

Abstract
Selenium (Se) is incorporated as the 21st amino acid selenocysteine (Sec) into the growing polypeptide chain of proteins involved in redox gatekeeper functions. Erythropoiesis presents a particular problem to redox regulation as the presence of iron, heme, and unpaired globin chains lead to high levels of free radical-mediated oxidative stress, which are detrimental to erythroid development and can lead to anemia. Under homeostatic conditions, bone marrow erythropoiesis produces sufficient erythrocytes to maintain homeostasis. In contrast, anemic stress induces an alternative pathway, stress erythropoiesis, which rapidly produces new erythrocytes at extramedullary sites, such as spleen, to alleviate anemia. Previous studies suggest that dietary Se protects erythrocytes from such oxidative damage and the absence of selenoproteins causes hemolysis of erythrocytes due to oxidative stress. Furthermore, Se deficiency or lack of selenoproteins severely impairs stress erythropoiesis exacerbating the anemia in rodent models and human patients. Interestingly, erythroid progenitors develop in close proximity with macrophages in structures referred to as erythroblastic islands (EBIs), where macrophage expression of selenoproteins appears to be critical for the expression of heme transporters to facilitate export of heme from macrophage stores to the developing erythroid cells. Here we review the role of Se and selenoproteins in the intrinsic development of erythroid cells in addition to their role in the development of the erythropoietic niche that supports the functional role of EBIs in erythroid expansion and maturation in the spleen during the recovery from anemia.
Introduction
Selenium (Se) exerts a wide range of pleiotropic effects in human health via incorporation into biologically functional selenoproteins as the 21st amino acid, selenocysteine (Sec). Twenty five selenoprotein genes have been discovered in human (twenty four in rodents) that partake a key role in redox homeostasis[1]. Other physiological functions range from mediating thyroid hormone production, benefiting cardiovascular health, to broader anti-inflammatory activities[2–7]. Insufficiency of Se and selenoproteins contributes to many pathophysiological conditions, including cardiomyopathy (Keshan disease), arthritis (Kashin-Beck disease) and defects in the immune response to viral infection[3, 4, 8, 9]. Groups that are at risk from Se inadequacy include people living in regions with endemically low levels of Se as well as those with single nucleotide polymorphisms in selenoproteins and related genes that affect selenoprotein synthesis[3, 4]. The role of Se and selenoproteins in various tissues and physical conditions have been well described in the past decade and reviewed elsewhere[1–4, 10–17].
The beneficial biochemical role of Se in erythrocytes was first defined in 1973 in the form of selenoenzyme glutathione peroxidases (Gpxs) that highlighted the important role of dietary Se, which contributed, in part, to cellular stabilization[18]. In the recent years, Se deficiency has been linked to anemia associated with aging and chronic inflammatory diseases in humans[19–22]. In addition, rodent dietary models and transgenic models have been developed to demonstrate the direct biological functions of Se and selenoproteins to erythrocytes and erythropoiesis in mammalian systems[23–27]. Se and selenoproteins have emerged to be crucial and beneficial to erythroid cells and erythropoiesis, where they are intricately involved in multiple ways. This review summarizes current perspectives on the contributions of Se and selenoproteins to erythroid cells and erythropoiesis.
Erythropoiesis
Erythrocytes are oxygen carriers that maintain normal tissue oxygenation. Erythropoiesis is the process by which new erythrocytes are generated and its regulation is intimately tied to tissue oxygen needs. Erythropoiesis can be divided into steady state erythropoiesis and stress erythropoiesis. Steady state erythropoiesis occurs primarily in the bone marrow and maintains erythroid homeostasis, resulting in erythrocyte generation at a rate of 1011 cells/day. It utilizes progenitors that are derived from common myeloid progenitors (CMPs) and megakaryocyte erythroid progenitors (MEPs) that are originally derived from hematopoietic stem cells (HSCs) in the bone marrow. However, under stress conditions, hypoxic response can increase the rate of erythrocyte production by ~10-fold. Stress erythropoiesis, which is extramedullary, has the capability to rapidly generate large numbers of erythrocytes in response to anemic stress, relying on stress erythroid progenitors (SEPs) that are derived directly from short-term reconstituting hematopoietic stem cells (ST-HSCs). The major difference between steady state and stress erythropoiesis is in the early stage, where they utilize distinct progenitors and response to unique signals[28–31]. Along with stem cell factor, stress erythropoiesis utilizes specific signals, including growth and differentiation factor 15 (Gdf15), bone morphogenetic protein 4 (Bmp4), and hedgehog (Hh), which do not regulate steady state erythropoiesis, to promote the rapid expansion of early SEPs[32–38].
As shown in Figure 1, the terminal differentiation of erythroid cells is hierarchical [28, 29], which are derived from erythroid lineage restricted progenitors, including erythroid burst-forming units (BFU-Es) and erythroid colony-forming units (CFU-Es). BFU-Es and CFU-Es are stages of pre-erythroblasts, and are characterized by their capability to form erythroid colonies in vitro, which is a quantitative reflection of erythroid progenitors (Figure 1). These processes are regulated by erythropoietin (Epo), the master regulator of erythropoiesis[39]. Erythroblast precursors include proerythroblasts (ProEs), basophilic (BasoE), polychromatophilic (PolyE), and orthochromatic (OrthoE) erythroblasts. They are morphologically identifiable and are characterized by the accumulation of hemoglobin (Hb), shrinkage in cell size, nuclear condensation, and reduced cellular mRNA. In addition, erythroblast terminal differentiation undergoes immunophenotypic alteration, as they downregulate the cell surface expression of transferrin receptor (CD71) and increase glycophorin A associated antigen (Ter119) expression. The final step is enucleation that leads to the formation of mature erythrocytes, which are then released into the blood stream. Erythropoiesis is Epo dependent and is also under the complex control of GATA binding factor-1 (GATA-1), the master erythroid transcription factor[40]. Defective erythropoiesis leads to anemia and serial pathophysiological issues, where in addition to genetic inherited pathologies, redox imbalance in erythrocyte precursors could further add insult to the injury.
Figure 1. Schematic depicting erythropoiesis.

HSCs give rise to erythroid lineage progenitors, which differentiate into regulatory erythroid progenitors or stress progenitors in response to steady state or stress signals, respectively. BFU-Es and CFU-Es are stages of pre-erythroblasts that are quantitative reflection of erythroid progenitors. These cells enter terminal differentiation, where they become morphologically identifiable erythroblasts and undergo series of maturation events before enucleation.
Susceptibility of erythroid cells and erythropoiesis to oxidative damage
Reactive oxygen species (ROS), including superoxide anion (O2·−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are natural byproducts of cellular metabolism. ROS have dual roles- beneficial in cell signaling such as cell proliferation and differentiation, or deleterious in causing damage to cells when their production is uncontrolled. Oxidative stress occurs when excess ROS accumulate due to ineffective cellular antioxidant capacity leading to dysregulated redox status causing damage to cell lipids, proteins, and DNA, which can be irreversible, as seen atherosclerosis, diabetes, cancer, neurodegeneration, and aging[41–44].
Erythrocytes and erythroid precursors are continuously exposed to oxidative stress, particularly during erythrocyte turnover. Erythropoiesis is a hierarchical process, where Hb synthesis and accumulation characterize erythrocyte maturation. Hb constantly carries and transports oxygen, maintaining normal tissue oxygenation. The assembling process of Hb can potentially generate excessive ROS if dysregulated, resulting in cellular oxidative stress[45]. Hemoglobin A (HbA) is a tetramer, comprising two α and β globin subunits, each bound to a heme moiety. Free α globin is structurally unstable, highly sensitive to oxidative stress, and tends to denature, which potentially produces damaging ROS via reactions catalyzed by the heme-bound iron[46, 47]. Unbound α globin aggregates on erythrocyte membrane to form Heinz bodies and eventually causes hemolysis due to membrane destruction and overexposure to oxidative stress[48, 49]. In erythroid cells, molecular chaperones sequester free α globin, preventing cytotoxic precipitation[48]. Alpha hemoglobin stabilizing protein (Ahsp) is an α globin chaperone protein, whose expression is induced by GATA-1[46, 47]. In Ahsp knockout mice, erythrocytes are short-lived, accompanied by Hb precipitates, and increased ROS, due to excess of free α globin. Erythroid precursors exhibit hyperplasia, but increased apoptosis[46, 47]. In β-thalassemia erythrocytes, α globin is expressed in excess over β globin due to genetically reduced production of β globin. As a result, the intracellular ROS is significantly increased by exacerbated autoxidation of α globin leading to increased membrane-bound globin and consequent perturbation of erythrocyte structure[49, 50]. Absence of Ahsp further exacerbates the phenotype of β-thalassemia[50–52].
The properties of erythroid cells make erythropoiesis and its regulation particularly sensitive to changes in redox status. Extensive studies have shown the profound influence of oxidative status on steady state and stress erythropoiesis, and are well-reviewed elsewhere[45, 50, 53–61]. In healthy individuals, erythroid cells contain a pool of antioxidant enzymes, including membrane oxidoreductases, superoxide dismutases, and catalase; antioxidant scavengers, such as glutaredoxins (Grxs), thioredoxins (Txns), and peroxiredoxins (Prxs); and transcription factors that regulate antioxidant genes including nuclear factor erythroid–derived 2 p45 unit (P45NF-E2), NF-E2-related factor (Nrf2), and forkhead box transcription factor O (FoxO) family, to control cellular redox balance, and protect the cells against pro-oxidant damage. Mice with deficiencies or deletion of these redox regulatory molecules exhibit pathological signs in erythrocytes and erythropoiesis, including embryonic lethality due to impaired fetal erythropoiesis as well as anemia in adulthood, accumulation of Heinz bodies, instability of Hb, and correspondingly increased ROS. These observations suggest that redox homeostasis occupies a pivotal role in erythropoiesis, where exacerbated ROS leads to pathological impairments in erythrocytes. In addition to the intrinsic sensitivity of erythrocytes to oxidative stress, pathological erythropoiesis sometimes occurs in cancer and chronic inflammatory diseases, in part, from reduced levels of Epo, or abnormal response of erythroid cells to Epo due to increased proinflammatory cytokines, which are also associated with accumulation of ROS[62–65].
Protective effects of Se and selenoproteins on erythroid cells
As efficient ROS scavengers, the role of selenoproteins in maintaining redox homeostasis in erythrocytes and erythropoiesis is expected to be critical. In fact, Se appears to play a significant role in pathological erythropoiesis. Long-term Se deficiency in rats was not associated with anemia; however, in a case of pathological erythropoiesis induced by methimazole, dietary Se supplementation showed protective properties[23]. Pregnant rats and their pups exhibited symptoms of anemia post methimazole challenge, and displayed signs of increased oxidative stress associated with low activities of antioxidant enzymes and scavengers. The symptoms were alleviated by dietary Se supplementation (0.5mg/kg of diet) in the form of sodium selenite[24, 25]. Thus, it appears that Se functions in a protective role and potentially serves as a primary antioxidant during erythropoiesis in rodents.
Recent studies have documented a more detailed mechanistic description underlying the protective effect of Se and selenoproteins. Kawatani et al.[27] elegantly demonstrated the primary role of selenoproteins and Nrf2 in regulating erythropoiesis utilizing a transgenic mouse model where the tRNA[Ser]Sec (Trsp) gene, which controls the selenoprotein synthesis, was deleted. Selenoprotein knockout mice (Trspfl/del:Mx1-Cre) developed anemia, and presented with decreased hematocrit, reduced serum Hb content, and increased mean corpuscular volume, which are all pathological signs of defective erythropoiesis. In addition, immature and damaged erythrocytes were found in the periphery and bone marrow of the knockout mice. Importantly, genes associated with oxidative stress were upregulated. These data provide strong support to the importance of selenoproteins in maintaining redox homeostasis within erythroid cells. On the other hand, Nrf2 and Trsp double knockout mice showed exacerbated pathological phenotypes. Consistently, in another study, it was reported that during Se deficiency Nrf2 target genes were induced, including many antioxidant enzymes, such as heme-oxygenase 1 (Hmox1), NADPH:quinone oxidoreductase (Nqo1), glutathione peroxidase 1 (Gpx1), peroxiredoxin 1 (Prx1), and glutamate-cysteine ligase catalytic subunit (Gclc)[66]. Thus, up-regulation of Nrf2 is considered as a compensatory mechanism, particularly in the absence of selenoproteins.
Studies in our laboratory have demonstrated the erythroid regulatory functions of selenoproteins in a Se dietary mouse model[26]. Se deficient (Se-D) mice were mildly anemic with increased BFU-Es in the spleen indicating increased hypoxia. Erythrocytes showed signs of oxidative damage of Hb. Furthermore, the transcription factor FoxO3a, which has been demonstrated to be required during erythropoiesis[65], was upregulated in G1E cells (a GATA-null murine erythroblast cell line) cultured under Se-D conditions. Recent studies have revealed a pivotal role for Se and selenoproteins in stress erythropoiesis [67]. Two acute anemia models were used in the study that included phenylhydrazine (PHZ)-induced hemolysis by oxidative damage of the erythrocyte membrane[68] and a short-term radioprotective model, where mice develop anemia post irradiation and new erythrocytes are rapidly generated following bone marrow transplantation[32]. PHZ-induced hemolytic anemia was lethal to Se-D mice, while Se adequacy was able to restore the hematocrit and SEPs in an effort to support efficient erythropoiesis. Interestingly, lack of selenoproteins (as in the Trsp mice) resulted in lethal anemia of mice in the short-term radioprotective model. In both the cases, stress erythropoiesis was severely impaired in mice. The defects occurred early in SEPs, which is the “rate limiting” step that determines the final erythrocyte output. The commitment towards erythroid lineage was likely compromised [67]. More interestingly, in the Trsp deleted chimeric mice, granulocytic and macrophage lineage commitment was also impaired, indicating broader HSC and/or multipotent progenitor defects. Analysis of terminal erythroblast maturation in Se-D mice showed that this stage of development was also compromised. We observed a blockage in the transition of ProEs to BasoEs, which was associated with functionally less active GATA-1 protein. Use of high-density transcriptomic arrays revealed significantly differences in Se-D and Se adequate (Se-A) ProEs on day 3 post PHZ treatment. Ingenuity pathway analysis indicated the involvement of pathways, including heme synthesis, oxidative stress control, cell death and cell cycle control, and erythroid cell growth and proliferation as underpinnings of Se-dependent protection [67]. Taken together, these studies greatly expand our understanding of the role of Se and selenoproteins in erythroid function that are not just limited to antioxidant protective roles in terminal maturation, but are critical in intrinsic programming of integrative cell development and cell-fate commitment that significantly highlights the functional role of selenoproteins in erythropoiesis. However, it remains to be seen which of the selenoproteins are important for these functions.
Se and selenoproteins and erythropoietic microenvironment
Erythroid cells develop within specialized niches referred to as erythroblastic islands (EBIs)[69, 70] that are formed by macrophages, which physically interact with the developing erythroblasts (Figure 2). Macrophages provide survival and proliferation signals to erythroblasts and are also responsible for the removal of extruded nuclei during erythroblast enucleation[69–72]. Recent studies report that mice with conditional deletion of Trsp in myeloid lineage (Trspfl/flLysMCre) resulted in a delayed stress recovery from anemia induced by PHZ, which demonstrates that selenoproteins within monocytes and macrophages are critical for stress erythropoietic microenvironment [67]. In Se-D mice and bone marrow chimeric mice with Trsp−/− bone marrow cells, we observed greatly reduced numbers of mature splenic macrophages. This observation coupled with decreased numbers of macrophages within the EBI with fewer erythroblasts attached to the central macrophages appears to contribute to the abnormality associated with terminal differentiation of erythroblasts during stress erythropoiesis.
Figure 2. Immunostaining visualization of EBIs.
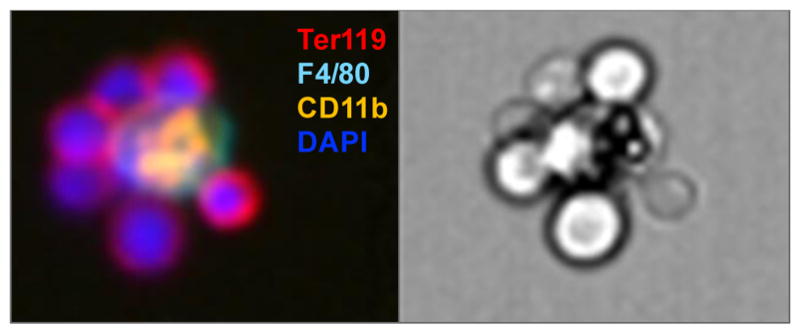
EBIs are “ring-shaped” structures that consists of a central macrophage marked by CD11b and F4/80, and surrounding erythroblasts that express Ter119 with large nuclei (DAPI+).
In addition, it is well known that macrophages play an important role in heme-iron-recycling in the mouse spleen and liver through phagocytosing senescent or damaged erythrocytes[73–76]. Heme homeostasis is pivotal in erythropoiesis, as heme is not only a core component of Hb, but also triggers several cellular signaling pathways. For example, BTB and CNC homology 1 (Bach1) is a transcriptional repressor that is degraded when it binds heme, which leads to the transcriptional activation of globins and promotes erythroid differentiation[77, 78]. Disruption of heme homeostasis causes excess oxidative stress leading to cell injury and impaired erythropoiesis[56, 79]. Heme import and export within macrophages relies on the heme transporter protein, Hrg1[80], and heme exporter, feline leukemia virus subgroup C receptor-related protein 1 (Flvcr1)[81], which are important in maintaining cellular heme homeostasis. Recent studies from our laborartory show that Hrg1 and Flvcr expression within the EBI macrophages was significantly decreased in Se-D mice compared to Se-A mice, which may lead to the accumulation of heme in the phagolysosome of these macrophages[80]. Decrease in Hrg1 and Flvcr expression could be potentially mediated through the inhibition of Bach1, whose expression is increased in Se-D ProEs when compared to Se-A controls [67]. Hmox1 releases iron from heme for storage and/or recycle, which maintains cellular heme-iron balance and in turn controls heme-associated toxicity[85]. Gene expression analysis showed that Hmox1 expression in EBIs was significantly decreased in the Se-D mice when compared to Se-A mice [67]. Potential issues with heme accessibility, heme degradation and export can all result in lower levels of heme supply to erythroblasts, leading to ineffective erythroid terminal maturation. The observations of association between Se and heme related pathways are interesting, particularly since iron is another essential dietary nutrient that is a key ingredient for heme synthesis. Further studies are underway to elucidate the exact control of Bach1 expression and regulation of downstream genes by selenoproteins.
The role of glutathione peroxidases, thioredoxin reductases, and selenoprotein W in erythropoiesis
Twenty-five selenoprotein genes have been identified in human (24 in rodents)[2, 86], but their role in erythropoiesis remains largely unclear. Currently known selenoproteins that are involved in erythroid cells include Gpxs and thioredoxin reductases (Txnrds) that constitute a major cellular redox buffer system[42, 60, 87]. In addition, our recent studies have suggested an intriguing role for selenoprotein W (SelenoW), in erythropoiesis [67] (Table 1).
Table 1.
Genetically modified mice with erythropoietic phenotypes
| Selenoprotein | Transgenic mouse model | Phenotype | Function in erythropoiesis | References |
|---|---|---|---|---|
| Gpx1 | Gpx1−/− | Normal | Eliminates endogenous hydrogen peroxide in erythrocytes and protects Hb from oxidative damage in vitro | [88–91] [89, 92] |
| Gpx4 | Gpx4−/− | Embryonic lethal | Unknown | [93] |
| Txnrd1 | Txnrd1−/− | Embryonic lethal | Unknown | [98] |
| Txnrd2 | Txnrd2−/− | Embryos are severely anemic | Protect cells from apoptosis induced by oxidative stress | [94, 95] |
| SelenoW | NA | mRNA decreased in Se-D ProEs from mice | Promotes erythroid expansion and erythroblast maturation potentially via 14-3-3 signaling pathways in vitro | [67] |
Gpx1 was first purified from erythrocytes and is one of the primary enzymes that eliminates endogenous hydrogen peroxide in erythrocytes and protects Hb from oxidative damage in vitro[88–91]. Surprisingly, erythropoiesis in Gpx1−/− mice was fairly normal[89, 92], which could be a result of the compensatory effects of other antioxidant scavengers or enzymes, indicating the possibility of dispensable role of Gpx1 in erythropoiesis under steady state in vivo. It is unclear if loss of Gpx1 contributes to stress or pathological erythropoiesis, such as thalassemia and some cases of inflammatory anemia, where redox balance is disrupted[50]. On the other hand, knockout of glutathione peroxidase 4 (Gpx4) in mice was lethal at embryonic stage due to the failure to form well-organized embryonic structures, but whether erythroid cells are affected remains unclear[93].
Thioredoxin (Txn)/Txnrd/peroxiredoxin (Prx) system is one of the primary redox buffer systems in the body[96] and is thought to play a critical role in redox signaling in human erythrocytes[97, 98]. Thioredoxin reductase 2 (Txnrd2) exerts a critical role in supporting erythropoiesis. Ubiquitous knockout of mitochondrial Txnrd2 was reported to embryonic lethal at E13. Txnrd2 knockout embryos were severely anemic with increased apoptosis in the liver, a primary erythropoietic tissue during fetal stage, which was also associated with significantly decreased size of CFU-Es[94]. Cardiac-specific conditional knockout of Txnrd2 resulted in fatal dilated cardiomyopathy, which resembles the pathological phenotype of Keshan disease[94, 95]. Txnrd1 knockout mice as well as Txn knockout mice are embryonic lethal with failure in embryogenesis[99–101]. But whether erythrocytes and erythropoiesis are affected in these models is unknown. The essential role of Prxs in protection from oxidative stress during erythropoiesis has been well elucidated using knockout animal models. Prx1 knockout mice are anemic and exhibit exacerbation of oxidative stress and increased ROS in erythrocytes, which result in protein oxidation, Hb instability, Heinz body formation, and decreased erythrocyte lifespan. In addition, both homozygotes and heterozygotes present increased malignancies, including lymphomas, sarcomas and carcinomas[102]. Prx2 functions as a noncatalytic-scavenger in erythrocytes[103], essential for stabilizing Hb and preventing hemolytic anemia[57], thereby sustaining a normal life span of erythrocytes[104]. In pathological cases, Prx2 and Prx3 play a cytoprotective role in β-thalassemic erythropoiesis in mice[59] and Fanconi anemia patients[105], respectively, by preventing oxidative stress in erythrocytes.
Recent studies in our laboratory have indicated regulatory effects of SelenoW in erythroid differentiation [67]. SelenoW was first identified from white muscle disease in lambs. SelenoW is expressed in many species including rodents, sheep, zebra fish, chicken, and primates in various tissues[106–110]. It belongs to the stress-related group of selenoproteins that are sensitive to the levels of Se in the diet[1, 107, 108]. SelenoW is a 10kDa protein containing a conserved–CXXU-motif (U= Sec), and its redox function has been suggested[111]. SelenoW has been reported to participate in diverse cellular signaling pathways via interaction with its target protein 14-3-3[112–114]. In our studies, the erythroid function of SelenoW emerged from the transcriptomic analysis of primary splenic ProEs and BasoEs sorted from Se-D and Se-A mice treated with PHZ, where SelenoW was the only selenoprotein that was highly regulated in the ProEs as a function of dietary Se status [67]. SelenoW protein was absent in primary ProEs, BasoEs, CD71+ erythroblasts from Se-D mice and Trsp conditional knockout bone marrow cells. SelenoW appears to be a potential target of GATA-1 as demonstrated by chromatin immunoprecipitation (ChIP) sequencing (ChIP-Seq) analysis. Our recent studies indicated that knockout of SelenoW in a murine erythroblast cell line G1E-ER4 and primary bone marrow cells impaired erythroid maturation [67]. Erythroblasts exhibited reduced expression of Ter119, while early progenitors exhibited decreased BFU-E colony formation. While the exact role of SelenoW and the mechanism remain unclear, we believe that suppression of PDZ-binding motif (TAZ) by 14-3-3 can be released by SelenoW sequestration of 14-3-3 to impact pathways of cellular differentiation, as reported in myocytes [113]. Needless to say, further work is necessary to elucidate the role of SelenoW in erythroid maturation.
Conclusions and future directions
Current literature supports the role of selenoproteins in erythropoiesis, especially during stress erythropoiesis in pathological cases such as anemia. In the absence of Se, oxidative stress directly targets erythrocytes by damaging Hb, which aggregates and precipitates on the membrane, leading to hemolysis. In addition to redox regulation, selenoproteins appear to have a broader influence, which not only affects erythroid cell development at multiple stages, but also impacts the erythropoietic microenvironment. Evidence suggests that selenoproteins may interfere erythroid cell-fate decision, but further research is needed to pinpoint the exact mechanism. Studies also suggest an intriguing link between Se and heme/iron homeostasis, particularly within the erythropoietic niche that supplies heme/iron to the developing erythrocytes, in which the ability to move heme/iron within the cells could be compromised by the lack of Se. Nonetheless, it is necessary to corroborate these results in humans. Future studies focusing on detailed mechanistic studies, via the generation of various tissue-specific transgenic mouse models to regulate expression of specific selenoproteins within the developing erythroid cells as well as the microenvironment, could provide novel insights.
Figure 3. Schematic depicting compromised erythropoiesis in Se and selenoprotein deficiency.

Anemia is a consequence of multiple disorders. Excessive oxidative stress may induce hemolysis and aggregation of Hb, which leads to stress erythropoiesis to produce massive erythrocytes to compensate the demand in erythrocytes. In the absence of Se and selenoproteins, stress erythropoiesis is dysfunctional. The defects occur early in the erythroid progenitor stage leading to the reduced erythrocyte output. Other defects are found in erythroblast terminal differentiation, which represent the cumulative result of defective erythroid cells and the microenvironment with disrupted heme signaling. Moreover, SelenoW emerges to be a novel regulator, which is a potential downstream target of GATA-1.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health (NIDDK) R01 DK077152, Office of Dietary Supplements (K.S.P), R01 DK080040 (R.F.P), USDA-NIFA Hatch project numbers 4605 (K.S.P) and 4581 (R.F.P). We thank members of the Prabhu and Paulson laboratories for their timely help and suggestions.
Footnotes
Declarations of interest: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94(3):739–77. doi: 10.1152/physrev.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schomburg L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol. 2011;8(3):160–71. doi: 10.1038/nrendo.2011.174. [DOI] [PubMed] [Google Scholar]
- 3.Rayman MP. Selenium and human health. Lancet. 2012;379(9822):1256–68. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 4.Rayman MP. The importance of selenium to human health. Lancet. 2000;356(9225):233–41. doi: 10.1016/S0140-6736(00)02490-9. [DOI] [PubMed] [Google Scholar]
- 5.Kudva AK, Shay AE, Prabhu KS. Selenium and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;309(2):G71–7. doi: 10.1152/ajpgi.00379.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayan V, Ravindra KC, Liao C, Kaushal N, Carlson BA, Prabhu KS. Epigenetic regulation of inflammatory gene expression in macrophages by selenium. J Nutr Biochem. 2015;26(2):138–45. doi: 10.1016/j.jnutbio.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diwakar BT, Finch ER, Liao C, Shay AE, Prabhu KS. The role of selenoproteins in resolution of inflammation. In: Hatfield DL, Berry MJ, Gladyshev VN, editors. Selenium: Its Molecular Biology and Role in Human Health. Springer-Verlag; New York: 2012. pp. 499–510. [Google Scholar]
- 8.Hoffmann PR, Berry MJ. The influence of selenium on immune responses. Mol Nutr Food Res. 2008;52(11):1273–80. doi: 10.1002/mnfr.200700330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck MA, Levander OA, Handy J. Selenium deficiency and viral infection. J Nutr. 2003;133(5 Suppl 1):1463S–7S. doi: 10.1093/jn/133.5.1463S. [DOI] [PubMed] [Google Scholar]
- 10.Roman M, Jitaru P, Barbante C. Selenium biochemistry and its role for human health. Metallomics. 2014;6(1):25–54. doi: 10.1039/c3mt00185g. [DOI] [PubMed] [Google Scholar]
- 11.Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39(3):112–20. doi: 10.1016/j.tibs.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fairweather-Tait SJ, Bao Y, Broadley MR, Collings R, Ford D, Hesketh JE, Hurst R. Selenium in human health and disease. Antioxid Redox Signal. 2011;14(7):1337–83. doi: 10.1089/ars.2010.3275. [DOI] [PubMed] [Google Scholar]
- 13.Papp LV, Lu J, Holmgren A, Khanna KK. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid Redox Signal. 2007;9(7):775–806. doi: 10.1089/ars.2007.1528. [DOI] [PubMed] [Google Scholar]
- 14.Brown KM, Arthur JR. Selenium, selenoproteins and human health: a review. Public Health Nutr. 2001;4(2B):593–9. doi: 10.1079/phn2001143. [DOI] [PubMed] [Google Scholar]
- 15.Weeks BS, Hanna MS, Cooperstein D. Dietary selenium and selenoprotein function. Med Sci Monit. 2012;18(8):RA127–132. doi: 10.12659/MSM.883258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellinger FP, Raman AV, Reeves MA, Berry MJ. Regulation and function of selenoproteins in human disease. Biochem J. 2009;422(1):11–22. doi: 10.1042/BJ20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diwakar BT, Korwar AM, Paulson RF, Prabhu KS. The Regulation of Pathways of Inflammation and Resolution in Immune Cells and Cancer Stem Cells by Selenium. Adv Cancer Res. 2017;136:153–172. doi: 10.1016/bs.acr.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179(4073):588–90. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 19.Semba RD, Ricks MO, Ferrucci L, Xue QL, Guralnik JM, Fried LP. Low serum selenium is associated with anemia among older adults in the United States. Eur J Clin Nutr. 2009;63(1):93–9. doi: 10.1038/sj.ejcn.1602889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Lettow M, West CE, van der Meer JW, Wieringa FT, Semba RD. Low plasma selenium concentrations, high plasma human immunodeficiency virus load and high interleukin-6 concentrations are risk factors associated with anemia in adults presenting with pulmonary tuberculosis in Zomba district, Malawi. Eur J Clin Nutr. 2005;59(4):526–32. doi: 10.1038/sj.ejcn.1602116. [DOI] [PubMed] [Google Scholar]
- 21.Natta CL, Chen LC, Chow CK. Selenium and glutathione peroxidase levels in sickle cell anemia. Acta Haematol. 1990;83(3):130–2. doi: 10.1159/000205188. [DOI] [PubMed] [Google Scholar]
- 22.Goncharova EV, Govorin AV, Scherbakova OA, Chistiakova MV. The dynamics of indicators of selenium, glutathione and anti-oxidant defense of blood in patients with anemic cardiomyopathy against the background of treatment with preparations of iron and selenium. Klin Lab Diagn. 2015;60(2):23–6. [PubMed] [Google Scholar]
- 23.Hu ML, Chung C, Spallholz JE. Hematologic data of selenium-deficient and selenium-supplemented rats. J Inorg Biochem. 1984;22(3):165–73. doi: 10.1016/0162-0134(84)80025-2. [DOI] [PubMed] [Google Scholar]
- 24.Amara IB, Hakim A, Troudi A, Soudani N, Makni FA, Zeghal KM, Zeghal N. Protective effects of selenium on methimazole-induced anemia and oxidative stress in adult rats and their offspring. Hum Exp Toxicol. 2011;30(10):1549–60. doi: 10.1177/0960327110392403. [DOI] [PubMed] [Google Scholar]
- 25.Ben Amara I, Troudi A, Garoui E, Hakim A, Boudawara T, Zeghal KM, Zeghal N. Protective effects of selenium on methimazole nephrotoxicity in adult rats and their offspring. Exp Toxicol Pathol. 2011;63(6):553–61. doi: 10.1016/j.etp.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Kaushal N, Hegde S, Lumadue J, Paulson RF, Prabhu KS. The regulation of erythropoiesis by selenium in mice. Antioxid Redox Signal. 2011;14(8):1403–12. doi: 10.1089/ars.2010.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawatani Y, Suzuki T, Shimizu R, Kelly VP, Yamamoto M. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood. 2011;117(3):986–96. doi: 10.1182/blood-2010-05-285817. [DOI] [PubMed] [Google Scholar]
- 28.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life. 2009;61(8):800–30. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 29.Palis J. Ontogeny of erythropoiesis. Curr Opin Hematol. 2008;15(3):155–61. doi: 10.1097/MOH.0b013e3282f97ae1. [DOI] [PubMed] [Google Scholar]
- 30.Paulson RF, Shi L, Wu DC. Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol. 2011;18(3):139–45. doi: 10.1097/MOH.0b013e32834521c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu DC, Paulson RF. Hypoxia regulates BMP4 expression in the murine spleen during the recovery from acute anemia. PLoS One. 2010;5(6):e11303. doi: 10.1371/journal.pone.0011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harandi OF, Hedge S, Wu DC, McKeone D, Paulson RF. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest. 2010;120(12):4507–19. doi: 10.1172/JCI41291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry JM, Harandi OF, Paulson RF. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood. 2007;109(10):4494–502. doi: 10.1182/blood-2006-04-016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry JM, Harandi OF, Porayette P, Hegde S, Kannan AK, Paulson RF. Maintenance of the BMP4-dependent stress erythropoiesis pathway in the murine spleen requires hedgehog signaling. Blood. 2009;113(4):911–8. doi: 10.1182/blood-2008-03-147892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porayette P, Paulson RF. BMP4/Smad5 dependent stress erythropoiesis is required for the expansion of erythroid progenitors during fetal development. Dev Biol. 2008;317(1):24–35. doi: 10.1016/j.ydbio.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang J, Wu DC, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood. 2015;125(11):1803–12. doi: 10.1182/blood-2014-07-591453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenox LE, Perry JM, Paulson RF. BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood. 2005;105(7):2741–8. doi: 10.1182/blood-2004-02-0703. [DOI] [PubMed] [Google Scholar]
- 38.Millot S, Andrieu V, Letteron P, Lyoumi S, Hurtado-Nedelec M, Karim Z, Thibaudeau O, Bennada S, Charrier JL, Lasocki S, Beaumont C. Erythropoietin stimulates spleen BMP4-dependent stress erythropoiesis and partially corrects anemia in a mouse model of generalized inflammation. Blood. 2010;116(26):6072–81. doi: 10.1182/blood-2010-04-281840. [DOI] [PubMed] [Google Scholar]
- 39.Hodges VM, Rainey S, Lappin TR, Maxwell AP. Pathophysiology of anemia and erythrocytosis. Crit Rev Oncol Hematol. 2007;64(2):139–58. doi: 10.1016/j.critrevonc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Martin DI, Tsai SF, Orkin SH. Increased gamma-globin expression in a nondeletion HPFH mediated by an erythroid-specific DNA-binding factor. Nature. 1989;338(6214):435–8. doi: 10.1038/338435a0. [DOI] [PubMed] [Google Scholar]
- 41.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–90. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–62. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 44.Covarrubias L, Hernandez-Garcia D, Schnabel D, Salas-Vidal E, Castro-Obregon S. Function of reactive oxygen species during animal development: passive or active? Dev Biol. 2008;320(1):1–11. doi: 10.1016/j.ydbio.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 45.Ghaffari S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal. 2008;10(11):1923–40. doi: 10.1089/ars.2008.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng L, Gell DA, Zhou S, Gu L, Kong Y, Li J, Hu M, Yan N, Lee C, Rich AM, Armstrong RS, Lay PA, Gow AJ, Weiss MJ, Mackay JP, Shi Y. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119(5):629–40. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 47.Feng L, Zhou S, Gu L, Gell DA, Mackay JP, Weiss MJ, Gow AJ, Shi Y. Structure of oxidized alpha-haemoglobin bound to AHSP reveals a protective mechanism for haem. Nature. 2005;435(7042):697–701. doi: 10.1038/nature03609. [DOI] [PubMed] [Google Scholar]
- 48.Weiss MJ, dos Santos CO. Chaperoning erythropoiesis. Blood. 2009;113(10):2136–44. doi: 10.1182/blood-2008-09-115238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scott MD, van den Berg JJ, Repka T, Rouyer-Fessard P, Hebbel RP, Beuzard Y, Lubin BH. Effect of excess alpha-hemoglobin chains on cellular and membrane oxidation in model beta-thalassemic erythrocytes. J Clin Invest. 1993;91(4):1706–12. doi: 10.1172/JCI116380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Franceschi L, Bertoldi M, Matte A, Santos Franco S, Pantaleo A, Ferru E, Turrini F. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid Med Cell Longev. 2013;2013:985210. doi: 10.1155/2013/985210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, Simon MC, Blobel GA, Weiss MJ. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature. 2002;417(6890):758–63. doi: 10.1038/nature00803. [DOI] [PubMed] [Google Scholar]
- 52.Kong Y, Zhou S, Kihm AJ, Katein AM, Yu X, Gell DA, Mackay JP, Adachi K, Foster-Brown L, Louden CS, Gow AJ, Weiss MJ. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114(10):1457–66. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao B, Mei Y, Yang J, Ji P. Erythropoietin-regulated oxidative stress negatively affects enucleation during terminal erythropoiesis. Exp Hematol. 2016;44(10):975–81. doi: 10.1016/j.exphem.2016.06.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hattangadi SM, Lodish HF. Regulation of erythrocyte lifespan: do reactive oxygen species set the clock? J Clin Invest. 2007;117(8):2075–7. doi: 10.1172/JCI32559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, Huber T, Ghaffari S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117(8):2133–44. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suragani RN, Zachariah RS, Velazquez JG, Liu S, Sun CW, Townes TM, Chen JJ. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood. 2012;119(22):5276–84. doi: 10.1182/blood-2011-10-388132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han YH, Kim SU, Kwon TH, Lee DS, Ha HL, Park DS, Woo EJ, Lee SH, Kim JM, Chae HB, Lee SY, Kim BY, Yoon DY, Rhee SG, Fibach E, Yu DY. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem Biophys Res Commun. 2012;426(3):427–32. doi: 10.1016/j.bbrc.2012.08.113. [DOI] [PubMed] [Google Scholar]
- 58.Kwon TH, Han YH, Hong SG, Lee DJ, Ha HL, Kang SW, Li W, Yoon DY, Yu DY. Reactive oxygen species mediated DNA damage is essential for abnormal erythropoiesis in peroxiredoxin II(−/−) mice. Biochem Biophys Res Commun. 2012;424(1):189–95. doi: 10.1016/j.bbrc.2012.06.113. [DOI] [PubMed] [Google Scholar]
- 59.De Franceschi L, Bertoldi M, De Falco L, Santos Franco S, Ronzoni L, Turrini F, Colancecco A, Camaschella C, Cappellini MD, Iolascon A. Oxidative stress modulates heme synthesis and induces peroxiredoxin-2 as a novel cytoprotective response in beta-thalassemic erythropoiesis. Haematologica. 2011;96(11):1595–604. doi: 10.3324/haematol.2011.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013;19(13):1539–605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leung L, Kwong M, Hou S, Lee C, Chan JY. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem. 2003;278(48):48021–9. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 62.Greenwald RA. Oxygen radicals, inflammation, and arthritis: pathophysiological considerations and implications for treatment. Semin Arthritis Rheum. 1991;20(4):219–40. doi: 10.1016/0049-0172(91)90018-u. [DOI] [PubMed] [Google Scholar]
- 63.Morceau F, Dicato M, Diederich M. Pro-inflammatory cytokine-mediated anemia: regarding molecular mechanisms of erythropoiesis. Mediators Inflamm. 2009;2009:405016. doi: 10.1155/2009/405016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maccio A, Madeddu C, Massa D, Mudu MC, Lusso MR, Gramignano G, Serpe R, Melis GB, Mantovani G. Hemoglobin levels correlate with interleukin-6 levels in patients with advanced untreated epithelial ovarian cancer: role of inflammation in cancer-related anemia. Blood. 2005;106(1):362–7. doi: 10.1182/blood-2005-01-0160. [DOI] [PubMed] [Google Scholar]
- 65.Mantovani G, Maccio A, Madeddu C, Mura L, Gramignano G, Lusso MR, Mulas C, Mudu MC, Murgia V, Camboni P, Massa E, Ferreli L, Contu P, Rinaldi A, Sanjust E, Atzei D, Elsener B. Quantitative evaluation of oxidative stress, chronic inflammatory indices and leptin in cancer patients: correlation with stage and performance status. Int J Cancer. 2002;98(1):84–91. doi: 10.1002/ijc.10143. [DOI] [PubMed] [Google Scholar]
- 66.Muller M, Banning A, Brigelius-Flohe R, Kipp A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010;5(4):297–307. doi: 10.1007/s12263-010-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liao C, Hardison RC, Kennett MJ, Carlson BA, Paulson RF, Prabhu KS. Selenoproteins regulate stress erythroid progenitors and spleen microenvironment during stress erythropoiesis. Blood. 2018 doi: 10.1182/blood-2017-08-800607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Itano HA, Hirota K, Hosokawa K. Mechanism of induction of haemolytic anaemia by phenylhydrazine. Nature. 1975;256(5519):665–7. doi: 10.1038/256665a0. [DOI] [PubMed] [Google Scholar]
- 69.An X, Mohandas N. Erythroblastic islands, terminal erythroid differentiation and reticulocyte maturation. Int J Hematol. 2011;93(2):139–43. doi: 10.1007/s12185-011-0779-x. [DOI] [PubMed] [Google Scholar]
- 70.Manwani D, Bieker JJ. The erythroblastic island. Curr Top Dev Biol. 2008;82:23–53. doi: 10.1016/S0070-2153(07)00002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohandas N, Chasis JA. The erythroid niche: molecular processes occurring within erythroblastic islands. Transfus Clin Biol. 2010;17(3):110–1. doi: 10.1016/j.tracli.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature. 2005;437(7059):754–8. doi: 10.1038/nature03964. [DOI] [PubMed] [Google Scholar]
- 73.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A. 2005;102(5):1324–8. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun. 2012;4(5–6):446–53. doi: 10.1159/000336423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood. 2005;106(12):3979–84. doi: 10.1182/blood-2005-06-2398. [DOI] [PubMed] [Google Scholar]
- 76.de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of macrophages and red blood cells; a complex love story. Front Physiol. 2014;5:9. doi: 10.3389/fphys.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, Fujita H, Igarashi K, Taketani S. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279(7):5480–7. doi: 10.1074/jbc.M302733200. [DOI] [PubMed] [Google Scholar]
- 78.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27(19):6962–71. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103(1):129–35. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, Ward DM, Liu P, Fleming MD, Hamza I. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013;17(2):261–70. doi: 10.1016/j.cmet.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–8. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 82.Khan AA, Quigley JG. Heme and FLVCR-related transporter families SLC48 and SLC49. Mol Aspects Med. 2013;34(2–3):669–82. doi: 10.1016/j.mam.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118(6):757–66. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 84.Warnatz HJ, Schmidt D, Manke T, Piccini I, Sultan M, Borodina T, Balzereit D, Wruck W, Soldatov A, Vingron M, Lehrach H, Yaspo ML. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J Biol Chem. 2011;286(26):23521–32. doi: 10.1074/jbc.M111.220178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood. 2010;116(26):6054–62. doi: 10.1182/blood-2010-03-272138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439–43. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 87.Hawkes WC, Alkan Z. Regulation of redox signaling by selenoproteins. Biol Trace Elem Res. 2010;134(3):235–51. doi: 10.1007/s12011-010-8656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mills GC. The purification and properties of glutathione peroxidase of erythrocytes. J Biol Chem. 1959;234(3):502–6. [PubMed] [Google Scholar]
- 89.Johnson RM, Goyette G, Jr, Ravindranath Y, Ho YS. Red cells from glutathione peroxidase-1-deficient mice have nearly normal defenses against exogenous peroxides. Blood. 2000;96(5):1985–8. [PubMed] [Google Scholar]
- 90.Johnson RM, Ho YS, Yu DY, Kuypers FA, Ravindranath Y, Goyette GW. The effects of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radic Biol Med. 2010;48(4):519–25. doi: 10.1016/j.freeradbiomed.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nagababu E, Chrest FJ, Rifkind JM. Hydrogen-peroxide-induced heme degradation in red blood cells: the protective roles of catalase and glutathione peroxidase. Biochim Biophys Acta. 2003;1620(1–3):211–7. doi: 10.1016/s0304-4165(02)00537-8. [DOI] [PubMed] [Google Scholar]
- 92.Cheng WH, Ho YS, Ross DA, Valentine BA, Combs GF, Lei XG. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J Nutr. 1997;127(8):1445–50. doi: 10.1093/jn/127.8.1445. [DOI] [PubMed] [Google Scholar]
- 93.Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34(4):496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 94.Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 2004;24(21):9414–23. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH, Schoensiegel F, Zischka H, Prehn C, Adamski J, Bekeredjian R, Ivandic B, Kupatt C, Brielmeier M. Heart-Specific Knockout of the Mitochondrial Thioredoxin Reductase (Txnrd2) Induces Metabolic and Contractile Dysfunction in the Aging Myocardium. J Am Heart Assoc. 2015;4(7) doi: 10.1161/JAHA.115.002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2013;18(10):1165–207. doi: 10.1089/ars.2011.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cha MK, Kim IH. Thioredoxin-linked peroxidase from human red blood cell: evidence for the existence of thioredoxin and thioredoxin reductase in human red blood cell. Biochem Biophys Res Commun. 1995;217(3):900–7. doi: 10.1006/bbrc.1995.2856. [DOI] [PubMed] [Google Scholar]
- 98.Benfeitas R, Selvaggio G, Antunes F, Coelho P, Salvador A. Is the Peroxiredoxin 2/Thioredoxin/Thioredoxin Reductase system in human erythrocytes designed for redox signaling? Free Radic Biol Med. 2014;75(Suppl 1):S24. doi: 10.1016/j.freeradbiomed.2014.10.741. [DOI] [PubMed] [Google Scholar]
- 99.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178(1):179–85. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 100.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23(3):916–22. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, de Angelis MH, Wurst W, Bornkamm GW, Brielmeier M, Conrad M. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25(5):1980–8. doi: 10.1128/MCB.25.5.1980-1988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–5. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 103.Low FM, Hampton MB, Winterbourn CC. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008;10(9):1621–30. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 104.Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, Dho SH, Kwon KS, Kwon HJ, Han YH, Jeong S, Kang SW, Shin HS, Lee KK, Rhee SG, Yu DY. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101(12):5033–8. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 105.Mukhopadhyay SS, Leung KS, Hicks MJ, Hastings PJ, Youssoufian H, Plon SE. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J Cell Biol. 2006;175(2):225–35. doi: 10.1083/jcb.200607061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yeh JY, Beilstein MA, Andrews JS, Whanger PD. Tissue distribution and influence of selenium status on levels of selenoprotein W. FASEB J. 1995;9(5):392–6. doi: 10.1096/fasebj.9.5.7896009. [DOI] [PubMed] [Google Scholar]
- 107.Whanger PD. Selenoprotein W: a review. Cell Mol Life Sci. 2000;57(13–14):1846–52. doi: 10.1007/PL00000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Whanger PD. Selenoprotein expression and function-selenoprotein W. Biochim Biophys Acta. 2009;1790(11):1448–52. doi: 10.1016/j.bbagen.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 109.Gu QP, Beilstein MA, Vendeland SC, Lugade A, Ream W, Whanger PD. Conserved features of selenocysteine insertion sequence (SECIS) elements in selenoprotein W cDNAs from five species. Gene. 1997;193(2):187–96. doi: 10.1016/s0378-1119(97)00113-3. [DOI] [PubMed] [Google Scholar]
- 110.Bellingham J, Gregory-Evans K, Fox MF, Gregory-Evans CY. Gene structure and tissue expression of human selenoprotein W, SEPW1, and identification of a retroprocessed pseudogene, SEPW1P. Biochim Biophys Acta. 2003;1627(2–3):140–6. doi: 10.1016/s0167-4781(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 111.Dikiy A, Novoselov SV, Fomenko DE, Sengupta A, Carlson BA, Cerny RL, Ginalski K, Grishin NV, Hatfield DL, Gladyshev VN. SelT, SelW, SelH, and Rdx12: genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry. 2007;46(23):6871–82. doi: 10.1021/bi602462q. [DOI] [PubMed] [Google Scholar]
- 112.Jeon YH, Park YH, Kwon JH, Lee JH, Kim IY. Inhibition of 14-3-3 binding to Rictor of mTORC2 for Akt phosphorylation at Ser473 is regulated by selenoprotein W. Biochim Biophys Acta. 2013;1833(10):2135–42. doi: 10.1016/j.bbamcr.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 113.Jeon YH, Park YH, Lee JH, Hong JH, Kim IY. Selenoprotein W enhances skeletal muscle differentiation by inhibiting TAZ binding to 14-3-3 protein. Biochim Biophys Acta. 2014;1843(7):1356–64. doi: 10.1016/j.bbamcr.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 114.Park YH, Jeon YH, Kim IY. Selenoprotein W promotes cell cycle recovery from G2 arrest through the activation of CDC25B. Biochim Biophys Acta. 2012;1823(12):2217–26. doi: 10.1016/j.bbamcr.2012.09.001. [DOI] [PubMed] [Google Scholar]