differentiated tumor mass to metastatic capacity in colorectal cancer. Cancer Res. 2014;74:6717–6730. doi: 10.1158/0008-5472.CAN-14-1645. [DOI] [PubMed] [Google Scholar]
- 143.Krehl S, Loewinger M, Florian S, Kipp AP, Banning A, et al. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis. 2012;33:620–628. doi: 10.1093/carcin/bgr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kasaikina MV, Kravtsova MA, Lee BC, Seravalli J, Peterson DA, et al. Dietary selenium affects host selenoproteome expression by influencing the gut microbiota. FASEB J. 2011;25:2492–2499. doi: 10.1096/fj.11-181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Esworthy RS, Aranda R, Martin MG, Doroshow JH, Binder SW, et al. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001;281 doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 146.Esworthy RS, Smith DD, Chu FF. A strong impact of genetic background on gut microflora in mice. Int J Inflam. 2010;1:986046. doi: 10.4061/2010/986046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen B, Rao X, House MG, Nephew KP, Cullen KJ, et al. GPx3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer Lett. 2011;309:37–45. doi: 10.1016/j.canlet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 148.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:8. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lee BC, Dikiy A, Kim HY, Gladyshev VN. Functions and evolution of selenoprotein methionine sulfoxide reductases. Biochim Biophys Acta. 2009;11:4. doi: 10.1016/j.bbagen.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De Luca A, Sacchetta P, Nieddu M, Di Ilio C, Favaloro B. Important roles of multiple Sp1 binding sites and epigenetic modifications in the regulation of the methionine sulfoxide reductase B1 (MsrB1) promoter. BMC Mol Biol. 2007;8:1471–2199. doi: 10.1186/1471-2199-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gladyshev VN. Selenium and methionine sulfoxide reduction. Free Radic Biol Med. 2014;75:10. doi: 10.1016/j.freeradbiomed.2014.10.848. [DOI] [PubMed] [Google Scholar]
- 152.Labunskyy VM, Lee BC, Handy DE, Loscalzo J, Hatfield DL, et al. Both maximal expression of selenoproteins and selenoprotein deficiency can promote development of type 2 diabetes-like phenotype in mice. Antioxid Redox Signal. 2011;14:2327–2336. doi: 10.1089/ars.2010.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee BC, Lee SG, Choo MK, Kim JH, Lee HM, et al. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci Rep. 2017;7:017–05230. doi: 10.1038/s41598-017-05230-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kudva AK, Shay AE, Prabhu KS. Selenium and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;309:4. doi: 10.1152/ajpgi.00379.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peters U, Bien S, Zubair N. Genetic architecture of colorectal cancer. Gut. 2015;64:1623–1636. doi: 10.1136/gutjnl-2013-306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Perez S, Talens-Visconti R, Rius-Perez S, Finamor I, Sastre J. Redox signaling in the gastrointestinal tract. Free Radic Biol Med. 2017;104:75–103. doi: 10.1016/j.freeradbiomed.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 157.Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;10:1477–3163. doi: 10.4103/1477-3163.78111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin. 2011;43:745–756. doi: 10.1093/abbs/gmr079. [DOI] [PubMed] [Google Scholar]
- 159.Coant N, Ben Mkaddem S, Pedruzzi E, Guichard C, Treton X, et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol Cell Biol. 2010;30:2636–2650. doi: 10.1128/MCB.01194-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deitrick J, Pruitt WM. Wnt/beta catenin-mediated signaling commonly altered in colorectal cancer. Prog Mol Biol Transl Sci. 2016;144:49–68. doi: 10.1016/bs.pmbts.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 161.de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci. 2007;12:471–491. doi: 10.2741/2076. [DOI] [PubMed] [Google Scholar]
- 162.Kipp A, Banning A, Brigelius-Flohé R. Activation of the glutathione peroxidase 2 (GPx2) promoter by beta-catenin. Biol Chem. 2007;388:1027–1033. doi: 10.1515/BC.2007.137. [DOI] [PubMed] [Google Scholar]
- 163.Gerashchenko MV, Su D, Gladyshev VN. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J Biol Chem. 2010;285:4595–4602. doi: 10.1074/jbc.M109.070532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dentice M, Luongo C, Ambrosio R, Sibilio A, Casillo A, et al. beta-Catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterology. 2012;143:1037–1047. doi: 10.1053/j.gastro.2012.06.042. [DOI] [PubMed] [Google Scholar]
- 165.Köhrle J, Gärtner R. Selenium and thyroid. Best Pract Res Clin Endocrinol Metab. 2009;23:815–827. doi: 10.1016/j.beem.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 166.Foster CB, Aswath K, Chanock SJ, McKay HF, Peters U. Polymorphism analysis of six selenoprotein genes: support for a selective sweep at the glutathione peroxidase 1 locus (3p21) in Asian populations. BMC Genet. 2006;7:1471–2156. doi: 10.1186/1471-2156-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hu YJ, Dolan ME, Bae R, Yee H, Roy M, et al. Allelic loss at the GPx-1 locus in cancer of the head and neck. Biol Trace Elem Res. 2004;101:97–106. doi: 10.1385/BTER:101:2:097. [DOI] [PubMed] [Google Scholar]
- 168.Hu Y, Benya RV, Carroll RE, Diamond AM. Allelic loss of the gene for the GPX1 selenium-containing protein is a common event in cancer. J Nutr. 2005;135:3021S–3024S. doi: 10.1093/jn/135.12.3021S. [DOI] [PubMed] [Google Scholar]
- 169.Peters U, Chatterjee N, Hayes RB, Schoen RE, Wang Y, et al. Variation in the selenoenzyme genes and risk of advanced distal colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2008;17:1144–1154. doi: 10.1158/1055-9965.EPI-07-2947. [DOI] [PubMed] [Google Scholar]
- 170.Goldberg M, Alberts DS, Buckmeier JA, Prasad AR, Krouse RS, et al. Loss of heterozygosity at the glutathione peroxidase 1 locus is not an early event in colon carcinogenesis. Genes Cancer. 2011;2:910–913. doi: 10.1177/1947601911431840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hansen RD, Krath BN, Frederiksen K, Tjonneland A, Overvad K, et al. GPX1 Pro(198)Leu polymorphism, erythrocyte GPX activity, interaction with alcohol consumption and smoking, and risk of colorectal cancer. Mutat Res. 2009;664:13–19. doi: 10.1016/j.mrfmmm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 172.Hansen R, Saebo M, Skjelbred CF, Nexo BA, Hagen PC, et al. GPX Pro198Leu and OGG1 Ser326Cys polymorphisms and risk of development of colorectal adenomas and colorectal cancer. Cancer Lett. 2005;229:85–91. doi: 10.1016/j.canlet.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 173.Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chu FF, Rohan de Silva HA, Esworthy RS, Boteva KK, Walters CE, et al. Polymorphism and chromosomal localization of the GI-form of human glutathione peroxidase (GPX2) on 14q24.1 by in situ hybridization. Genomics. 1996;32:272–276. doi: 10.1006/geno.1996.0115. [DOI] [PubMed] [Google Scholar]
- 175.Haug U, Poole EM, Xiao L, Curtin K, Duggan D, et al. Glutathione peroxidase tagSNPs: associations with rectal cancer but not with colon cancer. Genes Chromosomes Cancer. 2012;51:598–605. doi: 10.1002/gcc.21946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bermano G, Parmantidis V, Holloway N, Kadri S, Mowat NA, et al. Evidence that a polymorphism within the 3' UTR of glutathione peroxidase 4 is functional and is associated with susceptibility to colorectal cancer. Genes Nutr. 2007;2:225–232. doi: 10.1007/s12263-007-0052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Méplan C, Hesketh J. The influence of selenium and selenoprotein gene variants on colorectal cancer risk. Mutagenesis. 2012;27:177–186. doi: 10.1093/mutage/ger058. [DOI] [PubMed] [Google Scholar]
- 178.Méplan C, Hughes DJ, Pardini B, Naccarati A, Soucek P, et al. Genetic variants in selenoprotein genes increase risk of colorectal cancer. Carcinogenesis. 2010;31:1074–1079. doi: 10.1093/carcin/bgq076. [DOI] [PubMed] [Google Scholar]
- 179.Båvner A, Matthews J, Sanyal S, Gustafsson JA, Treuter E. EID3 is a novel EID family member and an inhibitor of CBP-dependent co-activation. Nucleic Acids Res. 2005;33:3561–3569. doi: 10.1093/nar/gki667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Leone A, Roca MS, Ciardiello C, Costantini S, Budillon A. Oxidative stress gene expression profile correlates with cancer patient poor prognosis: Identification of crucial pathways might delect novel therapeutic approaches. Oxid Med Cell Longev. 2017;2597581:9. doi: 10.1155/2017/2597581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hu YJ, Korotkov KV, Mehta R, Hatfield DL, Rotimi CN, et al. Distribution and functional consequences of nucleotide polymorphisms in the 3'-untranslated region of the human Sep15 gene. Cancer Res. 2001;61:2307–2310. [PubMed] [Google Scholar]
- 182.Sutherland A, Kim DH, Relton C, Ahn YO, Hesketh J. Polymorphisms in the selenoprotein S and 15-kDa selenoprotein genes are associated with altered susceptibility to colorectal cancer. Genes Nutr. 2010;5:215–223. doi: 10.1007/s12263-010-0176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 184.Barzi A, Lenz AM, Labonte MJ, Lenz HJ. Molecular pathways: Estrogen pathway in colorectal cancer. Clin Cancer Res. 2013;19:5842–5848. doi: 10.1158/1078-0432.CCR-13-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Polo A, Guariniello S, Colonna G, Ciliberto G, Costantini S. A study on the structural features of SELK, an over-expressed protein in hepatocellular carcinoma, by molecular dynamics simulations in a lipid-water system. Mol Biosyst. 2016;12:3209–3222. doi: 10.1039/c6mb00469e. [DOI] [PubMed] [Google Scholar]
- 186.Pitts MW, Hoffmann PR. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium. 2017;4:30047–30047. doi: 10.1016/j.ceca.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li M, Cheng W, Nie T, Lai H, Hu X, et al. Selenoprotein K mediates the proliferation, migration, and invasion of human choriocarcinoma cells by negatively regulating human chorionic gonadotropin expression via ERK, p38 MAPK, and Akt signaling pathway. Biol Trace Elem Res. 2017;6:017–1155. doi: 10.1007/s12011-017-1155-3. [DOI] [PubMed] [Google Scholar]
- 188.Lan X, Xing J, Gao H, Li S, Quan L, et al. Decreased expression of selenoproteins as a poor prognosticator of gastric cancer in humans. Biol Trace Elem Res. 2017;178:22–28. doi: 10.1007/s12011-016-0908-8. [DOI] [PubMed] [Google Scholar]
- 189.Méplan C, Rohrmann S, Steinbrecher A, Schomburg L, Jansen E, et al. Polymorphisms in thioredoxin reductase and selenoprotein K genes and selenium status modulate risk of prostate cancer. PLoS One. 2012;7:1. doi: 10.1371/journal.pone.0048709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ferguson AD, Labunskyy VM, Fomenko DE, Arac D, Chelliah Y, et al. NMR structure of the selenoprotein Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J Biol Chem. 2006;281:3536–3543. doi: 10.1074/jbc.M511386200. [DOI] [PubMed] [Google Scholar]
- 191.Reeves MA, Bellinger FP, Berry MJ. The neuroprotective functions of selenoprotein M and its role in cytosolic calcium regulation. Antioxid Redox Signal. 2010;12:809–818. doi: 10.1089/ars.2009.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Guerriero E, Accardo M, Capone F, Colonna G, Castello G, et al. Assessment of the Selenoprotein M (SELM) over-expression on human hepatocellular carcinoma tissues by immunohistochemistry. Eur J Histochem. 2014;58 doi: 10.4081/ejh.2014.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Korotkov KV, Novoselov SV, Hatfield DL, Gladyshev VN, et al. Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element. Mol Cell Biol. 2002;22:1402–1411. doi: 10.1128/mcb.22.5.1402-1411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Y, Zhou Y, Schweizer U, Savaskan NE, Hua D, et al. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J Biol Chem. 2008;283:2427–2438. doi: 10.1074/jbc.M707951200. [DOI] [PubMed] [Google Scholar]
- 195.Xu XM, Carlson BA, Irons R, Mix H, Zhong N, et al. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem J. 2007;404:115–120. doi: 10.1042/BJ20070165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dudaladava V, Jarzab M, Stobiecka E, Chmielik E, Simek K, et al. Gene Expression Profiling in Hereditary, BRCA1-linked Breast Cancer: Preliminary Report. Hered Cancer Clin Pract. 2006;4:28–38. doi: 10.1186/1897-4287-4-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Galecka E, Talarowska M, Maes M, Su KP, Gorski P, et al. Polymorphisms of iodothyronine deiodinases (DIO1, DIO3) genes are not associated with recurrent depressive disorder. Pharmacol Rep. 2016;68:913–917. doi: 10.1016/j.pharep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 198.Labunskyy VM, Hatfield DL, Gladyshev VN. The Sep15 protein family: roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life. 2007;59:1–5. doi: 10.1080/15216540601126694. [DOI] [PubMed] [Google Scholar]
- 199.Slattery ML, Pellatt DF, Wolff RK, Lundgreen A. Genes, environment and gene expression in colon tissue: a pathway approach to determining functionality. Int J Mol Epidemiol Genet. 2016;7:45–57. [PMC free article] [PubMed] [Google Scholar]
- 200.Tachibana M, Ohkura Y, Kobayashi Y, Sakamoto H, Tanaka Y, et al. Expression of apolipoprotein A1 in colonic adenocarcinoma. Anticancer Res. 2003;23:4161–4167. [PubMed] [Google Scholar]
- 201.Sengupta A, Carlson BA, Labunskyy VM, Gladyshev VN, Hatfield DL. Selenoprotein T deficiency alters cell adhesion and elevates selenoprotein W expression in murine fibroblast cells. Biochem Cell Biol. 2009;87:953–961. doi: 10.1139/o09-064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tanguy Y, Falluel-Morel A, Arthaud S, Boukhzar L, Manecka DL, et al. The PACAP-regulated gene selenoprotein T is highly induced in nervous, endocrine, and metabolic tissues during ontogenetic and regenerative processes. Endocrinology. 2011;152:4322–4335. doi: 10.1210/en.2011-1246. [DOI] [PubMed] [Google Scholar]
- 203.Han SJ, Lee BC, Yim SH, Gladyshev VN, Lee SR. Characterization of mammalian selenoprotein o. a redox-active mitochondrial protein. PLoS One. 2014;9 doi: 10.1371/journal.pone.0095518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Barrett CW, Reddy VK, Short SP, Motley AK, Lintel MK, et al. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J Clin Invest. 2015;125:2646–2660. doi: 10.1172/JCI76099. [DOI] [PMC free article] [PubMed] [Google Scholar]