

Abstract
Tumor-associated macrophages (TAM) are causally associated with tumorigenesis as well as regulation of anti-tumor immune responses and have emerged as potential immunotherapeutic targets. Recent evidence suggests TAM phagocytose apoptotic tumor cells within the tumor microenvironment (TME) through efferocytosis in an immunologically silent manner, thus maintaining an immunosuppressed microenvironment. The signal transduction pathways coupling efferocytosis and immunosuppression are not well known. Neuropilin-2 (NRP2) is member of the membrane-associated Neuropilin family and has been reported in different immune cells but is poorly characterized. In this study, we show that NRP2 is expressed during macrophage differentiation, is induced by tumor cells, and regulates phagocytosis in macrophages. Furthermore, NRP2 in TAM promoted efferocytosis and facilitated tumor growth. Deletion of NRP2 from TAM impaired the clearance of apoptotic tumor cells and increased secondary necrosis within tumors. This resulted in a break in the immune tolerance and re-initiated anti-tumor immune responses, characterized by robust infiltration of CD8+ T and NK cells. This suggests NRP2 may act as a molecular mediator that connects efferocytosis and immune suppression. Deletion of NRP2 in TAM downregulated several immunosuppressive and tumor-promoting genes and upregulated immunostimulatory genes in the myeloid compartment. Taken together, our study demonstrates that TAM-derived NRP2 plays a crucial role in tumor promotion through efferocytosis, opening the enticing option for the development of effective immunotherapy targeting TAM.
Keywords: NRP2, Tumor Associated Macrophages, efferocytosis, secondary necrosis, CD8, NK
Introduction:
Macrophages are multifaceted, highly plastic hematopoietic cells and extremely diverse in their functions. They are mononuclear phagocytes that maintain tissue homeostasis by clearing erythrocytes, apoptotic cells, and cellular debris (1, 2). They can also act as immune effector cells and bridge the innate and adaptive arms of the immune responses. Based on the nature of signals macrophages are exposed to in the tissue, they can either differentiate to classical type or alternative type macrophages and undergo profound changes in gene signature that we commonly refer to as ‘macrophage polarization’. The classical type may arise following exposure to GM-CSF, or TLR ligands like LPS and IFN-γ and mediates resistance against pathogens, produces immunostimulatory factors (IL-12) and is anti-tumorigenic. The alternatively activated type, which exists in many varieties, such as M2a, M2b or M2c, due to their stimulation by different extracellular signals (such as M-CSF, IL-4, IL-13, IL-10, glucocorticoids etc.), are collectively immunosuppressive in nature, upregulate the expression of CD163, CD206, IL-10, IL-4, TGF-β and other anti-inflammatory cytokines to blunt T cell responses (2, 3). Macrophages have also been causally associated with tumorigenesis and various stages of tumor progression and metastasis. High infiltration of tumor-associated macrophages (TAM) is a predictor of poor clinical outcome in various cancers (4, 5). Within the tumor, TAM behavior is dictated by microenvironment-derived factors that potentially suppresses antitumor immune responses and actively promote disease progression. TAMs exhibit complex molecular profile(s), resembling more of the alternatively-activated type macrophages and actively suppress anti-tumor immune responses. Therefore, understanding the origin and the mechanistic functioning of TAMs and molecular approaches to re-educate them towards a cytotoxic and immunostimulatory phenotype is important for developing anti-cancer therapy.
Efferocytosis is a strictly orchestrated process where phagocytes, such as macrophages, recognize, engulf and clear apoptotic cells in an immunologically silent manner. In adult humans, one million cells undergo apoptosis per second as part of the regular turnover process (6, 7). The apoptotic cellular debris is as efficiently removed as it is generated, to avoid inappropriately evoking the adaptive immune responses. If apoptotic cells are not engulfed, they release their cellular contents as secondary necrosis and this results in aberrant immune responses by exposure to self-antigens and a break in tolerance (8–13). Efferocytosis reprograms macrophages towards an anti-inflammatory phenotype, suppressing the production of inflammatory cytokines like, IL-12, TNF-α and IL-1 and upregulating the levels of immunosuppressive cytokines like, TGF-β and IL-10 (14, 15). Both TGF-β and IL-10 are potent in dampening effector helper T cell response by stimulating regulatory T cells and T helper 2 cells (16). The role of efferocytosis in cancer has remained elusive. However, there are now several examples to indicate that the non-immunogenic properties of efferocytosis that prevail under normal homeostasis conditions can be mimicked by malignant cells to create an environment of immunosuppression in the TME. In the tumor microenvironment, TAMs show increased efferocytosis of apoptotic tumor cells, which also facilitates immune tolerance by rendering the former more protumorigenic and increase the risk for metastasis (17–19). A protumoral role of efferocytosis was also observed where tumor progression was dampened following genetic deletion of the efferocytosis receptor MerTK (20). MerTK−/− CD11b+ cells isolated from the tumors showed signs of immune activation, characterized by elevated expression of IL-12 and reduced expression of the immunosuppressive cytokines like IL-10. In a separate study, dendritic cells with increasing efferocytic activity because of their exposure to liposomes containing the well-known ‘eat me’ signal Phosphatidylserine (PS) failed to upregulate several co-stimulatory molecules and had diminished capacity to produce IL-12 and activate T cell responses (21). Efferocytosis of apoptotic tumor cells by TAMs also facilitates prostate cancer metastases (22). However, the molecules that couple the two pathways, efferocytosis and immune modulation are still not well known.
Neuropilins (NRPs) are multifunctional cell surface, non-tyrosine kinase receptors that are expressed in all vertebrates and highly conserved across species. NRPs have been associated with cellular processes such as development, axonal guidance, angiogenesis, immunity, bone homeostasis as well as pathological conditions like cancer. The two major isoforms of NRPs, NRP1 and NRP2, are transmembrane glycoproteins comprising of an N-terminal extracellular domain followed by a transmembrane region and a short cytosolic tail of 43–44 amino acids. There is some degree of amino acid identities among the domains between NRP1 and NRP2, suggesting their overlapping cellular functions. However, functions unique to each isoform have also been reported. Work over the past few years have documented a role for NRPs in the immune responses. NRP1 is important for the formation of immune synapse between dendritic cells and T lymphocytes. It is also considered to be a marker for murine regulatory T cells (Treg) where its expression correlates with immunosuppression. NRP1 is also reported to be expressed in tumor infiltrating macrophages and microglia (23–26). Recently, Miyauchi et al. reported a tumor-promoting function of NRP1 in glioma infiltrating microglia and macrophages. Their study revealed that either genetic ablation or pharmacological manipulation of NRP1 expression in microglia or bone marrow-derived macrophages (BMDM) arrested glioma progression and increased antitumorigenic polarization in the microglia and macrophages (25, 26). NRP2 on the other hand is much less characterized in the immune cell compartments. It is constitutively expressed in human thymic developing DP (CD4+CD8+) T cells. NRP2 is also detected in dendritic cells and microglia where it is post-translationally modified by polysialylation (27).
In the present study, we sought to determine the role of NRP2 in macrophages and its implication in tumor progression. We detected expression of NRP2 in macrophages present in pancreatic cancer (PDAC) tissues. Our results indicate a novel function of NRP2 in promoting efferocytosis of apoptotic cells by macrophages and that in its absence, the clearance of the apoptotic cell corpse is delayed. We also found that NRP2 deletion in macrophages resulted in increased infiltration of cytotoxic CD8+ T lymphocytes and NK cells into the tumor and thus slowed pancreatic tumor growth. This could be attributable to delayed clearance of dying tumor cells by NRP2-deleted macrophages, which resulted in secondary necrosis leading to an anti-tumor immune response. Further, NRP2 deletion in TAMs has a direct effect on their ability to express several immunosuppressive and checkpoint inhibitor genes, like, IL-10, TGF-β, IL-4, MMPs and PDL2 as well as immunostimulatory genes like IL-12 and thereby provides an additional mechanism of anti-tumor immune response. Together, we believe, our observations will impact the therapeutic approaches for targeting TAMs in the treatment of cancer.
Materials and Methods:
Antibodies used
NRP2 (CST 3366 for mouse, R&D AF2215 for human), CD8 (CST 98941), CD68 (ebioscience 14–0681–82), F4/80 (ebioscience 14–4801–82), CD31 (ab28364), Rab5 (ab13253), Rab7 (ab50533), Rho GDI (Santa Cruz Biotechnology sc373724), β-actin (Cell Signaling Technology, 4970), Hsc 70 (Santa Cruz Biotechlogy, sc 7298), α,β tubulin (Cell signaling technology 2148), CD69 (Biolegend 104502, clone H1.2F3), NK1.1 (abcam, 25026), CD 163 594 PE-dazzle (Biolegend 333623, clone GHI/61).
Animals
Animals were housed at the University of Nebraska Medical Center facility. All animal experiments were performed according to the animal care guidelines, as approved and enforced by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center. The NRP2flox/flox mouse was developed by and a kind gift from Dr. Peter Mombaerts, Max Planck Research Unit for Neurogenetics (28). These mice were later bred to pure C57BL/6 background. The FVB- Tg(Csf1r-Mer-iCre-Mer)1Jwp/J mice (developed by Dr. Jeffrey W Pollard, Albert Einstein College of Medicine) were purchased from Jackson Laboratories. These transgenic mice express a Cre recombinase/mutant murine estrogen receptor double-fusion protein under the control of the mouse Csf1rpromoter. Tamoxifen-inducible cre activity was detected in bone-marrow-derived as well as yolk sac macrophages. CSF1R-iCre mice were bred with NRP2f/f mice to obtain CSF1R-iCre;NRP2f/f where NRP2 can be conditionally deleted from the myeloid lineage following administration of Tamoxifen intraperitoneally or adding (Z)-4-HudroxyTamoxifen into the cell culture medium. Genotyping was performed following Jackson Laboratories standard protocol.
Cell culture and generation of human PBMC and murine bone marrow-derived macrophages
Monocytes isolated from the peripheral blood of unidentified healthy human donors were obtained from the elutriation core facility at the University of Nebraska Medical Center. Cells obtained were >97% pure. PBMCs were then cultured at 37ºC with 5% CO2 in RPMI 1640 containing 10% fetal bovine serum, 2mM glutamine, 100 μg/mL streptomycin and 100U/mL penicillin for the indicated time points, with either 100ng/mL GM-CSF or M-CSF. For generation of mouse macrophages, bone marrow from either C57BL/6 or CSF1R-iCre;NRP2f/f mice was flushed from the femurs and tibia with ice cold PBS using a 25-gauge needle and 30cc needle. RBC was removed using Erythrocyte Lysis Buffer. Cells were resuspended in α-MEM media containing 10% fetal bovine serum, 2mM glutamine, 100 μg/mL streptomycin and 100U/mL penicillin and differentiated using either 50 ng/mL murine recombinant GM-CSF or 100ng/mL M-CSF for indicated time points. For some experiments, to knock out NRP2, 4-HydroxyTamoxifen (1mM stock in methanol) was added into the culture media at a concentration of 0.3 μM every alternate day for 3–5days.
Panc-1 cells were purchased from ATCC. UN-KC-6141 (derived from spontaneous pancreatic tumors arising in mice harboring a mutation in K-RasG12D in the pancreas) cells were generated by Dr. Surinder K. Batra, University of Nebraska Medical Center and were a kind gift to us from his laboratory. All these cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) media containing 10% fetal bovine serum, 2mM glutamine, 100 μg/mL streptomycin and 100U/mL penicillin at 37ºC with 5% CO2. For generation of conditioned media, cells were plated in 75mm2 tissue culture flasks and allowed to grow till 75% confluency. The complete media was then removed, and cells washed twice with PBS without Calcium or Magnesium and replaced with only DMEM for 48hours. The conditioned media was then collected, centrifuged at 100rpm for 5minutes, strained through 0.22μm filter and kept at -80ºC until use.
Jurkat cells were purchased from ATCC and cultured in RPMI-1640 medium containing 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4500 mg/L glucose, and 1500 mg/L sodium bicarbonate, 10% FBS, 100 μg/mL streptomycin and 100U/mL penicillin at 37ºC and 5% CO2.
Where mentioned, PBMC and BMDM were treated with conditioned media (50% v/v) collected from pancreatic cancer cell line Panc-1 (ATCC) and UN-KC-6141 cell lines respectively for the indicated periods of time.
Procurement of PDAC patient tissue and mouse pancreatic cancer tissue
Tissues from patients with Pancreatic Cancer but no previous history of treatment were procured from the Rapid Autopsy Pancreatic Program (RAPP) at University of Nebraska Medical Center.
Nucleofection of human monocytes or macrophages
siRNA transfection in PBMCs was performed using Human Monocyte Nucleofection kit (Lonza, VPA-1007) following manufacturer’s protocol. 25nM siNRP2 or Scrambled antisense RNA was used. Where mentioned, 40nM siNRP1 RNA was used. Cells were analyzed or subjected to any assay 48–72 hours after siRNA transfection.
Immunoblot analysis
Briefly, cells were washed with ice cold PBS followed by lysis with CHAPS lysis buffer (150mM KCl, 50mM HEPES (pH 7.4), 0.1% CHAPS, 2mM EDTA, 20μg/mL Leupeptin, 10μg/mL Aprotinin, 5mM DTT, 1mM PMSF and Halt protease and phosphatase inhibitor cocktail) on ice. Cells were gently scraped using cell scraper and sonicated on ice. Supernatant was separated by cold centrifugation (13000 rpm for 5 mins) and total protein was estimated using Bradford method (Biorad, Hercules, CA). SDS sample buffer was added and the sampled were heated at 95ºC for 8mins. The whole cell extracts were next run on 4–20% Mini-PROTEAN® TGX-Gel (BioRad) and transferred to polyvinylidene difluoride (PVDF) membrane (Life Technologies, Carlsbad, CA). The membrane was then blocked in either 5% non-fat dry milk or 5% Bovine Albumin Serum (BSA) in 1X TBST (1X Tris Buffered Saline, 0.1% Tween-20) and then incubated overnight in 1X PBS containing appropriate dilution of primary antibodies overnight at 4ºC with continuous shaking at low speed. The next day, membrane was washed with 1X TBST and then incubated for 1hr in 1X TBST containing appropriate dilution of horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Dallas, TX) with continuous shaking at low speed. The membranes were washed in 1X TBST and the protein bands were detected using SuperSignal™ West Femto Maximum Sensitivity Substrate. For successive immunoblots, membranes were stripped in Stripping Buffer (Thermo Fisher Scientific) for 20mins, re-blocked in 1X TBST containing either 5% BSA or 5% non-fat dry milk and probed as mentioned before.
Phagocytosis and pulse and chase in vitro clearance assay:
Phagocytosis to demonstrate phagosome maturation defect: The pHrodo™ Red E. coli BioParticles™ Conjugate or pH insensitive E.coli bioparticles (red or green) suspension was prepared following manufacturer’s protocol. An optimized dose of 65μg/mL bioparticles were used for phagocytosis.
Human PBMC-derived macrophages or murine BMDM were grown in two welled chambers as described. Human PBMC derived-macrophages were subjected to nucleofection on day5 to knock down NRP2 and the assay was performed on day7. Before the assay, cells were washed twice with 1X DPBS. The fluorescent E.coli particles were then added at a concentration of 65μg/mL and incubated at 37ºC for the indicated periods of time to allow adequate particle internalization. Where mentioned, pHrodo red and pH insensitive E.coli bioparticles were added simultaneously. Under conditions where we simultaneously added pHrodo red and pH insensitive green E.coli bioparticles, an optimized dose of 33μg/mL of either type was used. Phagocytosis was arrested by placing the cells on ice and washing vigorously 3–5 times with 1X DPBS to remove the excess particles. Cells were then fixed with 4% paraformaldehyde at 4ºC for 20 minutes and washed with DPBS to remove the excess fixative. Nuclei were stained using the nuclear dye Hoechst in PBS and analyzed by either Zeiss LSM 800 with Airyscan or Zeiss 710 Confocal Laser Scanning Microscope at UNMC confocal core facility, and data were analyzed and processed with the Zeiss Zen 2010 software. All confocal data were quantified using ImageJ software and graphical illustrations made using GraphPad Prism software as mentioned later.
For in vitro clearance assay, BMDM were grown in two well chambers as described earlier. On day of the experiment, cells were washed twice with 1X DPBS and added 500 μL of Opti-MEM media per well. pHrodo red E.coli or zymosan bioparticles were added at a concentration of 65 μg/mL for indicated time (pulse) to allow their internalization. Uptake was stopped by adding ice cold DPBS. Cells were washed vigorously with DPBS (x3) to remove any E.coli or zymosan particle that was not taken up or loosely adhered to cell surface. Fresh complete α-MEM media was added and the cells were monitored at different time points (chase). At each time point, chambers were taken out, cells vigorously washed with DPBS (x5) and fixed with 4% PFA for 20mins at 4ºC, nuclei stained with Hoechst and analyzed using confocal microscopy as described earlier.
Efferocytosis assay
Apoptosis was induced in Jurkat or UNKC-6141 cells using 50 and 75 μM Etoposide (Abcam) respectively and kept overnight at 37ºC and 5% CO2 for 12hrs. This treatment routinely yielded 80–90% apoptotic cells. Cells were centrifuged at 2000 rpm for 10mins and the pellet was washed twice with PBS. The cells were incubated with pHrodo™ Red succinimidyl ester (Thermo Scientific) following manufacturer’s instructions. Cells were centrifuged at 2000rpm for 10mins. The cell pellet was washed twice with 1X DPBS and resuspended in Opti-MEM medium (Gibco). Mouse BMDM were plated in two well chambers and grown in the presence of M-CSF or CM collected from UNKC-6141 cells. (Z)-4-HydroxyTamoxifen was added to knock out NRP2 as described earlier. Apoptotic Jurkat or UNKC-6141 cells were added to the macrophages (10:1 target to effector ratio) for 1hr (pulse) at 37ºC. Uptake was stopped by adding ice cold DPBS; cells were vigorously washed with ice cold DPBS to remove any cell that was not phagocytosed or remained loosely bound to the cell surface. Fresh complete α-MEM media was added to the cells and the clearance of apoptotic cells were monitored for the indicated time (chase). Nuclei were counterstained using Hoechst for 5mins and analysed using confocal microscopy as mentioned earlier.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry and immunofluorescence staining on histological sections were performed using the following procedure: slides containing tissue sections (4μm thick) were kept on heat block at 58ºC for 2hours. They were then rehydrated in a sequential passage of solutions starting with Xylene for 20mins, 100% ethanol for 15mins, 95%, 90%, 80%, 75%, 50% and 20% ethanol for 5mins each followed by immersion in double distilled water for 10mins. For IHC only, slides were next immersed in 3% Hydrogen Peroxide (H2O2) in methanol for 1hr at room temperature followed by incubation in double distilled water for 5mins. Antigen retrieval was performed using Dako antigen retrieval solution (either pH9 or pH6, depending on the antigen of interest). The antigen unmasking solution was first preheated at a high temperature in the microwave until boiling and then the slides were immersed into it and boiled on a 98ºC water bath for 45mins. Following this, the slides were gradually allowed to cool to room temperature and washed with 1X PBS. Slides were then blocked with 5% goat serum in 1XPBS containing 0.2% saponin at 4ºC for 1hr and incubated overnight with primary antibody in PBS containing 0.2% saponin and 3% BSA at 4ºC. Biotinylated antibodies (IHC) or 1:500 for fluorophore conjugated antibodies (IF) was added. For IHC staining, slides were next washed with 1X TBS and then incubated with Avidin-Biotin complex for 40mins at room temperature, following manufacturer’s instructions and added diaminobenzidine solution containing 0.3% H2O2 as a substrate for peroxidase (Dako) until the desired staining intensity was developed. Hematoxylin was used to counter staining. Slides were dehydrated by gradual passage of slides from double distilled water to xylene in a reverse order mentioned earlier for rehydration of slides and mounted with Permount and covered with glass cover slips. The whole slides were next digitally scanned at Tissue Science Facility, UNMC. For IF, following incubation with secondary antibody cells were washed and mounted with Vectashield mounting media containing DAPI (Vector Laboratories, Burlingame, CA).
Snap frozen tissues were fixed in 10% neutral buffered formalin for 20mins followed by permeabilization in 2% TritonX-100 in dPBS for 30mins. Slides were blocked in 5% goat serum in 0.2% TritonX-100 containing dPBS for 1hr and then incubated with primary antibody for 1hr. Slides were washed in 1X TBS and secondary antibody added. DAPI was used to stain the nuclei.
For monolayer culture, immunofluorescence staining was performed as described: briefly, cells were grown on poly-DL-lysine–coated coverslips (BD Biosciences) for indicated periods of time before fixation and analysis by confocal microscopy. Cells were rinsed with Dulbecco’s Phosphate-Buffered Saline (DPBS; Invitrogen), followed by fixation with ice cold 4% paraformaldehyde at 4ºC for 20 minutes. Cells were then washed with DPBS and blocked using 1% BSA and 0.2% saponin in PBS for 1hr at 4ºC in a moist chamber. The slides were then incubated overnight in the same blocking buffer containing appropriate concentration of primary antibodies at 4ºC. The next day, the coverslips were carefully washed in 1X PBS and fluorescent conjugated antibodies added (1:200) in 1X PBS containing 1% BSA and 0.2% saponin at 4ºC. Slides were washed and mounted with Vectashield mounting media containing DAPI (Vector Laboratories, Burlingame, CA).
Slides were viewed using either Zeiss LSM 800 with Airyscan or Zeiss 710 Confocal Laser Scanning Microscope at UNMC confocal core facility, and data were analyzed and processed with the Zeiss Zen 2010 software. All confocal data were quantified using ImageJ software and graphical illustrations made using GraphPad Prism software.
Flow Cytometric Analysis
Human peripheral blood-derived monocytes and murine bone marrow-derived cells (BMDC) were treated with either GM-CSF, M-CSF or CM from pancreatic cancer cell lines for 7days as described earlier. Cells were then harvested, washed and resuspended in FACS buffer (ice cold PBS containing 10% BSA and 1% sodium azide) at a concentration of 3×106 cells/mL. Cells were incubated with CD163-PE 594 dazzle antibody and kept for 1hr at 4ºC in the dark. Following this, cells were washed 3× at 400g for 5minutes and resuspended in FACS buffer at a concentration of 3×106 cells/mL and immediately analyzed. LIVE/DEAD™ Fixable Blue Dead Cell Stain (ThermoFisher Scientific) was added to quantify the viability of the cells.
Subcutaneous tumor implantation
500,000 or 2×106 UN-KC-6141 cells were mixed with equal volume of Matrigel (without growth factors) and implanted subcutaneously into the right flank of animals. For our studies, mice were divided into two groups, control and test (n=3 or 5). To deplete NRP2 from the macrophages, the test group was injected with Tamoxifen intraperitoneally (75mg/kg body weight from a 20mg/mL stock in corn oil) everyday till the end point of the experiment. The control group received corn oil as vehicle control. The tumor growth was manually monitored regularly using digital slide calipers and weight of the mice recorded. Once the end point of the experiment was reached, mice were euthanized by CO2 asphyxiation following IACUC protocol and tumors harvested. Harvested tumors were washed gently in ice cold DPBS, transferred to new tubes containing DPBS and kept on ice. Tumors were fixed in 10% formaldehyde overnight and transferred to 70% ethanol and given to Tissue Science Facility at UNMC for paraffin embedding, sectioning (4 μm) and Hematoxylin and Eosin (H&E) staining.
Isolation of CD11b+ cells from subcutaneous tumors
Subcutaneous mouse tumors were generated as mentioned earlier. The harvested tumors kept in ice cold RPMI media on ice, cut into small pieces. The digestion media was added (RPMI media containing the following: 10U/mL Collagenase I, 400U/mL Collagenase IV, 30U/mL DNAse I- all diluted in HBSS) and kept at 37ºC for 30mins. Following this, the tumor pieces were crushed with the plunger of a 10mL syringe, 5mL RPMI media was added and homogenized well. The tumor suspension was filtered by passing through a 70μm nylon gauze, the suspension centrifuged at 450g for 6min at 4ºC; the pellet was resuspended in 2mL erythrocyte lysis buffer to remove the RBCs. The tubes were allowed to stand for 2mins at room temperature, neutralized by the addition of 12mL of RPMI media, passed through a sterile 70μm nylon gauze and centrifuged again at 450g for 6mins at 4ºC. The pellet was resuspended in Lymphoprep solution at a concentration of 1–2X107 cells/mL and transferred to fresh tubes. To this was added 6mL RPMI media very cautiously to obtain a two-phase gradient. The gradients were centrifuged at 800 x g for 30 min at room temperature without acceleration or break. The interphase (enriched in myeloid cells and lymphocytes as well as the upper layer containing the RPMI media were collected into fresh tubes without disturbing the lymphoprep layer. The cells were washed once with MACS buffer, centrifuged at 800g for 5mins at 4ºC and the supernatant discarded. The cell pellet was finally resuspended in MACS buffer at a concentration of 108 cell/mL. TAMs were isolated using CD11b magnetic beads and LS columns (both from Miltenyi Biotech) following manufacturer’s instructions. The CD11b+ myeloid cells were arrested in the columns and collected for isolation of RNA and RNA-Sequencing analysis.
Visualization of Monosodium Urate crystals using Polarizing Microscope
Frozen tumor tissue sections (4μm) were fixed with neutral buffered formalin (10%) for 20mins, washed in PBS and mounted with Consulmount mounting media. MSU crystals were visualized using Velocity software and Polarizing filters at 5× magnification. The intensity of MSU crystals per field were quantified using Image J software and represented as a scatter plot.
Transcriptome analysis using RNA Sequencing and Bioinformatics analysis
CD11b+ cells were isolated from subcutaneous tumors. RNA was isolated from the control and test group with n=3 animals contributing to each pool. This was done in order to obtain sufficient amount of RNA. RNA-Seq service was obtained from Kelvin Chan and his team at Seqmatic. Extracted RNA was QC with Agilent TapeStation RNA screenTape. The RIN value of the control sample was 9.1 and that of the test was 9.4. A paired end read 2x75bp sequencing run of RNA libraries were performed using the Illumina NextSeq 500 instrument. For analysis, raw reads were demultiplexed by barcode and output into FASTQ format. Cutadapt was used to filter out adapter sequences and low-quality bases. Filtered sequence reads were aligned to mouse reference genome grch38 using HISAT2 aligner. Reads mapping to exon regions as defined by Ensembl gene annotations were counted using FeatureCounts. Data analysis was performed with the help of Seqmatic, California and the Bioinformatics Core at UNMC. Genes with a cutoff value log fold change (+/− 1) were selected and those with zero counts in either control or test samples were eliminated for stringency. The Ingenuity Pathway Knowledge Base (IPA) was used to identify the enriched cellular and molecular functions among the differentially expressed transcripts in the two samples. The Database for Annotation, Visualization and Integrated Discovery (DAVID), The Gene Ontology Project, Kegg pathway were used and extensive literature reviewed to annotate differentially regulated transcripts with a GO designation. Among the enriched functional annotation clusters, we selected representative clusters (as mentioned in the Results section) to analyze the effect of NRP2 deletion on phagocytosis, macrophage phenotype and secretion of chemokines and cytokines as well as interaction between macrophages and leucocytes/lymphocytes.
Statistical Analyses:
All the graphical illustrations statistical tests were performed using Prism-6 software (GraphPad software, Inc., La Jolla, CA). All data reported in graphs are expressed as mean ± standard error of mean (SEM) unless otherwise mentioned and were compared using unpaired student T-test, p values were considered statistically significant when less than 0.05. All experiments were repeated at least 3 times unless specified. *P < .05; **P < .005; ***P < .0005. ns=not significant.
Results
NRP2 is induced during differentiation of monocytes to macrophages and is expressed by TAMs
To evaluate the expression pattern of NRP2 in macrophages in response to differentiation stimuli, we obtained freshly isolated peripheral blood-derived human monocytes (PBMCs) from the Elutriation Core Facility. Immunoblot analysis revealed freshly isolated monocytes do not express NRP2 protein at a detectable level, however, its expression was induced within 24–72hrs following stimulation with M-CSF (Fig. 1A). NRP2 expression was maintained in the similar level when M-CSF induced macrophages were further treated with IL-4, IL-10 and IL-13 to promote differentiation to different alternative subtypes (Supplementary Fig. 1A). We corroborated our observations in human macrophages using BMDMs isolated from C57BL/6 mice. Immunoblot analysis revealed a similar expression pattern for NRP2 as in human monocytes/macrophages (Fig. 1B). Flow Cytometric analysis of human macrophages revealed 93.33% of M-CSF induced macrophages expressed CD163, a marker for alternatively activated macrophages, whereas 2.73% of GM-CSF treated macrophages were positive for CD163 (Supplementary Fig. 2A). Similarly, confocal microscopy showed a majority of CD163+ human and mouse macrophages expressed NRP2 following M-CSF treatment (Supplementary Fig. 2B). Further, M-CSF treatment induced the expression of immunosuppressive genes like IL-10 and MMP-9 but not inflammatory genes like IL-12b (Supplementary Fig. 2C). NRP2 was similarly upregulated in both human and mouse macrophages following its differentiation with GM-CSF (Supplementary Fig. 1B and 1C). TAMs are abundantly present in solid tumor microenvironment and are causally associated with various aspects of tumor progression, immune evasion, metastasis and therapy resistance (5). The infiltration of TAMs correlates with disease stage and worse prognosis in a wide variety of malignancies, including Pancreatic Cancer (PDAC) (4, 29, 30). TAMs have been shown to resemble more of M-CSF induced alternatively activated macrophages. To evaluate whether TAMs in PDAC express NRP2 in treatment naïve conditions, we obtained tissue sections from a cohort of 10 treatment naïve pancreatic cancer patients from the Rapid Autopsy Program at University of Nebraska Medical Center. Details about the disease stage and grade of the tissues are provided in Fig. 1. Our data revealed that 6 samples had high number of NRP2+ TAMs (on average 85% macrophages expressed NRP2) and 3 showed moderate to low number (on average 30%) of NRP2+ TAMs. One sample showed very scarce infiltration of CD68+ cells (not shown in graph) (Fig. 1C and 1D). However, currently we do not know how the function and immunophenotype of NRP2− TAMs differ from that of NRP2+ TAMs in PDAC. It is also difficult to conclude from our small tissue cohort if NRP2 expression in TAMs correlate with stage and grade of the disease, metastatic potential or overall survival of the patients. These aspects of NRP2 expression in TAMs merit further investigation using a bigger tissue cohort. Next, we differentiated human PBMCs with conditioned media (CM) from a human PDAC cell line, Panc-1. Immunoblot analyses showed a strong induction of NRP2 expression in the macrophages within 24hrs of treatment with CM (Fig. 1E). Similarly, treatment of mouse BMDMs with CM from UNKC-6141 cells induced NRP2 expression in the macrophages (Fig. 1F). Flow cytometric analysis with CM treated human macrophages revealed 94.9% cells expressed CD163, indicating Panc-1 CM drives the macrophages towards an alternatively activated phenotype (Supplementary Fig. 2D). Confocal microscopy revealed that majority of either Panc-1 or UNKC-6141 CM treated macrophages that expressed CD163 were also positive for NRP2 expression (Supplementary Fig. 2E and 2F). Also, Panc-1 and UNKC-6141 CM induced the expression of immunosuppressive and wound healing genes but not immunostimulatory genes in human macrophages and BMDM (Supplementary Fig. 2C and 2G), indicating an alternative type activation. Overall, these data indicate NRP2 is not detected in monocytes or bone marrow precursors, however is expressed in macrophages under GM-CSF, M-CSF as well as cancer cell-derived CM induced conditions.
Figure 1. Expression of NRP2 in macrophages.

(A, B) NRP2 expression in freshly isolated human monocytes and bone marrow cells from C57BL/6 mice differentiated to macrophages with M-CSF. (C) Representative confocal image showing the presence of NRP2 expressing CD68+ TAMs in human PDAC tissue. The first panel shows only NRP2 positive cells (red). The second panel shows CD68+ macrophages (green). The third panel represents merged image showing NRP2+CD68+ macrophage in the tumor. Scale bar, 20μm. Inset shows magnified image of part of the tissue. Dapi was used to stain the nucleus. (D) Graphical representation showing relative abundance of NRP2+ macrophages in a cohort of treatment naïve PDAC tissues derived from RAPP. Table shows details of patient tissues procured. (E, F) Immunoblot analyses showing NRP2 expression in human and mouse macrophages differentiated with pancreatic cancer cell line-derived Conditioned medium (CM) from Panc-1 or UNKC-6141 cells respectively in vitro.
NRP2 regulates phagosome maturation and degradation in macrophages without significantly affecting the uptake of phagocytic cargo
Phagocytosis is one of the key functions of macrophages and important for maintenance of physiological homeostasis as well as microbial clearance during infections (31). We wanted to test whether NRP2 regulates the phagocytic activity of macrophages. For this, we performed a pulse and chase phagocytosis assay using pH-sensitive red fluorophore-tagged E.coli bioparticles (pHrodo red). The E.coli bioparticles are non-fluorescent at neutral pH, however their fluorescence intensity increases with an increase in acidity of the vesicle they are localized in. Following engulfment, phagocytic cargo remains in nascent early phagosomes of higher pH, which sequentially mature to late phagosomes, becoming more and more acidic and finally fuse with the lysosomes for degradation and clearance. Therefore, a bright red fluorescence dot in the cell indicates a mature phagocytic vesicle containing the E.coli, which has or is going to be fused with lysosomes and thus can indicate an active phagocytic process. We have developed a transgenic mouse model by breeding NRP2f/f with CSF-1R-iCRE mice. In the resultant CSF1R-iCre;NRP2f/f mice, NRP2 can be selectively and conditionally deleted genetically from the monocytes/macrophages following administration of Tamoxifen or addition of (Z)-4-Hydroxytamoxifen into cell culture medium. For the phagocytosis assay, M-CSF treated BMDM from CSF1R-iCre;NRP2f/f mice were challenged with pHrodo red E.coli bioparticles for 15mins (pulse) and then excess E.coli bioparticles that were either not phagocytosed or remained loosely bound to macrophages were washed off. Next, the phagosome maturation, degradation and clearance of the already phagocytosed bioparticles from the cells were monitored at 15mins, 45mins, 1.5hrs, 2hrs and 4hrs (chase) using confocal microscopy. Images revealed that in the control macrophages, starting from 0 min up to 2hrs chase, there was a gradual increase in the intensity and number of red fluorescence indicating gradual maturation and fusion of the E.coli containing phagosomes with lysosomes. Interestingly, at 4hrs chase, there was once again a decrease in the intensity of the visible red puncta, suggesting degradation of the phagocytosed E.coli bioparticles (Fig. 2A). This means, the engulfed E.coli bioparticles containing phagosomes matured into late phagosomes or phagolysosomes and then the bioparticles underwent degradation in the control macrophages. In contrast, in the NRP2-deleted macrophages, we observed that till 90min (chase) there was no significant visible red fluorescence indicating a delayed maturation of the E.coli bioparticles. However, from 120mins chase time period, we did observe some phagosomes maturation in the NRP2-deleted cells, however the red intensity of these did not reach the maximal level as seen in WT macrophages. This indicated that the phagosome maturation in NRP2-deleted macrophages was significantly delayed compared to the WT macrophages. At 4hrs chase, higher value of total cellular fluorescence in the NRP2-deleted cells in comparison to the control macrophages at the same time point indicated that phagosomes have now started maturing in the former, whereas the bioparticles are almost degraded at this time in the control cells (Fig. 2B). This indicated that in control macrophages, phagosomes containing E.coli bioparticles matured and fused with lysosomes and degraded the phagocytic cargo, whereas in absence of NRP2, there was a defect in maturation of phagosomes and degradation of E.coli bioparticles. Overall, this data suggested that NRP2 regulates phagosome maturation and therefore degradation and clearance of engulfed cargo.
Figure 2. NRP2 regulates phagosome maturation without affecting uptake of phagocytic cargo.

(A) BMDM from NRP2fl/flCSF1R-iCre mice were assessed for phagosome maturation and degradation of internalized E.coli bioparticles at time = 0min, 15min, 45min, 90min, 120min and 240min. Scale bars, 10 μm. Magnified images within the inset show the phagosomes containing E.coli particles. (B) Phagosome maturation and degradation of bioparticles was assessed as corrected total cell fluorescence using Image J software, at times indicated at the graph abscissa and represented in bar graphs as mean ± SEM. (C,F,I,L) Western Blots showing knock out or deletion of NRP2 for each experiment. (C) shows NRP1 protein level following NRP2 knock out in macrophages. (D) M-CSF treated BMDM from NRP2fl/flCSF1R-iCre mice were analyzed for phagosome maturation and degradation of internalized zymosan particlesat time= 0hr, 6hr, 10hr, 14hr and 18hr. Scale bars, 10μm. Magnified images within the inset show the phagosomes containing zymosan particles. (E) Phagosome maturation and degradation of zymosan particles was scored as corrected total cell fluorescence using Image J software at times indicated and represented in bar graph as mean ± SEM. (G) Phagocytosis assay for assessing the ability of human macrophages to uptake E.coli bioparticles (green) and phagosome maturation (red) following NRP2 knockdown. The first column shows phagosome maturation (red) in the scr (upper) and siNRP2 treated (bottom) cells. The second column indicates the uptake efficiency (green) in the scr (upper) and siNRP2 treated (bottom) cells. The third column represents merged images showing the role of NRP2 on uptake and phagosome maturation. Scale bars, 20μm. Single cell magnified within the boxed region shows green or red E.coli particle. (H) Uptake efficiency was measured as green cellular fluorescence whereas the intensity of red fluorescence indicated phagosome maturation. Results were represented graphically, values as mean ± SEM. Dapi was used for staining the nucleus. (J) Phagocytosis assay to show the effect of NRP1 depletion on cargo uptake (green) as well as phagosome maturation (red) in human macrophages. Insets show magnified image of cell containing bacteria. Scale bars 10μm. (K) Phagosome maturation and uptake efficiency in the presence and absence of NRP1 were quantified as in (H), and represented graphically as mean ± SEM. (L) Western Blot showing NRP1 knockdown.
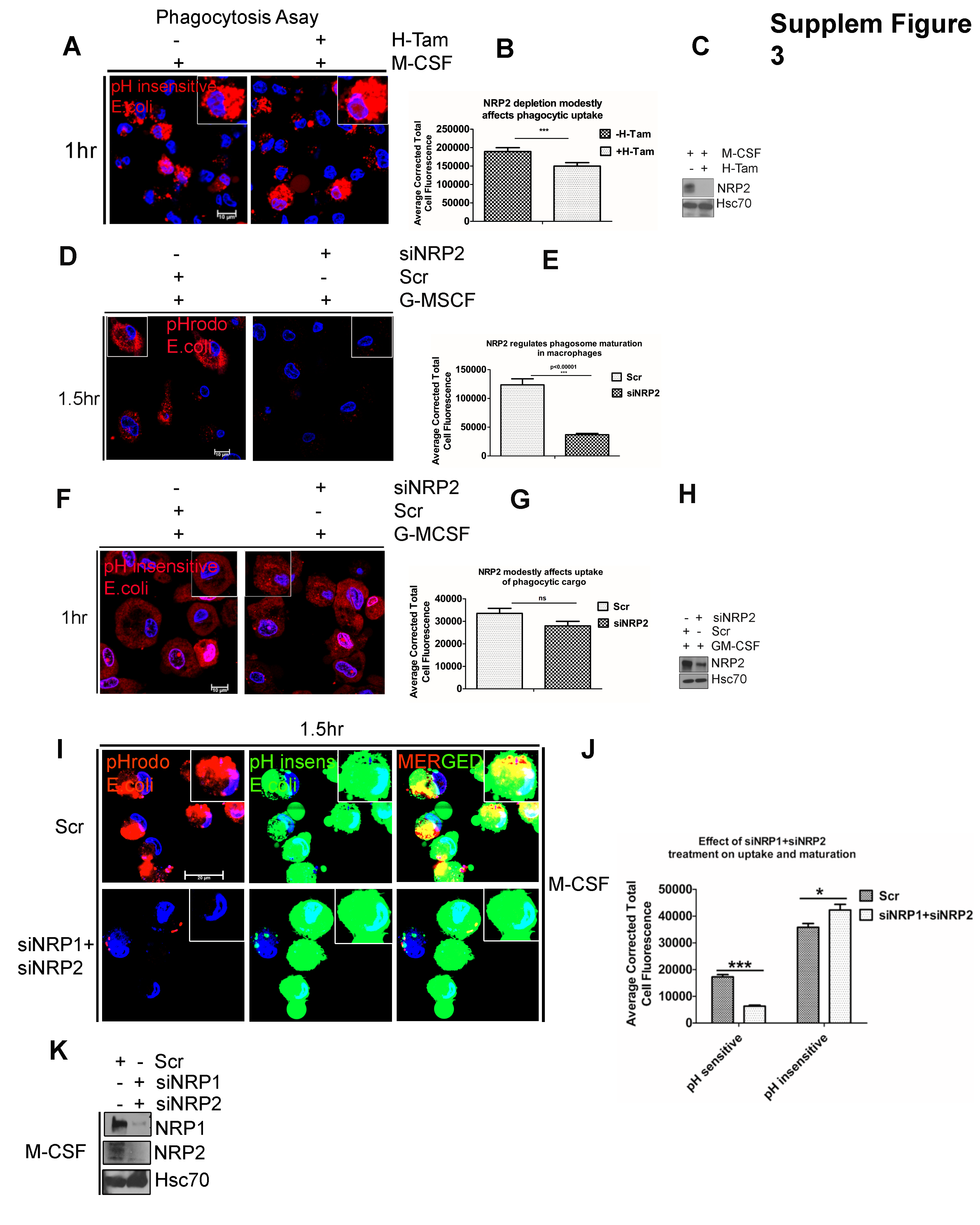
Following uptake of E.coli bioparticles, TLR4 pathway is activated. Therefore, we wanted to determine whether NRP2 selectively regulates TLR4 mediated phagosome maturation and degradation or whether it can regulate cellular mediators common to other types of phagosome maturation and degradation. To test this, we repeated the pulse and chase experiment using pHrodo zymosan particles (a yeast component that activates the TLR2 pathway) that are non-fluorescent at neutral pH but show bright red fluorescence in the acidic late phagosomes or phagolysosomes. Depending on the cargo, phagosome maturation and eventual clearance may follow different kinetics. Based on our preliminary observations, the degradation of internalized zymosan particle containing phagosomes was monitored at 6hrs, 10hrs, 14hrs and 18hrs (chase). After 15mins pulse (0hr time point of chase), both the control and NRP2-deleted macrophages showed similar number of zymosan containing phagosomes. At 6hrs, there was an increased red fluorescence in both the control and NRP2-deleted cells. This possibly indicated maturation of zymosan containing phagosomes in control and NRP2 knocked out cells and that in case of zymosan, NRP2 is not involved in the regulation of early stages of maturation. However, at 10hrs, 14hrs and 18hrs, the control macrophages gradually degraded and cleared the zymosan particles, as evident from the decreased intensity of visible red puncti structures of the zymosan particles. Interestingly, NRP2 knock out macrophages showed a significantly delayed degradation and clearance of zymosan particles. This was evident from the bright red fluorescence and bigger size of the zymosan particles that persisted within the cells, even at 18hrs, when the control cells had efficiently degraded individual particles (Fig. 2D and 2E). To further confirm whether NRP2 is involved in the uptake process, we repeated the phagocytosis uptake assay with BMDM using E.coli bioparticles tagged with a pH insensitive red fluorophore that shows uniform fluorescence intensity irrespective of the pH of the phagosome they are localized in. Therefore, the visible red puncta as observed using this dye indicate the uptake efficiency of the macrophages. We observed a modest difference in uptake efficiency in NRP2-deleted cells (Supplementary Fig. 3A). The slight decrease in the uptake efficiency in absence of NRP2 can arise as a secondary effect of delayed downstream maturation and degradation of the internalized cargo which subsequently dampened the uptake.
To confirm if NRP2 plays a similar role in phagocytosis in human macrophages, M-CSF treated macrophages were challenged with pHrodo red and pH insensitive green E.coli bioparticles simultaneously for 1.5hrs. This experimental approach enabled us to simultaneously test the ability of the macrophages to mature the phagosomes (red) as well as their uptake efficiency (green) in the presence and absence of NRP2. Quantification of the average total cell fluorescence revealed a significant decrease in the intensity of red puncta, indicating a delayed phagosomal maturation in siNRP2 treated cells. However, the uptake efficiency in mock and siNRP2 treated macrophages were similar, as evident from the intensity of the green puncta (Fig. 2G and 2H). This data therefore confirmed that similar to mouse macrophages, NRP2 regulates phagosome maturation in human macrophages, without significantly affecting the uptake process. Since, during infection or injury, GM-CSF induced inflammatory macrophages are predominantly found in tissues and phagocytose pathogens, we tested the role of NRP2 in phagocytosis in GM-CSF treated human macrophages. Our results indicated a similar maturation defect with no significant effect on the uptake efficiency in absence of NRP2 (Supplementary Fig. 3D and 3F). These data suggest that NRP2 regulates phagocytosis in macrophages through modulation of phagosome maturation.
NRP2 and NRP1 are both expressed in macrophages. Because of their structural similarity, both isoforms may have redundant as well as unique functions. Interestingly, the observed defect in phagosome maturation was more specific to the absence of NRP2 as we did not observe any significant difference in the phagosome maturation or uptake efficiency when NRP1 was knocked down in macrophages (Fig. 2J and 2K). Immunoblot analysis also did not reveal any change in NRP1 protein level following NRP2 deletion in BMDM, indicating further that our observation is possibly a NRP2 specific function (Fig. 2C). Additionally, we simultaneously knocked down NRP1 and NRP2 in human macrophages and repeated phagocytosis assay with pHrodo and pHinsensitive E.coli biopartciles. However, under our experimental conditions, we failed to observe any additive effect on cargo uptake or phagosome maturation in the absence of both NRP1 and NRP2 (Supplementary Fig. 3I and 3J). Nevertheless, with our experimental model, it is difficult to rule out the possibility that NRP1 may possibly regulate phagocytosis albeit with lesser efficiency. It is also possible that both NRP2 and NRP1 regulate non-overlapping although essential functions for efficient phagocytic activity in macrophages and the residual NRP1 present following siNRP1 treatment is sufficient to maintain the part of the function it regulates. This possibility will be tested in genetically engineered mouse system in our future studies.
NRP2 regulates early to late phagosome maturation
Nascent phagosome maturation occurs in a step-wise manner through sequential recruitment of Rab5 to the early phagosomes followed by Rab7 to the late phagosomes. To confirm our previous observations that NRP2 is involved in the regulation of phagosome maturation, we next assessed Rab5+ early phagosomes and Rab7+ late phagosomes in NRP2 proficient and deficient cells. In NRP2 knocked down human macrophages, there was an increased accumulation of Rab5+ early phagosomes and a concomitant decrease in the Rab7+ late phagosomes (Fig. 3A). Similar observations were made in mouse BMDM following NRP2 knock out (Fig. 3D). Interestingly, total cellular protein level of Rab5 and Rab7 remained unchanged (Fig. 3C and 3F). Together, these data suggest that NRP2 regulates phagosome maturation in macrophages. Deletion or knock down of NRP2 arrests phagosomes in the early stages and impairs their maturation to late phagosomes or phagolysosomes. Further, since we observed an increase in Rab5+ vesicles in NRP2 depleted macrophages, it indicates NRP2 acts downstream of Rab5.
Figure 3. Effect of NRP2 on early and late phagosomes.

Knockdown or deletion of NRP2 inhibits maturation of early to late phagosomes in macrophages. (A) Immunostaining of early and late phagosomal maturation markers in human macrophages following NRP2 knockdown. The upper panel represents early phagosome marker Rab5 (green). Scale bars 10μm. The lower panel shows representative the late phagosomal marker Rab7 (green). Scale bars 20μm. Magnified images of individual cell are shown in the inset for each condition. (B) Immunostaining data for Rab5 and Rab7 were quantified as cellular fluorescence using Image J software and represented graphically. The upper and lower panels show graphical representation of Rab5 and Rab7 respectively, in the presence and absence of NRP2. (D) Immunostaining of early and late phagosomal maturation markers in mouse BMDM following NRP2 deletion. The upper and lower panels represent early phagosome marker Rab5 (red) and the late phagosomal marker Rab7 (red) respectively. Scale bars 10μm. The insets are magnified image of individual cell for each condition. (E) Representative bar graphs showing quantification of Rab5 and Rab7 using Image J software. The upper and lower panels represent changes in Rab5 and Rab7 respectively, following NRP2 deletion. All values are shown as mean ± SEM. Dapi was used for staining the nucleus. (C,F) Western Blot showing total cellular Rab5 and Rab7 in whole cell lysates from human and mouse macrophages following knockdown or knockout of NRP2 respectively.
NRP2 regulates efferocytosis of apoptotic cells by macrophages
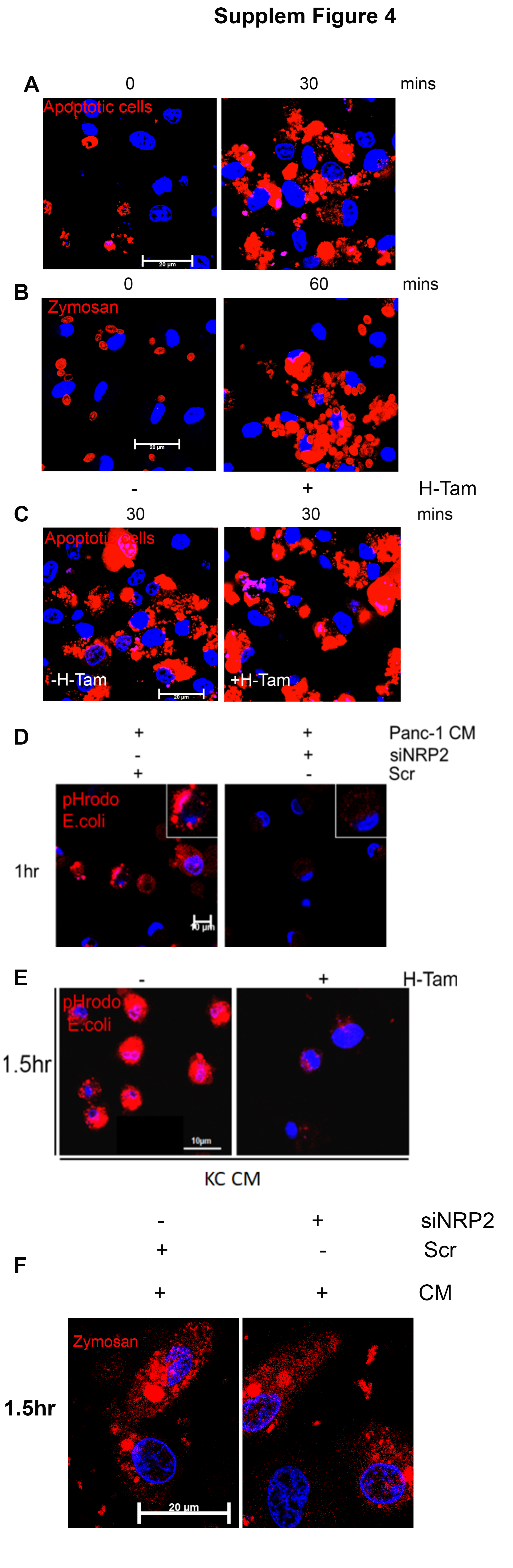
Macrophages efficiently phagocytose and clear apoptotic cells in tissues. This is indispensable for the maintenance of immunological homeostasis. Impaired efferocytosis results in aberrant immune activation (32). Maturation of nascent efferosome (phagosome containing apoptotic debris) and subsequent degradation share similarity with classical phagocytosis. Both the pathways often hire similar family of effector molecules (6, 7, 33, 34). Our previous data suggested that NRP2 regulates phagosome maturation and the degradation of phagocytosed cargo in macrophages. Using pHrodo E.coli bioparticles, we also observed a maturation defect in NRP2-deleted macrophages treated with cancer cell CM (Supplementary Fig. 4D and 4E). Based on these findings, we wanted to investigate the role of NRP2 in the clearance of dying cells by macrophages. To test this, BMDM were challenged with apoptotic Jurkat cells and then monitored for their ability to degrade the internalized apoptotic cargo for up to 8hrs. Degradation was assessed from the loss of fluorescence and disappearance of the apoptotic cells (red). At 2hrs chase, there was significant increase in the red fluorescence in both the control as well as NRP2 KO cells, indicating mature efferosomes. However, at 6hrs, there was a significant decrease in the red fluorescent intensity and disappearance of apoptotic cells in the control macrophages, indicating their degradation. In contrast, even after 8hrs, NRP2 KO cells exhibited significant delay in the clearance of apoptotic cell cargo (Fig. 4A and 4B). This was apparent from larger size of the cargo and higher amount of fluorescence that persisted in the cells even after 8hrs. Similar to our previous observation, NRP2 deletion did not result in the change in uptake efficiency of apoptotic cargo. No notable difference was observed when we quantified the average number of cells engulfed by macrophages at 0hr chase (immediately after the incubation or uptake phase) following NRP2 deletion (Fig. 4 B).
Figure 4. NRP2 regulates efferocytosis of apoptotic cells by macrophages in vitro.

Efferocytosis assay to assess the effect of NRP2 deletion on the ability of macrophages to degrade the apoptotic cells. (A,D,G) BMDM from NRP2fl/flCSF1R-iCre mice were treated with either M-CSF (A) or UNKC-6141 CM (D,G) and then assessed for degradation of internalized apoptotic Jurkat cells (A,D) or UKC-6141 cells (G) at time points= 0hr, 2hr, 6hr, and 8 or 12hr. Scale bars, 10μm (A,D), 20μm (G). Insets show single macrophage containing apoptotic cells. (B,E,H) Corrected total cellular fluorescence (red) was analyzed at the time points indicated using Image J software as a measure for efferosome maturation and degradation of the apoptotic cargo. Results are represented graphically as mean ± SEM. (C,F,I) Immunoblot to show knock out of NRP2 for A,D,G. Dapi was used for staining the nucleus.
Recent studies have highlighted the importance of efferocytosis in tumor progression and metastasis (17, 20, 22). Infiltrating TAMs efficiently phagocytose and remove the dying cancer cells from the tumor milieu. This induces the expression of tumor promoting immunosuppressive and wound healing genes in TAMs and suppresses the antitumor immune responses, while actively supporting tumor growth. Based on our previous data that NRP2 deletion affects the clearance of apoptotic cells, we further wanted to investigate if NRP2 regulates the clearance of apoptotic debris by TAMs. Our preliminary observations revealed NRP2 was expressed by F4/80+ TAMs in subcutaneous mouse pancreatic cancer tissues (Supplementary Fig. 5A). To test the role of NRP2 in apoptotic cell clearance, UNKC-6141 CM-treated BMDM from NRP2f/f;CSF-1R-iCre mice were subjected to efferocytosis pulse and chase assay At the beginning of chase, there was no significant difference in the intensity of red fluorescence in the control and NRP2 KO cells. At 2hr chase, the intensity increased in the control and NRP2 KO macrophages, indicating the efferosomes are maturing and becoming acidic. However, at 6 and 12 hrs of chase, there was a significant decrease in the intensity of red fluorescence as well as size of the internalized apoptotic debris in the control macrophages, suggesting efficient degradation and clearance of apoptotic cells. In contrast, the higher red fluorescent intensity in the NRP2 KO macrophages at the above-mentioned time points indicated a delayed clearance of apoptotic cargo in the absence of NRP2 (Fig. 4D and 4E). Similar defect in the degradation of phagocytosed apoptotic UNKC-6141 cells was observed in NRP2 KO macrophages treated with CM derived from the same cell line (Fig. 4G and 4H). Overall, these results suggested that NRP2 regulates efferocytosis of apoptotic cells in macrophages, during physiological homeostasis (M-CSF) as well as cancer cell CM induced conditions.
NRP2 in macrophages affects tumor growth and antitumor adaptive immune response
Previous studies have demonstrated the tumor promoting role of efferocytosis (17). That clearance of dying cells is an immunologically silent process is mimicked by cancer cells to promote disease progression and metastasis (17). To examine the role of macrophage NRP2 mediated efferocytosis on tumor growth, we used a subcutaneous pancreatic cancer mouse model where we implanted either 2×106 or 500,000 UNKC-6141 cells into the right flank of NRP2f/f;CSF1R-iCre mice. Once the tumors became palpable, animals were randomly divided into control and test groups (n=3 or 5). Tamoxifen was administered intraperitoneally to selectively knock out NRP2 from the macrophages. Tumors were regularly measured, and tumor growth monitored over a period of 21, 25 or 15 days (Fig. 5A). Under all conditions, deletion of NRP2 in macrophages reduced the tumor size (Fig. 5C, 5E and 5G) as well as the relative tumor volume (Fig. 5B). Importantly, Tamoxifen administration into tumor bearing Cre− mice did not have any effect on tumor growth, although we observed a trend towards higher tumor volume in Cre− animals that received Tamoxifen compared to Cre+ mice that did not receive Tamoxifen (Fig. 5B). This suggests that the reduction in tumor growth we observed is because of deletion of NRP2 from macrophages. Although not statistically significant, we also observed a reduction in the weight of the tumors following NRP2 deletion in the macrophages (Supplementary Fig. 6A–6C). Recent studies have shown that BMDMs are actively recruited to feed the pool of TAMs in tumors. To confirm that NRP2 was efficiently knocked out from TAMs, we isolated bone marrow from control and test animals and differentiated them to macrophages with M-CSF. Immunoblot analysis with lysates from BMDM as well as well as RNA-Seq data from CD11b+ myeloid cells isolated from tumors (mentioned later) indicated tamoxifen efficiently delete NRP2 not only from BMDM but also from intratumoral macrophages (Fig. 5D, 5F and 5H), suggesting the effect on tumor growth arises due to NRP2 deletion in macrophages. Further confocal staining indicated an efficient deletion of NRP2 from the intratumoral F4/80+ macrophages whereas NRP2 was still detected in surrounding tissue (Supplementary Fig. 7A). It has been reported that efferocytosis of apoptotic tumor cells has a protumorigenic effect on TAMs and blockade of this process may hinder tumor growth and metastasis (17). To assess if the decrease in tumor growth was due to inefficient efferocytosis by NRP2-deleted TAMs, TUNEL staining was performed. An increase in the number of necrotic foci in test tumors following NRP2 deletion from macrophages suggested apoptotic tumor cells were not efficiently removed and underwent secondary necrosis. (Fig. 6A and 6B). Secondary necrotic cells release uric acid (derived from degradation of nuclear DNA) into the extracellular space, which then in the presence of high sodium concentration forms monosodium urate (MSU) crystals and can be visualized using a polarizing microscope. As hypothesized, we detected significantly higher amount of MSU crystal deposition in test tumors, indicating impaired efferocytosis and increased secondary necrosis in the tissues (Fig. 6C and 6D). NRP2 is well characterized for its role in migration. To test if the inefficient efferocytosis was a consequence of decreased recruitment of macrophages to the site of the tumor, we stained histological sections of tumors with anti F4/80. Our data revealed no significant change in the average number of macrophages per field in control and test tumors, indicating NRP2 is dispensable for TAM recruitment (Figure 6E, 6F). Therefore, we concluded that the increased accumulation of late apoptotic or necrotic cells in the tumor tissue was a consequence of dampened efferocytosis ability of NRP2 KO TAMs. Further, staining with anti CD31 antibody revealed no significant difference in average vessel density between the control and test tumors, suggesting intratumoral angiogenesis was unaffected following NRP2 deletion in TAMs (Supplementary Fig. 7A and 7B).
Figure 5. NRP2 in macrophages affects tumor growth.

(A) Schematic diagram for subcutaneous tumor progression model. 2×106 or 500,000 UNKC-6141 cells were subcutaneously implanted into NRP2fl/flCSF1R-iCre mice. Tumor progression was monitored for the indicated time periods. (B) Graph showing relative tumor volume for control and test tumors (2×106 cells implanted). (C) Scatter plot representation of the final volume of the harvested tumors (2×106 cells implanted). (D,F) Immunoblot analysis showing efficient knock out of NRP2 from macrophages for experiment B and E. (E, G) Graphical representation of final volume of harvested tumors (n=3 or 5, 500,000 cells implanted). (H) RT-PCR showing efficient knock out of NRP2 from CD11b+ myeloid cells isolated from test tumors for G.
Figure 6. NRP2 in TAMs regulates the efferocytosis of apoptotic tumor cells and immune responses.

(A) Representative image showing TUNEL+ cells in control and test tumors. (B) The number of necrotic foci relative to tumor volume shown graphically. (C) Representative images showing deposition of MSU crystals in control and test tumors. Scale bar, 500 μm. (D) Scatter plot comparing the formation of MSU crystals in control versus test tumors. (E) Representative image showing role of NRP2 in the migration of macrophages (F4/80+, red) to the tumor. Scale bar, 20μm. Inset shows magnified image of a single F4/80+ macrophage. (F) the number of F4/80+ cells per field were counted using Image J software and represented graphically. (G) Representative immunohistochemistry image showing CD8+ T cell infiltration in control and test tumors. Image Scale, 20× magnification. (H) Number of CD8+ T cells per field was counted using Image J software, and represented graphically. (I) Representative confocal microscopy image for CD69+ T cell infiltration (green) in the control and test tumors. Scale bar 20μm. (J) Number of CD69+ T cells per field was counted using Image J software and represented as a scatter plot. (K) Representative confocal microscopy image for NK1.1+ cells (green) in control and test tumors. Scale bar 20μm. (L) Number of NK1.1+ cells per field was counted using Image J software, and represented graphically. Dapi was used to stain the nucleus. All values are mean± SEM.
Inefficient efferocytosis leads to secondary necrosis, which can activate adaptive immune response. Indeed, we observed a ~3-fold increase in intratumoral infiltration of cytotoxic CD8+ T cells in the test tumors following NRP2 deletion. This data suggests NRP2 in macrophages suppresses anti-tumor immune response and that its deletion in macrophages results in enhanced recruitment of CD8+ T cells into the tumor (Fig. 6G and 6H). Staining with early activation marker CD69 indicated an active CD8 T cell response following NRP2 deletion (Fig. 6I and 6J). Interestingly CD69 is also expressed by other immune cells, like NK cells. Staining with NK cell marker NK1.1 revealed a significant increase in NK cell infiltration in test tumors following NRP2 deletion in macrophages (Fig. 6K). Overall, these data suggest a protumorigenic role of NRP2 in TAMs in suppression of antitumor adaptive immune response through efferocytosis of apoptotic tumor cells.
Transcriptome analysis from CD11b+ myeloid cells by next generation RNA-Seq
Extensive efferocytic activity of macrophages present in tumor promotes M2-polarization. We therefore speculated that inhibition of efferocytic activity can directly affect the polarization of TAMs towards anti-tumorigenic macrophages. We isolated CD11b+ myeloid cells from the control and test tumors and determined their polarization by analyzing gene expression using Next-Gen RNA-Sequencing. Fig. 7A shows the schematic diagram of the experiment. We considered transcripts which were differentially expressed more than 2-fold (log2 fold change 1) in either control or test sample and eliminated all transcripts with zero counts in either sample for stringency. After applying the cutoff, of the 3616 differentially expressed genes, 1567 genes were up-regulated whereas 2049 genes were downregulated following NRP2 deletion. These differentially expressed transcripts were uploaded to the IPA database to identify the major enriched cellular and molecular functions in the absence of NRP2 in macrophages. We observed that pathways related to immune responses such as leucocyte extravasation signaling, role of cytokines in mediating communication between immune cells as well as phagosome formation were significantly affected. Some of the representative IPA pathways with gene enrichment and statistical significance is shown in Fig. 7B.
Figure 7. Transcriptome analysis from CD11b+ myeloid cells by next generation RNA-Seq.

(A) Schematic diagram showing experimental design. (B) Representative canonical pathways from Ingenuity Pathway Analysis are shown with gene counts and -log(p-value). (C) Representative functional annotation clusters from DAVID database are shown. (D) Representative list of genes related to macrophage polarization whose expressions were significantly altered in the CD11b+ myeloid population, following NRP2 deletion in macrophages in test tumors. (E) RT-PCR analysis in separate biological replicates (pooled, n=3 in either control or test) showing altered expression of genes in CD11b+ cells following NRP2 deletion in macrophages in test tumors.
For the current study, we were mainly interested in understanding the intrinsic immune responsive gene signature changes occurring in the myeloid compartment as a consequence of impaired efferocytosis in absence of NRP2 in macrophages. To gain a better understanding for the functional processes affected by NRP2 deletion, we determined the biological process gene ontological classification for each altered transcript using the DAVID and KEGG databases in combination with extensive review of published literature. As with IPA analysis, using DAVID, we observed that genes related to immune response and leucocyte/lymphocyte regulation were abundantly regulated. Also, clusters comprising of genes functionally annotated to cytokine and chemokine signaling were enriched (Fig. 7C). Genes from IPA, KEGG and DAVID clusters as well as from extensive review of curated literature for T and NK cell related immune responses, cytokine/chemokine signaling pathways and phagocytosis and phagosome maturation were compiled and a unique gene list was created for each function and compared with our dataset of 3616 differentially regulated genes.
Our analysis from IPA software, DAVID and KEGG pathways (Fig. 7D) revealed many of the immunosuppressive genes and those associated with EMT and metastasis, like, IL-4, IL-10 (3), IL-21R (35), IL-33 (36), IL-34 (37), IL-1β (38, 39) were downregulated in the CD11b+ myeloid cells following NRP2 deletion. MMPs are associated with cancer progression (40–42). Genes associated with ECM remodeling like MMP9, MMP13, MMP11, MMP23, MMP25 were also downregulated following NRP2 deletion. Among the inflammatory genes, we observed an upregulation of IFN-β1, IL-12a, Gr-K, Gr-F. Further, by RT-PCR we validated the altered expression of IL-10, IL-4 MMP9, MRC2, checkpoint inhibitor like PDL2 and immunostimulatory gene such as IL12a in separate biological replicates (RNA pooled from n=3 in either control or test groups) (Fig. 7E). Macrophages also secrete TGF-β to suppress immune responses. It also acts as a strong inducer of tumor-promoting TAMs. Interestingly, RT-PCR revealed a significant downregulation of TGF-β in NRP2-deleted myeloid cells (Fig. 7E). These observations are in support of our hypothesis and indicate that NRP2 can act as a molecular mediator that can couple efferocytosis and immunosuppression in macrophages.
Overall, we believe that the components of the secondary necrotic cells along with altered expression of several cytokines, chemokines and other signaling molecules in the myeloid compartment following NRP2 deletion in monocytes/macrophages act synergistically and result in a robust infiltration of CD8+ T and NK cells in to the tumors and impede tumor growth (Graphical Abstract).
Discussion:
Limited information is currently available about the role of NRP2 in macrophages. Our results indicated that NRP2 is expressed during the differentiation of classical and alternatively activated type macrophages. Interestingly, tumor secreted factors are also capable of inducing NRP2 expression in macrophages. These observations have raised the question what are the potential function(s) of NRP2, which is not only required in normal physiology but also may play a role in pathological conditions such as cancer. One particularly important question in this context is, whether the expression of NRP2 in macrophages present in a tumor is a host response due to the presence of tumor and thus anti-tumorigenic or its expression in macrophages facilitates tumor growth. The finding would be significant especially in the context of pancreatic cancer, where we detected NRP2+ macrophages in human pancreatic cancer tissues. Studies have indicated potent anti-tumor effect for drugs that can modulate TAMs in pancreatic cancer to classical inflammatory type (4, 43). We were therefore interested in understanding whether the presence of NRP2+ macrophages in pancreatic tumor microenvironment has a tumor promoting or tumor inhibiting function.
Our results presented in this study have answered some of these important questions. We have identified a novel function of NRP2 in macrophages, which is its ability to regulate phagocytosis. We observed a significantly delayed phagosome maturation and degradation of phagocytic cargo in macrophages following NRP2 deletion. Earlier literature suggested that neuropilins influence cellular locomotion. Interestingly, recent report also indicated the presence of a specific polysialylated form of NRP2 in dendritic cells, which is required for their movement to lymph nodes (44). Although NRP2 can potentially promote the migration, studying its other important functions will be crucial for the comprehensive understanding of its role in macrophages and how it can be targeted for the development of novel therapies against aggressive malignancy. In this respect, our finding that NRP2 regulates the phagocytic activity of macrophages is significant. We speculate that the regulation of phagocytic activity is dominantly regulated by NRP2. Although as discussed in the result section, it is difficult to rule out the possibility that NRP1 may also regulate phagocytosis in macrophages albeit with lesser efficiency. Interestingly, there are some reports where the authors have shown Tuftsin promoted phagocytosis in microglia in NRP1 dependent manner (45). This needs to be tested in future studies using a mouse model where NRP1 can be genetically deleted.
To further understand how NRP2 regulates the phagocytic activity of macrophages, we tested whether NRP2 knockdown or deletion leads to a defect in the uptake of phagocytic cargo or delays the maturation of phagosomes. Our experiments indicated that NRP2 knockdown or deletion did not result in any significant decrease in the uptake of phagocytic cargo and suggested a defect in the maturation processes. A conclusive proof for the involvement of NRP2 in regulating the maturation process of phagosomes came when we observed an increase in Rab5+ early vesicles with a concomitant decrease in Rab7+ late vesicles following NRP2 knockdown or deletion, suggesting a defect during the exchange of Rab5 to Rab7 in phagosomes. Generation of Rab7+ phagosomes is crucial for phagosome maturation and is a prerequisite step for phagosomes during their fusion with lysosomes to form phagolysosomes (33, 34, 46–49) (47–49). We therefore concluded that the molecular effectors downstream of Rab5 are regulated by the NRP2 axis to promote fusion between phagosome and lysosome. This is what we have observed for zymosan and apoptotic cells, where deletion of NRP2 resulted a significant delay in cargo degradation, indicating a problem in lysosome fusion during phagosome maturation. We also noticed that for apoptotic cells and zymosan, the pH sensitive dye fluorescence increased within 30 or 60 mins respectively of adding the phagocytic cargo to macrophages (Supplementary Fig. 4A and 4B), which was significantly shorter than what we observed for E.Coli particle (~90 min). This could be either due to faster time for maturation of apoptotic cell and zymosan-containing phagosomes or due to more acidic environment in those phagosomes even when they were in their earlier stages of maturation. Nevertheless, there was no detectable change in maturation kinetics for apoptotic cell (Supplementary Fig. 4C) and zymosan (Supplementary Fig. 4F) containing phagosomes in NRP2-KO versus control macrophages, thus indicating that NRP2 is not involved during their early maturation. However, the dye-intensity for E.Coli containing phagosomes did not reach its peak even after 4 hour of chase suggesting a maturation defect that could happen earlier than Rab5-Rab7 exchange during phagosome maturation. Currently, it is unknown whether NRP2 can regulate any other steps of phagosome maturation when E.Coli particle is phagocytosed. Interestingly, by carefully analyzing the expression pattern of genes detected by IPA (canonical pathway: phagosome formation) and DAVID (annotation clusters 91 and 92) as well as additional genes from our RNA-seq dataset, we have identified some potential molecular mediators of phagosome maturation process. We detected altered expression of several genes belonging to the Rab family of proteins following NRP2 deletion in macrophages, including those associated with phagocytosis or maturation such as Tubb4a, TubA8, Rufy4a. One microRNA, miR-24 was significantly upregulated in NRP2-deleted TAMs. This miRNA has been reported to attenuate phagocytosis as well modulate inflammatory cytokines in macrophages and DCs (50–53). The contribution of these NRP2-regulated genes in the phagosome maturation and degradation of different cargo is currently unknown and merits further investigation. Also, since macropinocytosis and phagocytosis are similar in many aspects especially during their maturation, NRP2 may regulate macropinocytosis as well. The conditions we chose for our experiments predominantly favor phagosome formation, although it did not rule out the possibility that in other conditions NRP2 regulates maturation of macropinosomes and thus control some important biological functions.
One of the most important functions of macrophages is the efferocytic clearance of apoptotic cells in an immunologically silent manner. Efferocytosis generates soluble and cell-bound signals that result in an anti-inflammatory state necessary for immune homeostasis (14). Under conditions of impaired efferocytosis, apoptotic cells undergo secondary necrosis releasing an array of cell-derived factors that can potentially activate immune responses and result in autoimmunity and lupus like conditions. Our in vitro assay with apoptotic cells suggested a role of NRP2 in regulating efferocytosis in macrophages. Deletion of NRP2 significantly delayed the degradation of apoptotic cells in the macrophages. Emerging evidences now suggest a potential protumoral role of efferocytosis in tumor progression and metastases (17). Efferocytosis by TAMs is potentially tumorigenic as it induces excessive pro-tumorigenic polarization and production of wound healing and immunosuppressive cytokines. This helps suppress antitumor adaptive responses and actively support tumor growth. In this respect, it is important to note that radiation or chemotherapy induced apoptosis in tumor cells triggers defective repair macrophages and thus activates a vicious feed-forward loop for cancer progression. We therefore hypothesized that deletion of NRP2 in macrophages in a tumor microenvironment can hinder their ability to phagocytose dying tumor cells, which should lead to an increase in secondary necrosis and thus a reduction in tumor growth. Indeed, deleting NRP2 from macrophages in mice bearing subcutaneous pancreatic tumors significantly impaired their efferocytic activity and increased secondary necrosis in the tumors. This re-initiated an anti-tumor immunogenic response characterized by a robust infiltration of active CD8+ T and NK cells into the tumors following NRP2 deletion in TAMs and reduced tumor growth. Although, reports have documented a role for NRP1 in inducing a pro-tumorigenic phenotype in macrophages (25, 26), its role in efferocytosis has not been studied. As previously mentioned, future studies with NRP1 KO mice will answer this interesting question.
Efferocytosis promotes an immunosuppressive microenvironment by cellular mechanisms not completely understood. The release of cellular contents from a secondary necrotic cell in the extracellular milieu in the absence of efferocytosis induces anti-tumor immune responses. Indeed, we observed a significant infiltration of CD8+ T and NK cells along with inhibition of growth in test tumors suggesting an anti-tumor immune response. Delayed efferocytosis could also suppress the expression of immunosuppressive genes in macrophages and activate immunogenic responses (12, 14, 54). Indeed, our RNA sequencing analysis in NRP2-deleted myeloid cells indicated additional changes in gene signatures for enhanced CD8+ T and NK cell activation and reduced tumor growth. We identified suppression of several wound healing and immunosuppressive genes that can support tumor promoting TAM phenotype or recruit and activate Tregs or blunt T and NK cell activation, whereas inflammatory genes or those with immune stimulatory potential were upregulated in NRP2-deleted condition. Taken together, we conclude that NRP2 deletion in macrophages affects a wide array of molecules associated with the immune response in the myeloid compartment that act in consort and result in robust CD8+ T and NK cell responses.
The expansion and recruitment of anti-tumor immune cells of adaptive or innate arms to the tumor microenvironment can significantly control tumor growth. This has been exemplified by the recent demonstration that administration of anti-CD40 antibody resulted in the mobilization of tumor-killing peripheral macrophages in pancreatic tumors (55). We speculate that NRP2-expressing TAMs due to their high efferocytic activity are immunosuppressive and therefore actively promote tumor progression. It is plausible that NRP2 driven efferocytosis of apoptotic tumor cells is a major contributing factor for the failure of cytotoxic therapies, that causes widespread apoptotic tumor cell death. Since therapy induced tumor cell apoptosis will increase efferocytosis, we speculate it will further exaggerate the immune suppressive microenvironment and result in more aggressive tumors with higher metastatic potential. Thus, inhibiting the NRP2 axis in TAMs would reduce pancreatic tumor progression and enhance the efficacy of established treatment modalities when used in combination with either chemotherapy or other immunotherapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Significance:
Neuropilin-2 (NRP2) in macrophages promotes tumor growth by regulating efferocytosis of apoptotic tumor cells and orchestrating immune suppression.
Acknowledgments
The authors thank Karen Yip and the team at Seqmatic (via Science Exchange) for the RNA Seq services, Janice A. Taylor and James R. Talaska at the Confocal Laser Scanning Microscope Core Facility, Victoria Smith and Samantha Wall at the Flow Cytometry facility and the animal care facility at the University of Nebraska Medical Center for their assistance and Dr. Paul Grandgenett of the RAP program at UNMC for providing the PDAC tissues.
Funding details: The study was supported by R01-NIH: 1R01CA182435–01A1, The Nebraska Center for Cellular Signaling CoBRE Developmental Grant: P30 GM106397, Pancreatic Tumor Microenvironment Network (TMEN) U54CA163120–03, Pancreas SPORE Developmental Research Program 2013 RFA, UNMC P50 CA127297, German Research Foundation DFG (Grant MU2687/5–1) and Rudolf Becker Foundation for Prostate Cancer Research.
Footnotes
Conflict of interest: No potential conflicts of interest were disclosed.
References
- 1.Okabe Y, Medzhitov R. Wormhole Travel for Macrophages. Cell. 2016;165(3):518–9. 10.1016/j.cell.2016.04.005. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 2.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. PubMed PMID: ; PMCID: PMC2724991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13 10.12703/P6-13. PubMed PMID: ; PMCID: PMC3944738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. 10.1038/nrclinonc.2016.217. PubMed PMID: ; PMCID: PMC5480600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. 10.1016/j.immuni.2014.06.010. PubMed PMID: ; PMCID: PMC4137410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189(7):1059–70. 10.1083/jcb.201004096. PubMed PMID: ; PMCID: PMC2894449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207(9):1807–17. 10.1084/jem.20101157. PubMed PMID: ; PMCID: PMC2931173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franz S, Gaipl US, Munoz LE, Sheriff A, Beer A, Kalden JR, Herrmann M. Apoptosis and autoimmunity: when apoptotic cells break their silence. Curr Rheumatol Rep. 2006;8(4):245–7. Epub 2006/07/15. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 9.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):191–201. . PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 10.Munoz LE, Janko C, Chaurio RA, Schett G, Gaipl US, Herrmann M. IgG opsonized nuclear remnants from dead cells cause systemic inflammation in SLE. Autoimmunity. 2010;43(3):232–5. 10.3109/08916930903510930. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 11.Munoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, Herrmann M. Autoimmunity and chronic inflammation - two clearance-related steps in the etiopathogenesis of SLE. Autoimmun Rev. 2010;10(1):38–42. 10.1016/j.autrev.2010.08.015. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 12.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6(5):280–9. 10.1038/nrrheum.2010.46. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 13.Munoz LE, Lauber K, Schiller M, Manfredi AA, Schett G, Voll RE, Herrmann M. [The role of incomplete clearance of apoptotic cells in the etiology and pathogenesis of SLE]. Z Rheumatol. 2010;69(2):152, 4–6. 10.1007/s00393-009-0603-7. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 14.Henson PM. Cell Removal: Efferocytosis. Annu Rev Cell Dev Biol. 2017;33:127–44. 10.1146/annurev-cellbio-111315-125315. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 15.Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166(11):6847–54. Epub 2001/05/22. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 16.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9(5):353–63. 10.1038/nri2545. PubMed PMID: ; PMCID: PMC2818721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanford JC, Young C, Hicks D, Owens P, Williams A, Vaught DB, Morrison MM, Lim J, Williams M, Brantley-Sieders DM, Balko JM, Tonetti D, Earp HS 3rd, Cook RS. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J Clin Invest. 2014;124(11):4737–52. 10.1172/JCI76375. PubMed PMID: ; PMCID: PMC4347249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar S, Calianese D, Birge RB. Efferocytosis of dying cells differentially modulate immunological outcomes in tumor microenvironment. Immunol Rev. 2017;280(1):149–64. 10.1111/imr.12587. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–78. 10.1038/cdd.2016.11. PubMed PMID: ; PMCID: PMC4987730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, Redente E, Sandahl M, Hunter DM, Strunk KE, Graham DK, Earp HS 3rd., MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. 2013;123(8):3231–42. 10.1172/JCI67655. PubMed PMID: ; PMCID: PMC3726162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Doffek K, Sugg SL, Shilyansky J. Phosphatidylserine regulates the maturation of human dendritic cells. Journal of immunology. 2004;173(5):2985–94. Epub 2004/08/24. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 22.Roca H, Jones JD, Purica MC, Weidner S, Koh AJ, Kuo R, Wilkinson JE, Wang Y, Daignault-Newton S, Pienta KJ, Morgan TM, Keller ET, Nor JE, Shea LD, McCauley LK. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J Clin Invest. 2018;128(1):248–66. 10.1172/JCI92466. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. 10.1016/j.ccr.2013.11.007. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 24.Dai X, Okon I, Liu Z, Wu Y, Zhu H, Song P, Zou MH. A novel role for myeloid cell-specific neuropilin 1 in mitigating sepsis. FASEB J. 2017;31(7):2881–92. 10.1096/fj.201601238R. PubMed PMID: ; PMCID: PMC5471517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyauchi JT, Caponegro MD, Chen D, Choi MK, Li M, Tsirka SE. Deletion of Neuropilin 1 from Microglia or Bone Marrow-Derived Macrophages Slows Glioma Progression. Cancer research. 2018;78(3):685–94. 10.1158/0008-5472.CAN-17-1435. PubMed PMID: ; PMCID: PMC5887044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyauchi JT, Chen D, Choi M, Nissen JC, Shroyer KR, Djordevic S, Zachary IC, Selwood D, Tsirka SE. Ablation of Neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget. 2016;7(9):9801–14. 10.18632/oncotarget.6877. PubMed PMID: ; PMCID: PMC4891085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy S, Bag AK, Singh RK, Talmadge JE, Batra SK, Datta K. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front Immunol. 2017;8:1228 10.3389/fimmu.2017.01228. PubMed PMID: ; PMCID: PMC5641316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walz A, Rodriguez I, Mombaerts P. Aberrant sensory innervation of the olfactory bulb in neuropilin-2 mutant mice. J Neurosci. 2002;22(10):4025–35. Epub 2002/05/23. doi: 20026374. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013;46(3):131–8. Epub 2013/03/27. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Protti MP, De Monte L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front Physiol. 2013;4:210 10.3389/fphys.2013.00210. PubMed PMID: ; PMCID: PMC3738865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon S Phagocytosis: An Immunobiologic Process. Immunity. 2016;44(3):463–75. 10.1016/j.immuni.2016.02.026. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 32.Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, Linkermann A, Virgin HW, Green DR. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. 10.1038/nature17950. PubMed PMID: ; PMCID: PMC4860026. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9(10):781–95. doi: 10.1038/nrm2515. PubMed PMID: ; PMCID: PMC2908392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5(1):a008748. doi: 10.1101/cshperspect.a008748. PubMed PMID: ; PMCID: PMC3579390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF Jr., Cheever AW, Young DA, Collins M, Grusby MJ, Wynn TA. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. 2006;116(7):2044–55. 10.1172/JCI27727. PubMed PMID: ; PMCID: PMC1479424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, Pena MM. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 2017;56(1):272–87. 10.1002/mc.22491. PubMed PMID: ; PMCID: PMC5630136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segaliny AI, Mohamadi A, Dizier B, Lokajczyk A, Brion R, Lanel R, Amiaud J, Charrier C, Boisson-Vidal C, Heymann D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int J Cancer. 2015;137(1):73–85. 10.1002/ijc.29376. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer. 2012;11:87 10.1186/1476-4598-11-87. PubMed PMID: ; PMCID: PMC3532073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, Wu TJ, Lin BR, Yang MH, Hortobagyi GN, Hung MC. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72(5):1290–300. 10.1158/0008-5472.CAN-11-3123. PubMed PMID: ; PMCID: PMC3350107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16–27. 10.1111/j.1742-4658.2010.07919.x. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 41.Cathcart J, Pulkoski-Gross A, Cao J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015;2(1):26–34. 10.1016/j.gendis.2014.12.002. PubMed PMID: ; PMCID: PMC4474140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shay G, Lynch CC, Fingleton B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015;44–46:200–6. 10.1016/j.matbio.2015.01.019. PubMed PMID: ; PMCID: PMC4564058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thind K, Padrnos LJ, Ramanathan RK, Borad MJ. Immunotherapy in pancreatic cancer treatment: a new frontier. Therap Adv Gastroenterol. 2017;10(1):168–94. 10.1177/1756283X16667909. PubMed PMID: ; PMCID: PMC5330603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rey-Gallardo A, Escribano C, Delgado-Martin C, Rodriguez-Fernandez JL, Gerardy-Schahn R, Rutishauser U, Corbi AL, Vega MA. Polysialylated neuropilin-2 enhances human dendritic cell migration through the basic C-terminal region of CCL21. Glycobiology. 2010;20(9):1139–46. 10.1093/glycob/cwq078. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 45.Nissen JC, Tsirka SE. Tuftsin-driven experimental autoimmune encephalomyelitis recovery requires neuropilin-1. Glia. 2016;64(6):923–36. 10.1002/glia.22972. PubMed PMID: ; PMCID: PMC4833601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitano M, Nakaya M, Nakamura T, Nagata S, Matsuda M. Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature. 2008;453(7192):241–5. doi: 10.1038/nature06857. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 47.Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16(9):907–17. doi: 10.1038/ni.3253. PubMed PMID: ; PMCID: PMC4826466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, Ravichandran KS. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10(5):556–66. doi: 10.1038/ncb1718. PubMed PMID: ; PMCID: PMC2851549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010;464(7289):778–82. 10.1038/nature08853. PubMed PMID: ; PMCID: 2901565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fordham JB, Naqvi AR, Nares S. miR-24 Regulates Macrophage Polarization and Plasticity. J Clin Cell Immunol. 2015;6(5). 10.4172/2155-9899.1000362. PubMed PMID: ; PMCID: PMC4721581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fordham JB, Naqvi AR, Nares S. Regulation of miR-24, miR-30b, and miR-142–3p during macrophage and dendritic cell differentiation potentiates innate immunity. J Leukoc Biol. 2015;98(2):195–207. 10.1189/jlb.1A1014-519RR. PubMed PMID: ; PMCID: PMC4501676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naqvi AR, Fordham JB, Ganesh B, Nares S. miR-24, miR-30b and miR-142–3p interfere with antigen processing and presentation by primary macrophages and dendritic cells. Sci Rep. 2016;6:32925 10.1038/srep32925. PubMed PMID: ; PMCID: PMC5017188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naqvi AR, Fordham JB, Nares S. miR-24, miR-30b, and miR-142–3p regulate phagocytosis in myeloid inflammatory cells. Journal of immunology. 2015;194(4):1916–27. 10.4049/jimmunol.1401893. PubMed PMID: ; PMCID: PMC4323870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. 2001;11(19):R795–805. Epub 2001/10/10. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 55.Vonderheide RH, Bajor DL, Winograd R, Evans RA, Bayne LJ, Beatty GL. CD40 immunotherapy for pancreatic cancer. Cancer Immunol Immunother. 2013;62(5):949–54. 10.1007/s00262-013-1427-5. PubMed PMID: ; PMCID: PMC3731141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.