

Abstract
Treatment for Acute myeloid leukemia (AML) has remained unchanged for past 40 years. Targeting cell metabolism is a promising avenue for future cancer therapy. We found that enzymes involved in metabolic hexosamine biosynthetic pathway (HBP) are increased in AML patients. Inhibiting GFAT (the rate limiting enzyme of HBP) induced differentiation and apoptosis in AML cells, sparing normal cells. UDP-GlcNAc, the end product of HBP, is the substrate for O-GlcNAcylation, a post-translational modification. O-GlcNAc transferase (OGT) is the enzyme which transfers GlcNAc from UDP-GlcNAc to target proteins. Inhibition of O-GlcNAcylation, using OGT inhibitors as well as genetic knockdown of OGT, also led to cell differentiation and apoptosis of AML cells. Finally, HBP inhibition in vivo, reduced the tumor growth in a sub-cutaneous AML xenograft model and tumor cells showed signs of differentiation in vivo. A circulating AML xenograft model also showed clearance of tumor load in bone marrow, spleen and blood, after HBP inhibition, with no signs of general toxicity. This study reveals an important role of HBP/O-GlcNAcylation in keeping AML cells in an undifferentiated state and sheds light into a new area of potential AML therapy by HBP/O-GlcNAc inhibition.
Keywords: Acute Myeloid Leukemia, HBP pathway, O-GlcNAcylation, metabolism, differentiation therapy, cell death
Acute myeloid leukemia (AML) is a heterogeneous disorder characterized by differentiation arrest of myeloid cells. This leads to uncontrolled clonal proliferation of immature myeloid blasts (1, 2). It is the most common acute leukemia in adults. AML can be classified based on different criteria including its cytogenetics, morphology, antigen markers and cytochemistry. AML can be divided into eight different subclasses (M0-M7) based on the differentiation status, according to the French-American-British (FAB) classification (3–7). The incidence of AML increases with age and over 20,000 cases per year is reported in the United States (8, 9). Current long-term survival rates in older patients (5–15%) is worse compared to younger patients (25–50%) (10). Toxicities associated with current treatment strategies and failure to tolerate those side effects, especially for older patients, are the two main reasons for this poor outcome. Although a number of targeted therapeutics have been proposed for treating AML, chemotherapy and allogeneic bone marrow transplantation still remains to be the first-line standard treatment option for most of the AML patients (11, 12).
Differentiation arrest and uncontrolled proliferation of myeloid cells are the two main features of AML, which needs to be targeted together for a better treatment efficacy. Current AML treatment strategies including All-trans retinoic acid (ATRA) and chemotherapy often fail due to their inefficacy to target both key issues. Chemotherapeutic drugs Cytarabine and Adriamycin mostly target the highly proliferative cells and relapse is a common problem associated with it. Unfortunately, ATRA is successful as a differentiation therapy for only one rare subtype of AML, acute promyelocytic leukemia (APL) (5–10% of AML) (13–16). ATRA leads to long term survival in 75–85% of APL patients and is more tolerable for older patients. Therapeutic progress for AML has been very slow when compared to other cancers. Therefore, more effective and less toxic therapy strategies to target both differentiation arrest and proliferation, represent an unmet need in the management of AML.
Targeting of cellular metabolism has emerged as an attractive strategy in the treatment of different cancers including AML. Inhibition of isocitrate dehydrogenase (IDH) have shown promising results in clinical trials in AML patients with IDH mutations (17, 18). Moreover, inhibiting PI3K/AKT/mTOR signaling pathway in AML cells received much attention as this pathway is overexpressed in AML cells. Anti-cancer potential of direct inhibitors of this pathway and inhibitors of mediators upstream in this pathway are also extensively studied (19, 20). Cancer cells depend on less efficient glycolysis known as Warburg effect. This results in increased uptake of glucose (21–25). Cancer cells also consume large amounts of glutamine, a precursor amino acid for the synthesis of glucosamine, initiator in the hexosamine biosynthetic pathway (HBP) (26, 27). Glutamine–fructose-6-phosphate amidotransferase (GFAT), a rate limiting enzyme of HBP combines Fructose-6-phosphate from the glycolytic pathway with glutamine to produce glucosamine-6-phosphate (28–30). UDP-GlcNAc is the end-product of HBP, which serves as the substrate for O-GlcNAcylation. OGT and OGA are enzymes with opposite functions, OGT adds O-GlcNAc residue to the hydroxyl groups of serine and/or threonine residues and OGA removes O-GlcNAc from target proteins (31) O-GlcNAcylation is a rapidly cycling posttranslational modification similar to phosphorylation. Because it happens primarily at serine and threonine residues or occurs at sites adjacent to phosphorylation, O-GlcNAcylation often competes with phosphorylation (32–34). Unlike many different types of kinases and phosphatases existing in a cell, there is only one OGT and OGA controlling O-GlcNAcylation of all cellular proteins.
Higher amounts of glucose or glutamine flowing through the HBP results in increased production of UDP-GlcNAc, which is substrate for O-GlcNAcylation. Increased O-GlcNAcylation is implicated in many cancers especially in transcriptional regulation, cell proliferation and survival (35–37). Recent studies have shown increased levels of O-GlcNAcylation in both systemic and solid cancers (36, 38–42). Clinical behavior of Chronic Lymphocytic Leukemia (CLL) cells is reported to correlate with intensity of O-GlcNAcylated protein levels in those cells and O-GlcNAcylation is increased in these cells after cytokine stimulations and Toll-like Receptor activation (41). Conversely, inhibition of HBP has been recently reported to promote castration-resistant prostate cancer (43). Thus O-GlcNAcylation could be involved in the pathogenesis of several types of cancers.
In this study, we found elevated levels of HBP enzymes and O-GlcNAcylation in AML cells compared to normal cells. We demonstrated that HBP or OGT inhibition leads to AML cell differentiation and apoptosis. Finally, using two different xenograft AML mouse models, we show inhibition of growth and differentiation of tumor cells after HBP inhibition. Our findings suggest that targeting HBP or OGT is an attractive potential novel therapeutic strategy for all subtypes of AML.
Materials and Methods
Supplementary information provides a detailed account of all material and methods used in this study.
Patient samples.
Human samples from normal donors and AML patients were obtained from Hematopoietic Stem Cell Core Facility located in Case Western Reserve University.
Cell culture.
OCI-AML3 (DSMZ), HL-60 (ATCC) and HEK293T (ATCC) cells were maintained according to standard protocols. Cell lines were initially tested for mycoplasma and then maintained in the presence of Normocin (invivoGen, USA). Cell lines were used up to 10 passages after thawing. PBMCs and AML patients were cultured in RPMI medium containing 10% FBS.
Cell death assay.
Cell death was estimated either by staining cells with propidium iodide (PI) followed by flow cytometry analysis or by trypan blue stain based cell counting. In experiment to determine PBMCs vs AML patient samples sensitivity towards DON treatment, cell death was analyzed by gating cells on a forward-scatter vs side-scatter plot in a flow cytometer.
Apoptosis assay.
Cells were stained with Annexin V and PI stain and analyzed by flow cytometer.
Cell proliferation assay.
The proliferation was monitored by measuring the cell confluence in an IncuCyte ZOOM (Essen Bioscience) instrument or by trypan blue stain based cell counting.
Cell differentiation.
Cells were plated at a density of 105 cells per ml and treated as indicated till 5 days. DON was added every 24 hr. On day 5 cells were imaged for changes in the cell morphology, NBT reduction assay (NBT (0.25 mg/ml) and PMA (500 ng/ml)) and surface expression of differentiation markers CD11b and CD14.
shRNA interference and lentiviral packaging.
The shRNA against OGT were validated for the knockdown efficiency. Lentiviral particles were produced using HEK293T cells and transduced by spinofection.
Ethics.
All mice experiments were done according to Institutional Animal Care and Use Committees (IACUC) guide lines and approved protocol.
In vivo AML xenograft models.
In solid AML xenograft tumor model, NSG mice were sub-cutaneously injected with 5×106 HL-60 cells and i.p. administered either with PBS or DON (0.65 mg/kg body weight) as shown in figure 6a. Tumor volume was calculated using formula (width × width × length × 0.5). Tumor cells were analyzed for surface expression of differentiation markers CD11b and CD14. In another liquid AML xenograft model, sub-lethally irradiated (200 cGy) NSG mice were i.v. injected with 3×106 OCI-AML3 cells, followed by i.p. administration of PBS or DON (0.65 mg/kg body weight) or cytarabine (50 mg/kg body weight) as shown in figure 6f. Bone marrow, spleen and blood were analyzed for the tumor burden by staining with anti-human CD45.
Figure 6. In vivo DON administration reduces AML.
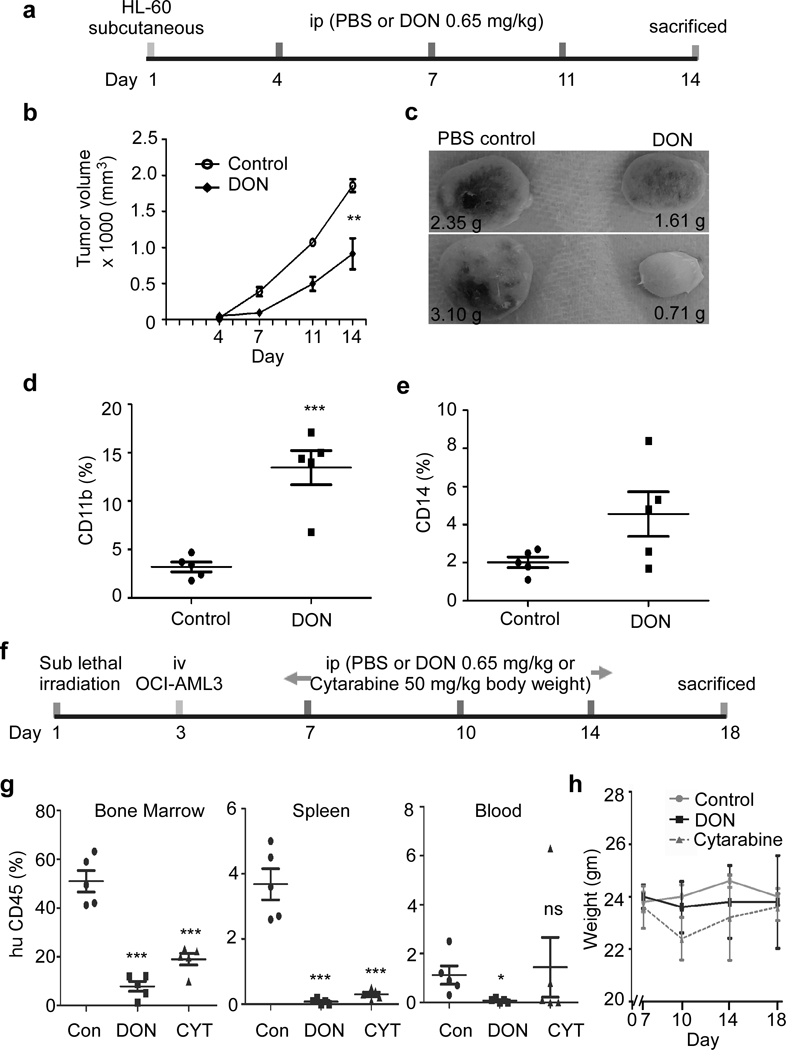
(a) Schematic representation of in vivo administration of DON or PBS in HL-60 based AML mice xenograft model. NSG mice were sub-cutaneous injected with HL-60 cells to generate solid AML mice xenograft model, followed by i.p. injection treatment as shown. (b) Tumor volume was monitored on day 4, 7, 11 and 14. (c) Representative image of harvested tumor is shown from control and DON treated mice group at day 14. Number indicates the weight of tumor in grams. Harvested tumors were analyzed for the presence of surface expression of differentiation marker (d) CD11b and (e) CD14. (f) Schematic representation of in vivo administration of DON/ cytarabine/ PBS in OCI-AML3 based AML xenograft mice model. Sub-lethally irradiated NSG mice were i.v. injected with OCI-AML3 cells to generate liquid AML xenograft mice model, followed by i.p. injection treatment as shown. (g) Bone marrow, spleen and blood cells isolated from indicated experimental groups were analyzed for the presence of human CD45 surface expression. Control group was i.p. injected with PBS and treated group was i.p. injected with DON (0.65 mg/kg). (h) Weight profile of animals from different experimental group during treatment regime at indicated days in AML mice model. N=5; Statistical significance was calculated using unpaired Student’s t-test. ns= not significant, *p<0.05, **p<0.01, ***p<0.001.
Statistical analysis.
Applicable data were analyzed using unpaired Student’s t-test. All experiments were done in triplicates (N=3) unless mentioned otherwise. P values assigned were ns= not significant, *p<0.05, **p<0.01, ***p<0.001. In vivo experiments has 5 animals in each experimental group (N=5).
Results
AML cells express higher levels of HBP enzymes and are hyper O-GlcNAcylated
We checked the levels of O-GlcNAcylated proteins in AML patient cells vs normal PBMCs and found that overall protein O-GlcNAcylation was significantly elevated in AML patient subtypes (M1, M2, M4, M5) and one relapsed AML-M1 patient cells compared to normal PBMCs (Figure. 1a). To understand the molecular events regulating O-GlcNAcylation in AML, we used the web-based microarray database called Oncomine to assess the relative gene expression levels of enzymes involved in O-GlcNAcylation. We analyzed 74 normal and 542 AML patient samples using Haferlach Leukemia statistics and it showed a significant fold increase in GFAT1 (1.156, p=4.11E-7) and OGT (1.360; p=2.01E-16) gene expression in AML cells compared with normal -samples (Figure 1b, c). Analysis of fold change of different enzymes involved in O-GlcNAcylation was performed using datasets available at Oncomine database (Supplementary Figure S1). Furthermore, we found that HBP pathway enzymes including GFAT1, PGM3, UAP1, GNA1 and OGT were overexpressed in AML cell lines OCI-AML3 and HL-60 compared to normal PBMCs (Figure 1d).
Figure 1. AML patient cells are hyper O-GlcNAcylated.
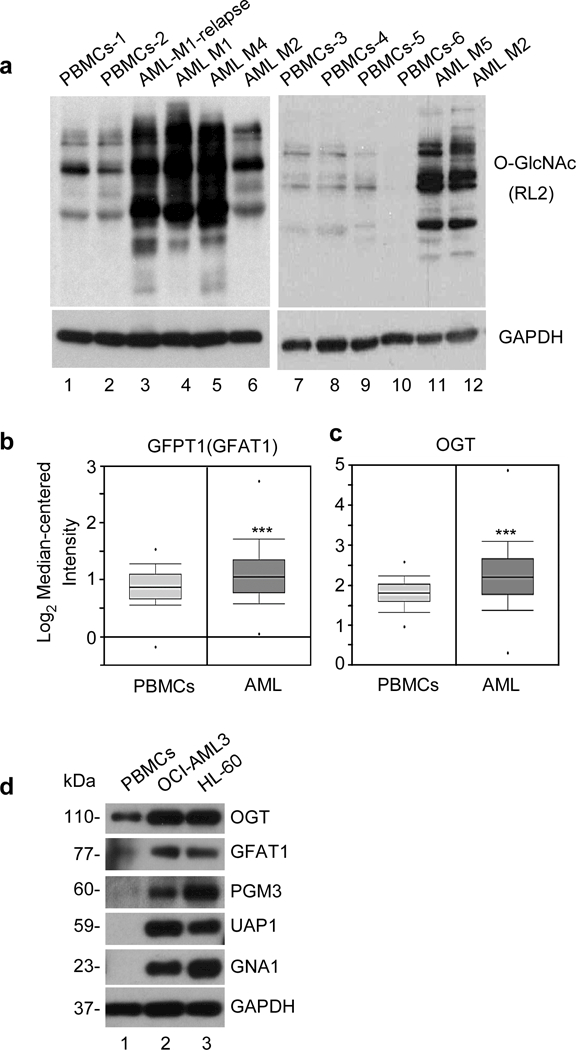
(a) Western blot analysis of normal PBMCs and AML patient samples using O-GlcNAc (RL2) antibody. GAPDH served as an endogenous loading control. mRNA expression levels from normal Peripheral Blood Mononuclear cells (74) were compared to Acute Myeloid Leukemia (542) samples for the gene (b) GFAT1 or GFPT1 (Glutamine-Fructose-6-Phosphate transaminase 1, a rate limiting enzyme of hexosamine biosynthetic pathway) and (c) OGT (O-GlcNAc transferase) enzyme, using Haferlach Leukemia statistics from Oncomine database. AML vs normal PBMCs sample fold change for GFPT1 is 1.156 (p-value 4.11E-7) and for OGT is 1.360 (p-value 2.01E-16). Statistical significance of the expression was calculated by t-test, ***p < 0.001. (d) Western blot analysis of enzymes involved in hexosamine biosynthetic pathway (GFAT1, PGM3, UAP1, GNA1), and OGT in normal PBMCs, OCI-AML3 and HL-60. GAPDH was used as an endogenous control.
Inhibition of HBP leads to AML cell death
The significant increase in expression of HBP enzymes and O-GlcNAcylation in AML cells prompted us to study the effect of HBP inhibitor on AML cell growth. DON (6-Diazo-5-oxo-L-nor-Leucine) inhibits GFAT1 (the enzyme that converts fructose-6-P to glucosamine-6-P), and thereby inhibits HBP and O-GlcNAcylation of proteins. We performed a dose and time kinetics study to determine the concentration and duration at which DON inhibit the proliferation of AML cells with minimal toxic effects on normal cells. We found a dose dependent increase in the killing potential of DON with 1 μM DON causing 15% cell death, while 50 μM killing up to ~60% of AML cells post 72 hours of incubation (Figure 2a). Next, we incubated OCI-AML3 cells in presence of 50 μM DON for 0–72 hours of treatment and found that at 24 hours about 30% of cells were killed and a plateau is achieved around 72 hours (Figure 2b). We treated normal PBMCs and monocytes cells also with 50 μM DON for 24 hours to study the survival response of these cells to HBP inhibition (Figure 2c). Surprisingly, DON had only minor effects on the viability of normal cells, while AML patient cells belonging to different subtypes M1, M4 and M5 showed significant killing (Figure 2d). Significant cell death (~ 35%) was also observed in OCI-AML3 and HL-60 cell lines at 24 hours post DON treatment (Figure 2e). We confirmed the decrease of O-GlcNAcylation after DON treatment in normal PBMCs and AML cells (Figure 2f)
Figure 2. Blocking protein O-GlcNAcylation kills AML cells.
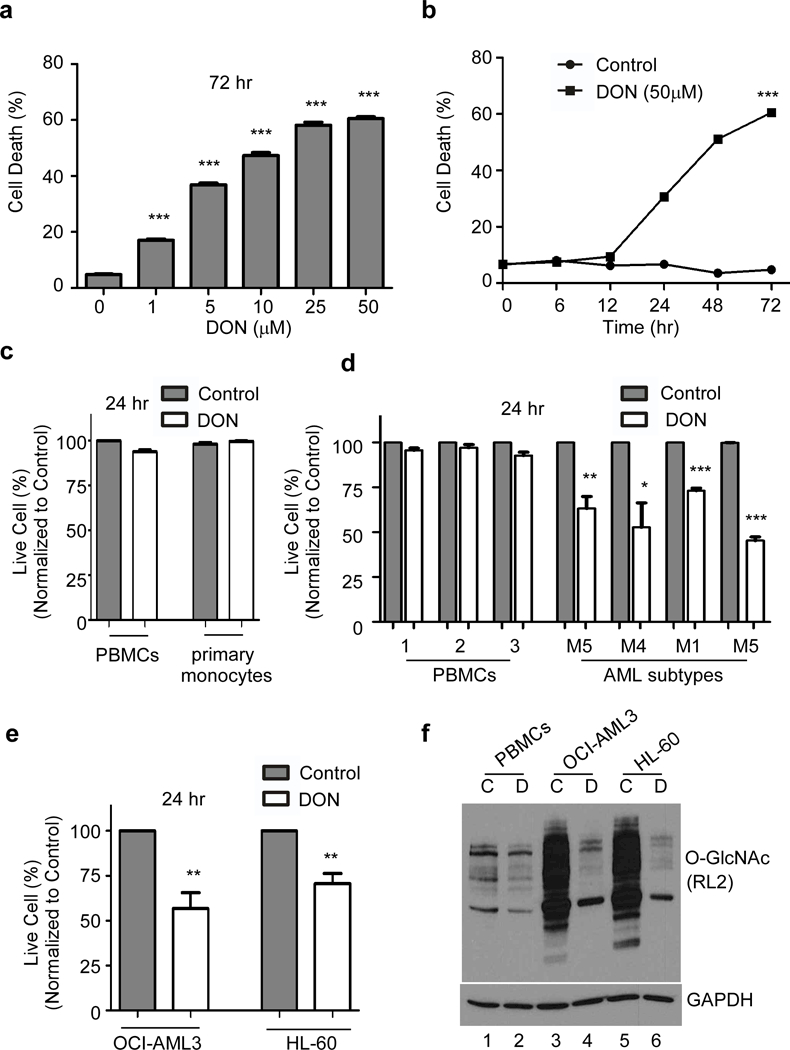
DON treatment blocks O-GlcNAcylation and subsequent cell death in OCI-AML3 cells was monitored (a) in a dose-dependent manner after 72 hr treatment and (b) in a time-dependent manner with DON (50 μM) treatment. (c) Cell viability of normal PBMCs and primary monocytes 24 hr after DON (50 μM) treatment compared to the untreated control. (d) Cell viability of PBMCs and AML patient blast samples treated with DON or untreated control after 24 hr. (e) Effect of DON (50 μM) on the cell viability of OCI-AML3 and HL-60 cells after 24 hr treatment. (f) Western blot showing O-GlcNAc profile of PBMCs, OCI-AML3 and HL-60 using O-GlcNAc (RL2) antibody. Cells were incubated (16 hr) as indicated. C-untreated control or D-DON (50μM). Actin was used as an endogenous loading control. Statistical significance was calculated using unpaired Student’s t-test. N=3; *p<0.05, **p<0.01, ***p<0.001.
To gain insights into the effect of DON on AML proliferation, we used IncuCyte ZOOM technology for automation of imaging and quantification of cell confluence and nuclear count data. Cell confluence decreased about 90% in both OCI-AML3 cells and HL-60 cells after 72 hours of DON treatment confirming the inhibition of cell proliferation in DON treated AML cells (Supplementary Figure S2a, b). Exposure of AML cells to DON (50 μM), induced apoptosis as evidenced by Annexin V positivity of these cells (Supplementary Figure S2c, d). DON treated cells also showed an increase in the cleaved caspase-3 and cleaved PARP proteins, confirming DON induced apoptosis in AML cells (Supplementary Figure S2e). We further confirmed this finding using alternate methods. We used OGT inhibitors OSMI-1 (44, 45) and BADGP (45) to inhibit O-GlcNAcylation in AML cells. Both OSMI-1 and BADGP inhibited cell proliferation of OCI-AML3 and HL-60 (Figure 3a, b) cells as evident by viable cell count.
Figure 3. OSMI-1 and BADGP mediated O-GLcNAc inhibition reduces proliferation and induces differentiation of AML cells.
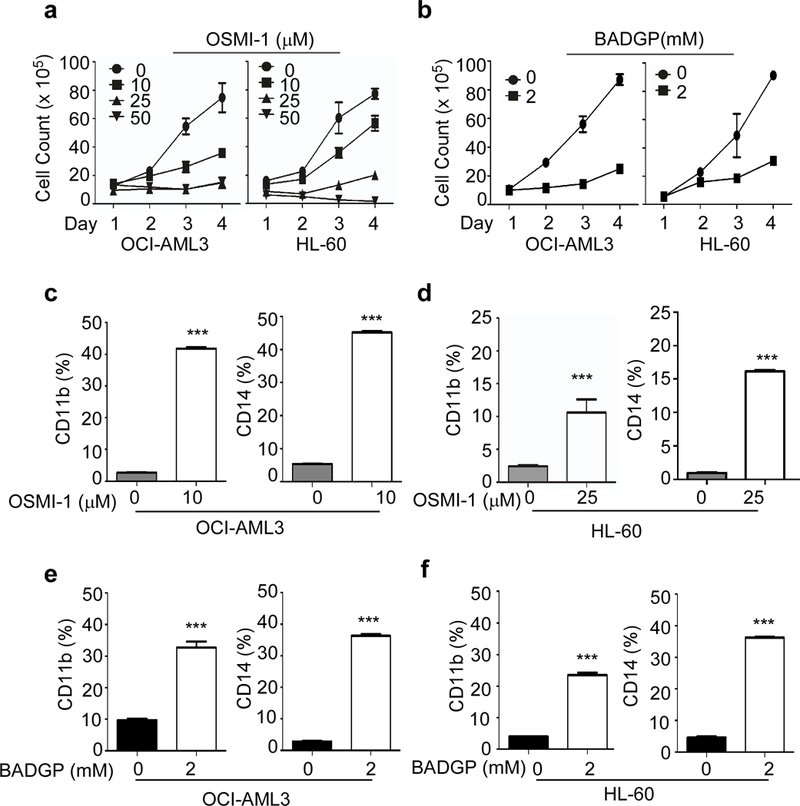
Proliferation of OCI-AML3 and HL-60 cells was monitored in the presence or absence of (a) OSMI-1 and (b) BADGP in OCI-AML3 and HL-60 cells, as indicated. Surface expression of differentiation marker CD11b and CD14 is shown in (c) OCI-AML3 and (d) HL-60 cells at day 5 after treatment with indicated concentration of OSMI-1. Surface expression of CD11b is shown at day 5 after treatment with BADGP in (e) OCI-AML3 and (f) HL-60 cells. Statistical significance was calculated using unpaired Student’s t-test. N=3; *p<0.05, **p<0.01, ***p<0.001.
Inhibition of HBP pathway leads to AML differentiation
AML cells undergo terminal differentiation and apoptosis when treated with ATRA and is used as a therapeutic strategy in APL patients. We studied whether DON induced apoptosis of AML involves cell differentiation. We found that low dose DON (12.5 μM) treatment for 5 days induce differentiation of AML cells, similar to as seen after ATRA treatment, as evidenced by the morphology changes (Figure 4a) and Nitroblue Tetrazolium (NBT) staining (Figure 4b). ATRA was used as a positive control and a combination of DON and ATRA caused differentiation similar to DON or ATRA alone (Figure 4a). Morphologically, DON induced monocytic differentiation results in an increased cytoplasmic to nuclear ratio. In addition, these cells exhibited a condensed and indented nucleus along with significant enlargement in cell size (Figure 4a). OCI-AML3 cells undergo monocytic differentiation, while HL-60 cells typically undergo granulocytic or monocytic differentiation. CD11b is used as a marker for myelomonocytic differentiation and CD14 as a marker for monocytic differentiation. Both OCI-AML3 and HL-60 cells showed increase in CD11b expression from 5% to 65% and 7% to 35% respectively on 5th day after DON treatment (12.5 μM), whereas CD14 expression increased from 4% to 80% in OCI-AML3 and from 3% to 30% in HL-60 cells (Figure 4c, d). Besides, morphological and immunophenotypic changes, DON exhibits high respiratory activity in OCI-AML3, a characteristic of differentiated cells as measured by NBT reduction (Figure 4b). Alternatively, OSMI-1 and BADGP treatments also show increase in CD11b and CD14 expression on 5th day. OCI-AML3 cells exhibit ~ 40 % and HL-60 ~12 % CD11b expression upon O-GlcNAc inhibition using OSMI-1 10 μM and 25 μM, respectively (Figure 3c, d). BADGP (2 mM) also induced CD11b expression ~32% in OCI-AML3 and ~22% in HL-60 cells (Figure 3e, f). CD14 expression increased to 45% in OCI-AML3 and 16% in HL-60 upon O-GlcNAc inhibition using OSMI-1 (10 μM) (Figure 3c, d) and up to 35% in OCI-AML3 and HL-60 upon inhibition with BADGP (2 mM) (Figure 3e, f). As inhibition of HBP pathway and O-GlcNAcylation by DON pushed AML cells to differentiation, we also tested what happens to O-GlcNAcylation status when AML cells are induced to differentiate. We used ATRA to induce differentiation and found that AML cells post ATRA treatment has decreased O-GlcNAcylation (Figure 4e). These experiments demonstrate that AML cell differentiation is associated with a decreased O-GlcNAcylation.
Figure 4. At low dose, DON induces AML differentiation.
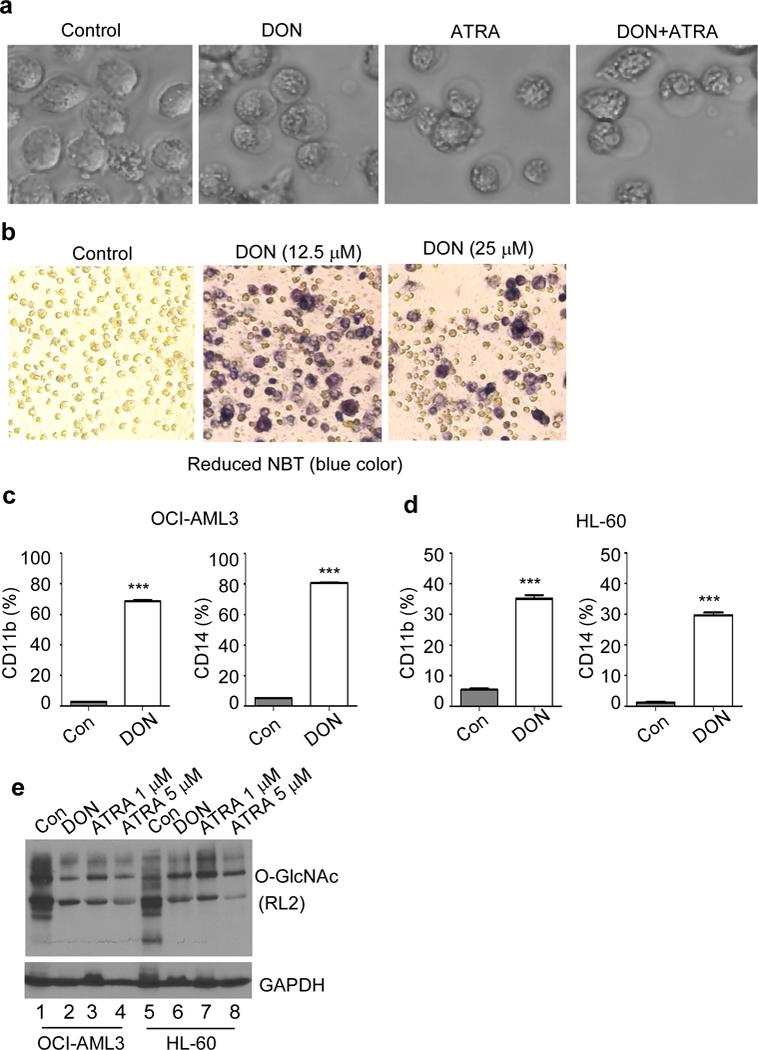
(a) Image of OCI-AML3 cells treated with DON (12.5 μM), ATRA (1 μM) either alone or in combination at day 5. Cells show a change in morphology and cytoplasm to nuclear ratio. ATRA is known to induce APL differentiation. (b) OCI-AML3 cells were treated with or without DON till day 5, as indicated. Differentiated cells reduces nitroblue tetrazolium (NBT) in the assay, indicated by the development of bluish/purple color. Surface expression of differentiation marker CD11b and CD14 was determined in either control or DON (12.5 μM) treated (c) OCI-AML3 and (d) HL-60 cells on day 5 by flow cytometry. (e) Western blot analysis of differentiating AML cells show reduction in O-GlcNAc (RL2) protein modification. OCI-AML3 and HL-60 cells were treated with indicated compounds for 3 days and probed for O-GLcNAc. GAPDH served as an endogenous loading control. ATRA was used to induce differentiation and DON was used to inhibit protein O-GlcNAcylation. Statistical significance was calculated using unpaired Student’s t-test. N=3; ***p<0.001.
O-GlcNAcylation and phosphorylation are two post-translational modifications that often occur on the same protein competing for the same serine/threonine site(s) and bear a reciprocal relationship. The interplay between O-GlcNAc and O-phosphate has been implicated in regulating protein function and plays an important role in cancer. It has been previously shown that inhibiting O-GlcNAcylation enhances phosphorylation of kinases such as Akt and GSK3β (46, 47). These proteins are extensively studied in AML cells (48, 49) and are shown to play important role in survival of AML cells. However, the cell type–specific regulation of protein phosphorylation by O-GlcNAcylation in AML cells has not been explored. Hence, we checked the phosphorylation of these proteins in AML cells after DON treatment. It has been reported that O-GlcNAcylation of Akt at Thr 305 and 312 inhibit its phosphorylation at Thr 308. We found that DON treatment and thereby inhibition of O-GlcNAcylation, increased phosphorylation of AKT at Thr308 and Ser473, and GSK3 phosphorylation at Ser9 (Supplementary Figure S3a). These results suggest that alteration in cellular O-GlcNAcylation levels could affect phosphorylation of AKT and GSK-3β in AML cells.
AML involves deregulation of critical transcription factors such as c-myc and c-myb, which are crucial for normal hematopoiesis. Transcription factors c-myc and c-myb are involved in diverse cellular functions including role in proliferation and differentiation (50, 51). However, they may have increased expression or aberrant activation in AML. Interestingly, downregulation of transcription factors such as c-myc and c-myb occurs in AML cells undergoing differentiation (50–52). We found that transcription level of c-myc and c-myb was reduced upon treatment with DON in OCI-AML3 cells (Supplementary Figure S3b). These results suggests that down regulation of transcription factor c-myc and c-myb are important early changes involved in initiation of DON mediated cellular differentiation.
HBP is a key metabolic pathway initiated by glucose or glutamine flux in the cell and can have profound effect on cellular metabolism. We studied whether DON induced HBP inhibition affect oxidative and glycolytic pathways in AML cells. We examined oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) of OCI-AML3 cells using a Seahorse XF instrument. We observed only modest changes in OCR and ECAR (Supplementary Figure S3c, d), at the indicated time points, after exposure to DON and were not indicative of any drastic metabolic changes in these cells.
OGT downregulation also leads to terminal cell differentiation in AML cells
Since transfer of GlcNAc to proteins is mediated by a single enzyme OGT and blocking OGT will inhibit O-GlcNAcylation, OCI-AML3 cells were transiently transfected with OGT-siRNA or scrambled siRNA. Percent cell confluence showed significant drop after 96 hours when compared to scrambled siRNA transfected cells (Supplementary Figure. S4a). Western blot analysis of these siRNA transfected cells shows inhibition of OGT and O-GlcNAcylated proteins compared to scrambled siRNA transfected cells (Supplementary Figure S4b) and these cells gradually underwent apoptosis as evidenced by caspase-3 cleavage and PARP cleavage (Supplementary Figure S4b).
OCI-AML3 cells transduced with three different OGT shRNA showed decrease in OGT levels directly proportional to the decrease in the overall levels of O-GlcNAcylation of cellular proteins (Figure 5a). The decrease in the protein O-GlcNAcylation was maximum in shRNA-OGT3, followed by shRNA-OGT1 and minimal in shRNA-OGT2 transduced cells. At day 5, viable cell count shows decreased rate of cell proliferation in OGT-ShRNA OCI-AML3 cells (Figure 5b). Moreover, staining with Annexin V/PI confirms apoptosis of these cells (Figure 5c) in shRNA-OGT3 (~55% cells) and shRNA-OGT1 (~32% cells) transduced cells at day 5 (Figure 5c, d). shRNA-OGT2 with minimal effect on OGT served as an internal control, show only ~7% apoptosis (Figure 5c, d). Furthermore, increase in surface expression of differentiation markers CD11b (shRNA-OGT3 ~42%; shRNA-OGT1~12%) and CD14 (shRNA-OGT3 ~75%; shRNA-OGT1~30%) in OCI-AML3 suggests cells were undergoing differentiation upon downregulation of OGT (Figure 5e, f). Interestingly, shRNA-OGT2, did not show increase in expression of differentiation markers CD11b and CD14.
Figure 5. OGT downregulation leads to apoptosis and differentiation of OCI-AML3 cells.
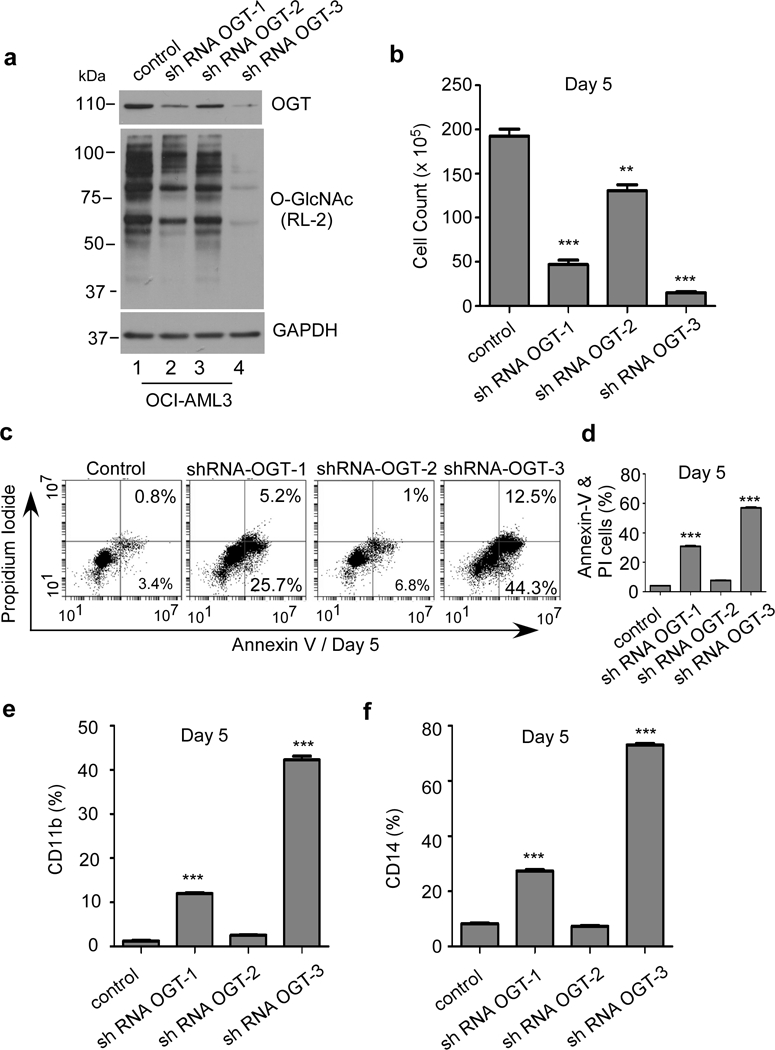
OCI-AML3 cells transduced with either control shRNA or shRNAs against OGT were analyzed on day 5. (a) Western blot analysis for OGT and O-GlcNAc levels after lentiviral transduction with 3 different shRNA directed against OGT in OCI-AML3 cells. GAPDH was used as an endogenous loading control. (b) Cell count of OCI-AML3 cells on day 5 after lentiviral transduction with either control or shRNA against OGT. (c) OCI-AML3 cells were stained with Annexin V and propidium iodide (PI) to determine apoptotic cell population, on day 5 after lentiviral transduction. (d) Graph showing total population of early and late apoptotic OCI-AML3 cells under indicated condition on day 5. Surface expression of differentiation markers (e) CD11b and (f) CD14 in OCI-AML3 transduced with different shRNA at day 5. Statistical significance was calculated using unpaired Student’s t-test. N=3; ***p<0.001.
Pharmacological inhibition of HBP pathway eliminates AML cells in vivo
To determine whether DON can be utilized as a potential therapeutic agent for AML, we used an AML xenograft NSG mice model. HL-60 cells were subcutaneously injected in NSG mice. At day 4 tumors were palpable, mice were treated with DON as schematically represented in Figure 6a. Following treatment significant decrease in AML tumor volume was observed in DON treated mice compared to vehicle treated xenograft mice (Figure 6b, c). O-GlcNAc levels were found to be moderately reduced in tumor extract from mice administered with DON when compared to vehicle treated (Supplementary Figure S5a). In addition to reduced tumor volume in DON treated mice increase in expression of differentiation marker CD11b and CD14 was observed (Figure 6d, e).
In another circulating tumor in vivo xenograft model, NSG mice were i.v. injected with OCI-AML3 cells and followed by three i.p injection of DON (Figure 6f). At day 18, control (PBS) and DON treated mice were sacrificed to analyze the tumor burden in bone marrow, spleen and blood as detected by human CD45 positive cells. As shown in figure 6g, a significant reduction of AML tumor load was observed in bone marrow, spleen and blood in DON treated mice compared to the control mice. We also compared the effect of DON to existing chemotherapy drug cytarabine and the effect was comparable.
All vehicle-treated xenografts showed nearly 40–60% positivity for human CD45+ cells in bone marrow, 3–5% in spleen and 0.5–2.5% in blood at 18 days post-transplantation, While DON treated mice showed dramatic reduction in CD45+ cells, 1–15% in BM, 0–0.5% in spleen and 0–0.5% in blood (Figure 6g). Mice weight did not significantly vary during the treatment regime (Figure 6h).
In order to evaluate toxicity of DON, C57BL mice were similarly i.p. injected with DON. Western blot of harvested organs confirmed the decrease in levels of O-GlcNAc in spleen, liver and kidney in DON administered mice (Supplementary Figure S5b) compared to control. However, bone marrow did not show any major alteration in O-GlcNAc levels. Their weight profile (Supplementary Figure S5c) and H&E stain of kidney, liver and spleen (Supplementary Figure S5d) did not show any significant difference from PBS injected control mice. Hematological profile of DON injected mice showed a marginal decrease in hemoglobin (HB) and hematocrit (HCT) values compared to normal range. DON treatment also cause a decrease in platelet count suggesting thrombocytopenia. However, WBC, neutrophils, lymphocytes, monocytes, eosinophils, basophils, RBC were in normal range (Supplementary Figure S6). Overall these experiments show specific killing of AML cells with no major toxicity to normal cells.
Discussion
Cancer cells generally use higher amounts of glucose and glutamine, channeled to different pathways, in order to maintain energy generation pathways, cell survival and proliferation. Glutamine metabolism has developed as an attractive target for treatment of different cancers. Glutamine and glucose channeled to HBP is utilized for making UDP-GlcNAc, the substrate for O-GlcNAcylation (53). Increased O-GlcNAcylation of proteins has been reported in many cancers (38–42). We report here that there is an upregulation of key enzymes involved in HBP and it results in an increased O-GlcNAcylation of proteins in AML cells. Inhibition of this pathway resulted in AML cell death. Interestingly, the death of AML cells was preceded by their differentiation (Figure 4). This shows that O-GlcNAcylation is a necessary event for AML cell survival.
Hyper O-GlcNAcylation in AML cells points to a greater dependency of these cells on HBP for their survival compared to normal cells. We used DON, a GFAT inhibitor to inhibit HBP and O-GlcNAcylation in AML cells. In agreement with existing literature we found that HBP inhibition leads to increase in specific phosphorylation of Akt and GSK3-β. To our surprise, moderate inhibition of HBP using low dose of DON led to AML cell differentiation, similar to ATRA, a commonly used differentiating agent for APL (16). It will take 4–7 days for the AML cells to die in presence of low concentration of DON, while they undergo apoptosis within 24 hours in presence of high concentration of DON. Based on this, we speculate that depending on degree of inhibition of HBP, AML cell death mechanisms differ. ATRA binds to the retinoic acid receptor (RAR) and activates transcription of genes necessary for AML differentiation. ATRA treatment fails to induce differentiation in cells with dysfunctional RAR signaling (54). ATRA treatment is successful only in one subset of AML patients with APL fusion protein as it induces activation and degradation of the APL fusion protein leading to re-expression of the myeloid differentiation program, terminal differentiation and apoptosis of leukemic cells (55). Since HBP was found activated in all AML subtypes tested including AML-1, AML-2, AML-4 and AML-5, inhibition of this pathway might be promising to treat all AML subtypes. Other AML subtypes also need to be analyzed for HBP enzymes and O-GlcNAcylation status in future studies.
Because inhibiting O-GlcNAcylation results in AML cell differentiation, we hypothesized that hyper O-GlcNAcylation keeps AML cells in un-differentiated state. To test this hypothesis, we analyzed the levels of O-GlcNAcylation in AML cells before and after ATRA induced differentiation. Interesting enough, we found that ATRA induced differentiation is associated with a decrease in O-GlcNAcylation, confirming our hypothesis that differentiation is connected with O-GlcNAcylation or HBP activation status of cells. Moreover, we found that similar to ATRA induced differentiation, HBP inhibition by DON also leads to early downregulation of c-myc and c-myb transcript levels which is linked to directing AML cells towards differentiation. Further work needs to be done to explore the actual mechanism behind this phenomenon. It is intriguing to see that both DON and ATRA behaves in a similar manner in causing AML cell differentiation in an O-GlcNAcylation dependent manner. Little is known about the role of ATRA in O-GlcNAcylation. However, one previous study shows interaction between OGT and nuclear receptors including RAR-alpha. Biological effects of ATRA are considered to be mainly mediated through RAR-alpha (56). Thus, it is conceivable that both DON and ATRA yields similar phenotypic effect and suppression of O-GlcNAcylation. Detecting the specific O-GlcNAcylated proteins associated with differentiation and delineating their function has the potential to lead to new arenas of targeted AML therapy.
DON is a GFAT inhibitor and acts upstream in HBP. To confirm that the observed effects are indeed due to HBP inhibition and not an artefactual outcome of DON treatment, we used alternate methods to confirm or refute our findings. Incubation of AML cells with small molecule OGT inhibitors (OSMI-1 and BADGP) as well as OGT knock down using shRNA inhibited AML cell proliferation and induces differentiation confirming that the phenotypes we observe are indeed due to the inhibition of O-GlcNAcylation. Interestingly, DON was found most effective of all these strategies for anti-leukemic activity. These findings suggest that the development of pharmacological agents that target HBP or OGT enzyme could have a significant therapeutic effect on AML. There are no existing OGT inhibitors tested in the clinic currently for treating cancer patients, indicating a need to develop novel and more effective OGT inhibitors. However, we should not forget the fact that mammalian OGT is a single copy (X-linked) gene and its knockout has been shown to be lethal (57). Therefore, targeting OGT in cancer cells needs a detailed dosing study to define optimal dose of OGT inhibitors showing minimal toxicity to normal cells.
Since GFAT and OGT are both nutrient-sensing enzymes, it is plausible that inhibiting them may affect normal metabolic state of cell. Metabolic profiling of prostate cancer cells after OGT inhibition decreases glucose consumption and lactate production, which results in the inhibition of proliferation of these cells (58). We analyzed oxidative phosphorylation indicator OCR and glycolysis indicator ECAR in AML cells after DON incubation and observed only some modest changes at the time points analyzed. A clear global picture of metabolic changes occurring in AML cells after HBP inhibition is essential to understand metabolic link and the mechanism behind AML cell differentiation and cell death post HBP inhibition.
Finally, our in vivo data is a clear evidence of therapeutic potential of DON and opens door for the use of many other HBP inhibitors for AML therapy. Sub-cutaneous AML xenograft model shows reduction in tumor burden accompanied with in vivo cell differentiation. In yet circulating AML model, eradication of AML cells from blood, spleen and bone marrow was noticed after DON treatment. Immunocompromised NSG mice did not show any weight loss, indicative of non-toxicity. AML killing was significant even after three DON injections which made us to think about its toxic effect on all other hematopoietic populations. Since NSG mice do not possess an intact immune system, we injected DON in C57BL6 mice for toxicity studies. We sacrificed all mice one day after 3rd injection to include both immediate toxic effects as well as long term effects. Besides reduced platelet count, DON injected C57BL6 mice did not show any significant changes in hematopoietic cell populations or organ histology.
Altogether, pharmacological targeting of HBP enzymes or OGT, could have a significant therapeutic effect on AML patients irrespective of their subtype. Since HBP inhibition targets both cell proliferation and differentiation of AML cells simultaneously, this strategy will be more effective than existing treatment methods. AML cells seems to be selectively sensitive to HBP inhibition without major toxicities to normal cells, hence developing new HBP or OGT inhibitors also will be a good bet for AML therapy.
Supplementary Material
Acknowledgements
We thank Dr. Rafick Sekaly and Dr. Jeffrey Tomalka for their help with Sea horse analysis. This research was supported by the Athymic Animal and Hemapoietic Biorepository and Cellular therapy core facilities, Shared Resources of the Case Comprehensive Cancer Center (P30CA043703) for their support and funding. We also thank NIH 1R21CA201775–01A1, 1R01AI116730–01A1, St.Baldrick’s Foundation, Mizutani foundation for Glycoscience, B positive foundation and Children’s Leukemia association for their funding support.
Financial support: NIH 1R21CA201775–01A1 (RP), 1R01AI116730–01A1 (PR), St.Baldrick’s Foundation (RP), Mizutani foundation for Glycoscience (RP), B positive foundation (RP) and Children’s Leukemia association (RP).
Footnotes
There is no conflict of interest for authors to disclose.
References
- 1.Estey EH. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am J Hematol 2014;89(11):1063–81. [DOI] [PubMed] [Google Scholar]
- 2.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366(12):1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976;33(4):451–8. [DOI] [PubMed] [Google Scholar]
- 4.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 1985;103(4):620–5. [DOI] [PubMed] [Google Scholar]
- 5.Cheson BD, Cassileth PA, Head DR, Schiffer CA, Bennett JM, Bloomfield CD, et al. Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol 1990;8(5):813–9. [DOI] [PubMed] [Google Scholar]
- 6.Casasnovas RO, Campos L, Mugneret F, Charrin C, Bene MC, Garand R, et al. Immunophenotypic patterns and cytogenetic anomalies in acute non-lymphoblastic leukemia subtypes: a prospective study of 432 patients. Leukemia 1998;12(1):34–43. [DOI] [PubMed] [Google Scholar]
- 7.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med 1999;341(14):1051–62. [DOI] [PubMed] [Google Scholar]
- 8.Almeida AM, Ramos F. Acute myeloid leukemia in the older adults. Leuk Res Rep 2016;6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J 2016;6(7):e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010;115(3):453–74. [DOI] [PubMed] [Google Scholar]
- 11.Stone RM. Targeted agents in AML: much more to do. Best Pract Res Clin Haematol 2007;20(1):39–48. [DOI] [PubMed] [Google Scholar]
- 12.Yates JW, Wallace HJ Jr., Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep 1973;57(4):485–8. [PubMed] [Google Scholar]
- 13.Tallmann MS. Curative therapeutic approaches to APL. Ann Hematol 2004;83 Suppl 1:S81–2. [DOI] [PubMed] [Google Scholar]
- 14.Stein EM, Tallman MS. Provocative pearls in diagnosing and treating acute promyelocytic leukemia. Oncology (Williston Park) 2012;26(7):636–41. [PMC free article] [PubMed] [Google Scholar]
- 15.Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 2011;117(22):5795–802. [DOI] [PubMed] [Google Scholar]
- 16.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood 2009;113(16):3655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reikvam H, Hauge M, Brenner AK, Hatfield KJ, Bruserud O. Emerging therapeutic targets for the treatment of human acute myeloid leukemia (part 1) - gene transcription, cell cycle regulation, metabolism and intercellular communication. Expert Rev Hematol 2015;8(3):299–313. [DOI] [PubMed] [Google Scholar]
- 18.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17(3):225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010;1(2):89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood 2008;111(1):379–82. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Lazaro M The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem 2008;8(3):305–12. [DOI] [PubMed] [Google Scholar]
- 22.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 2008;134(5):703–7. [DOI] [PubMed] [Google Scholar]
- 23.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927;8(6):519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324(5930):1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008;13(6):472–82. [DOI] [PubMed] [Google Scholar]
- 26.Pham LV, Bryant JL, Mendez R, Chen J, Tamayo AT, Xu-Monette ZY, et al. Targeting the hexosamine biosynthetic pathway and O-linked N-acetylglucosamine cycling for therapeutic and imaging capabilities in diffuse large B-cell lymphoma. Oncotarget 2016;7(49):80599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 2010;24(24):2784–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornfeld R Studies on L-glutamine D-fructose 6-phosphate amidotransferase. I. Feedback inhibition by uridine diphosphate-N-acetylglucosamine. J Biol Chem 1967;242(13):3135–41. [PubMed] [Google Scholar]
- 29.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 1991;266(8):4706–12. [PubMed] [Google Scholar]
- 30.McKnight GL, Mudri SL, Mathewes SL, Traxinger RR, Marshall S, Sheppard PO, et al. Molecular cloning, cDNA sequence, and bacterial expression of human glutamine:fructose-6-phosphate amidotransferase. J Biol Chem 1992;267(35):25208–12. [PubMed] [Google Scholar]
- 31.Hart GW, Akimoto Y. The O-GlcNAc Modification In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al. , editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory PressThe Consortium of Glycobiology; Editors, La Jolla, California.; 2009. [Google Scholar]
- 32.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 2011;80:825–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett 2010;584(12):2526–38. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A 2008;105(37):13793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Z, Vosseller K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 2014;289(50):34457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol 2016;428(16):3282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh JP, Zhang K, Wu J, Yang X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett 2015;356(2 Pt A):244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang M, Qiu Z, Zhang S, Fan X, Cai X, Xu B, et al. Elevated O-GlcNAcylation promotes gastric cancer cells proliferation by modulating cell cycle related proteins and ERK 1/2 signaling. Oncotarget 2016;7(38):61390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun 2017;8:15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Front Endocrinol (Lausanne) 2013;4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 2010;24(9):1588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Queiroz RM, Carvalho E, Dias WB. O-GlcNAcylation: The Sweet Side of the Cancer. Front Oncol 2014;4:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaushik AK, Shojaie A, Panzitt K, Sonavane R, Venghatakrishnan H, Manikkam M, et al. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat Commun 2016;7:11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, et al. A small molecule that inhibits OGT activity in cells. ACS Chem Biol 2015;10(6):1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trapannone R, Rafie K, van Aalten DM. O-GlcNAc transferase inhibitors: current tools and future challenges. Biochem Soc Trans 2016;44(1):88–93. [DOI] [PubMed] [Google Scholar]
- 46.Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, et al. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res 2008;314(11–12):2238–48. [DOI] [PubMed] [Google Scholar]
- 47.Shi J, Wu S, Dai CL, Li Y, Grundke-Iqbal I, Iqbal K, et al. Diverse regulation of AKT and GSK-3beta by O-GlcNAcylation in various types of cells. FEBS Lett 2012;586(16):2443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martelli AM, Nyakern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006;20(6):911–28. [DOI] [PubMed] [Google Scholar]
- 49.Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, et al. The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest 2012;122(3):935–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delgado MD, León J. Myc Roles in Hematopoiesis and Leukemia. Genes Cancer 2010;1(6):605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh I-H, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene 1999;18(19):3017. [DOI] [PubMed] [Google Scholar]
- 52.Dimberg A, Bahram F, Karlberg I, Larsson LG, Nilsson K, Oberg F. Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1). Blood 2002;99(6):2199–206. [DOI] [PubMed] [Google Scholar]
- 53.Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol 2016;17(6):712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson DE, Redner RL. An ATRActive future for differentiation therapy in AML. Blood Rev 2015;29(4):263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999;93(10):3167–215. [PubMed] [Google Scholar]
- 56.Li MD, Ruan HB, Singh JP, Zhao L, Zhao T, Azarhoush S, et al. O-GlcNAc transferase is involved in glucocorticoid receptor-mediated transrepression. J Biol Chem 2012;287(16):12904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, et al. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 2000;97(11):5735–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res 2013;73(16):5277–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.