

Abstract
RNA structural flexibility underlies fundamental biological processes, but there are no methods to explore the multiple conformations adopted by RNAs in vivo. We developed Crosslinking Of Matched RNAs And Deep Sequencing (COMRADES) for in-depth RNA conformation capture, and a pipeline for retrieval of RNA structural ensembles. Using COMRADES, we determined the architecture of the Zika virus RNA genome inside cells, and revealed multiple site-specific interactions with human non-coding RNAs.
RNA conformational flexibility is essential for splicing, translation, and posttranscriptional regulation. Recent methods have utilized proximity ligation to reveal RNA base-pairing within cells1–6, but because of insufficient probing depth and lack of appropriate computational algorithms, it has been difficult to assess the in vivo structural dynamics of RNAs. We developed a method—Crosslinking Of Matched RNAs And Deep Sequencing (COMRADES)—that couples in vivo probing of RNA base-pairing with selective RNA capturing. We additionally established an algorithm for assessing the structural complexity of RNA inside cells. (Fig 1a).
Fig. 1: COMRADES methodology.

a, Outline of COMRADES experimental workflow and the associated computational pipeline. B: biotin. b, % of chimeric reads in COMRADES and control datasets. Mean and s.d. of 3 independent experiments are shown. c, Probed interactions among cytoplasmic and mitochondrial ribosomal RNA subunits. Mean and s.d. of 3 independent experiments are shown. rRNA: cytoplasmic ribosomal RNA; mtrRNA: mitochondrial ribosomal RNA. d, Heat map of ZIKV RNA-RNA interactions. Each dot represents an interaction between the genomic coordinates on the x and y axes. Chimeras ligated in 5'-3' and 3'-5' orientations are plotted above and below the diagonal respectively. e, Zoom-in on a selected 1,500 nt region from (d).
COMRADES utilizes a cell-permeable, azide modified psoralen derivative (Psoralen-triethylene glycol azide) to facilitate coupling of two effective affinity capturing steps, while overcoming the limited cell permeability of biotin labelled psoralen (Supplementary Fig. 1a). The azide group does not affect the psoralen crosslinking properties (Supplementary Fig. 1a). Following in vivo crosslinking, an RNA of interest is selectively captured, allowing nearly 1,000-fold enrichment (Supplementary Fig. 1b-c). The RNA is then fragmented and a copper free click-chemistry reaction links a biotin moiety to in vivo crosslinked regions, enabling a second streptavidin-based affinity selection of crosslinked regions (Supplementary Fig. 1d). Half of resulting RNA is proximity ligated to create RNA chimeras, following reversal of the crosslink to enable high-throughput sequencing and assessment of the base-pairing (Supplementary Fig. 1e). The other half is used as a control, in which reversal of the crosslink precedes the proximity ligation. COMRADES and control samples contain essentially identical RNA composition, ensuring accurate assessment of artificial chimeric reads originated from random ligation or reverse transcription errors. COMRADES’s dual enrichment substantially increases structure probing depth of selected RNA, thus enabling an unbiased and global view of coexisting conformations. COMRADES yields high levels of ligated chimeric reads, whereas these are kept 4 fold lower in the control, as well as in non-crosslinked samples (Fig. 1b). We successfully reported on the known ribosomal RNA structure with high sensitivity (Supplementary Fig. 2), while spurious interactions between cytoplasmic and mitochondrial ribosomal RNA subunits occurred at a very low level (Fig. 1c). The robustness of COMRADES is further demonstrated by its high reproducibility (Fig. 1d-e).
RNA viruses utilize RNA base-pairing to regulate various aspects of their life cycle7–12. Inside the host cell however, the full-length architecture of RNA genomes and their interactions with the host transcriptome are largely unknown. We used COMRADES to determine RNA base-pairing along the 10.8 kilobases-long single-stranded RNA genome of Zika virus (ZIKV) from the Flavivirus genus inside human cells. We identified 1.7 million non-redundant chimeric reads corresponding to the structure of the ZIKV genome (Fig. 1d-e, Supplementary Fig. 3). This high probing coverage is valuable for analysing multiple coexisting conformations. Previous work mainly identified RNA structures in the untranslated regions (UTRs) of flaviviruses, while 95% of the genome remained unexplored7–10. COMRADES identified base-pairing along the entire genome and between the open reading frame (ORF) and the UTRs (Supplementary Fig. 4a). Nearly 80% of the identified interactions spanned a distance of less than 1,000 nucleotides (nt), implying local structure with a certain degree of three-dimensional compaction (Supplementary Fig. 4b). Both short- and long-range interactions were supported by reproducible, well-defined clusters of chimeric reads, ligated in 5'-3' and 3'-5' orientations (Supplementary Data 1), and showed strong evidence of base-pairing when analyzed with the hybrid-min RNA-folding algorithm as compared to a shuffled-chimeras control (Wilcoxon test p-value < 0.0001). COMRADES therefore enables a deep and comprehensive analysis of RNA base-pairing inside cells.
During replication, the genome of flaviviruses undergoes a global conformational change mediated by a long-distance base-pairing between the 5' and 3' cyclization sequences7,13 (5' CS and 3' CS respectively). Additional elements contributing to genome cyclization are the upstream and downstream of AUG regions (UAR and DAR respectively)8,9. COMRADES detected extensive and highly specific base-pairing between the known cyclization elements, therefore demonstrating genome cyclization inside cells (Fig. 2a-c, Supplementary Fig. 5a). COMRADES further refined the nature of the base-pairing associated with genome cyclization, by identifying contact regions upstream of the 5' UAR and downstream of the 3' UAR (Fig. 2a,c). We confirmed the existence of previously defined functional RNA pseudoknots including the dumbbell (DB) pseudoknot14, the downstream of 5' cyclization sequence-pseudoknot (DCS-PK)15, and the SL1 pseudoknot16 (Supplementary Fig. 5b). We additionally detected an alternative 5' UTR conformation where stem-loops A and B (SLA and SLB respectively) are not formed but rather engaged in long-distance base-pairing with the downstream envelope coding sequence (Supplementary Fig. 5c-e). The essential role of SLA during replication suggests that this structure is more likely involved in virus translation or packaging. Overall, COMRADES identified nearly all previously known flavivirus RNA structures and has further defined critical base-pairing involving the UTRs.
Fig. 2: The ZIKV genomic structure inside human cells.

a, Heatmap of RNA-RNA interactions between cyclization elements. Exp: experiment; cont: control. b, Viewpoint histograms showing binding positions of the cyclization sequences along the ZIKV genome. Viewpoint regions are marked by dashed red lines. c, Probed interactions along the circular genome conformation. Colour code indicates the number of non-redundant chimeric reads supporting each base-pair. New: newly identified base-pairing. d, Folding energy (dG) and experimentally supporting evidence (chimera reads) for each of the 1,000 computationally predicted structures corresponding to ZIKV genome nt 2,288 - 3,323. r: Pearson correlation coefficient. e, Clustering and prediction of alternative conformations for the region shown in (d). Colour code as described in (c).
Our intra-viral RNA-RNA interaction map revealed the presence of multiple mutually exclusive RNA structures, where one region alternately base-pairs with several other regions. The averaged Shannon entropy per nucleotide was 5.9 bits, implying high folding plasticity (Supplementary Fig. 6a). We found a strong inverse correlation between the degree of experimental support for base paired regions and their entropy (i.e., strong base-pairing correlates with low entropy, Supplementary Fig. 6b-d, 7). To explore the ensemble of alternative structures, we developed an algorithm to computationally fold ~1,000 nucleotide-long regions using randomly selected subsets of high-confidence mutually compatible folding constraints derived from COMRADES data. For each region, a set of 1,000 structures was generated (Supplementary Data 2-11). The validity of this approach is demonstrated by the clear correlation between the thermodynamic stability and the number of reads supporting each structure (Fig 2d, Supplementary Fig. 8). Nevertheless, the most thermodynamically favoured structures gained only moderate experimental support, implying the additional impact of the cellular environment on RNA folding17–19.
We further computed the degree of similarity between all pairs of structures and applied multidimensional scaling to cluster structures based on their similarity (Supplementary Data 12). The presence of separated well defined clusters reflects the occurrence of alternative conformations (Fig. 2e, Supplementary Fig. 9). As a control, we randomly shuffled the interacting RNA partners between the chimeric reads; the resulting shuffled structures clustered separately from the structures recovered by COMRADES (Supplementary fig. 10a,b). A single ZIKV structure typically accounted for ~30% of the in vivo observed interactions, whereas a reduced set of 5 structures was sufficient to capture 80-90% of the in vivo data (Supplementary Fig. 10c). Our analysis suggests that the intracellular folding complexity of the ZIKV genome might be explained by postulating the coexistence of a small set of alternative conformations.
Viral RNAs have an inherent capacity to form specific interactions through base-pairing with host RNAs20, but little is known about the prevalence of such interactions. COMRADES revealed multiple interactions between the ZIKV genome and human small regulatory RNAs (Fig. 3a). We found site-specific interactions between the ZIKV ORF and the U1 small nuclear RNA (snRNA, Supplementary Fig. 11a), plausibly affecting host splicing. We similarly detected site-specific interactions with certain human tRNAs (Supplementary Fig. 11b). We identified several interactions between the ZIKV genome and human microRNAs such as miR-21, miR-19, miR-512, miR-515, and miR-1323, while no interactions with microRNAs were detected in control datasets. (Fig. 3b, Supplementary Fig. 11c). COMRADES indicated non-canonical base-pairing between the 5' CS of ZIKV and the seed region of miR-21 (Fig. 3c, Benjamini-Hochberg adjusted p-value 1.0E−13). The miR-21 interaction with the ZIKV genome was further proved significant using an independent analysis pipeline (FDR 3.0E−25, quasi-likelihood moderated F-test). In vitro synthesized miR-21 failed to bind the ZIKV 5' CS on its own, while preloading miR-21 onto purified Argonaute 2 (AGO2) facilitated a strong and sequence specific interaction (Supplementary Fig. 12), supporting the involvement of AGO2 in this base-pairing. CRISPR-Cas9 deletion of MIR21 or antisense inhibition of mature miR-21 in human cells decreased the intracellular level of the ZIKV genome (Supplementary Fig. 13a-d). Abrogating the miR-21 binding ability of a ZIKV replicon through point mutations rendered it insensitive to miR-21 antisense inhibition (Supplementary Fig. 13e), indicating that miR-21 acts through direct interaction with the 5' CS. The ZIKV envelope protein was similarly affected in the MIR21 deficient cells (Supplementary Fig. 14). Although the effect size was relatively small, the strong evidence for the miR-21 - 5' CS interaction presented here suggests that miR-21 might assume a greater pro-viral role in the physiological context.
Fig. 3: Host-virus RNA-RNA interactions.
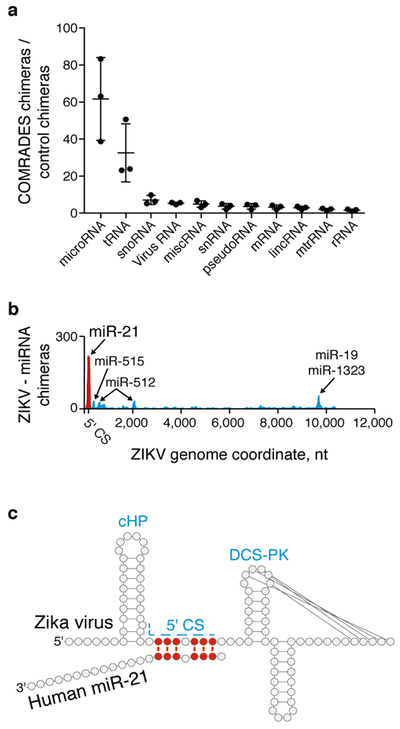
a, Human RNA species interacting with the ZIKV genome. Mean and s.d. of 3 independent experiments are shown. b, Probed interactions between the ZIKV genome and specific miRNAs in COMRADES and control samples. c, COMRADES determined base-pairing between ZIKV and miR-21. d, A model of the ZIKV 5' CS engaged in three separate functions. Ribosome and nascent polypeptide are marked in green.
COMRADES revealed the highly dynamic nature of an RNA genome inside cells, and its ability to engage in base-pairing with multiple host regulatory RNAs. The involvement of the conserved 5' CS of ZIKV in genome cyclization, capsid translation and miR-21 binding further demonstrates the intracellular structural complexity of viral RNA genomes. The general applicability of COMRADES provides an opportunity to undertake an unbiased analysis of the dynamic nature of RNA inside cells and can be utilized to investigate the structure and interacting partners of any cellular or foreign RNA in any species.
Online methods
COMRADES.
Each independent experiment was carried out on a different day, and included 3 sequencing-libraries: COMRADES, control, and a non-crosslinked sample.
Psoralen crosslink.
JEG-3 cells (~50 million cells per experiment) were inoculated with ZIKV isolate PE243, at MOI: 2 TCID50/cell. 20 hours post inoculation cells were washed 3 times in PBS and were incubated for 20 minutes with 0.4 mg/ml Psoralen-triethylene glycol azide (psoralen-TEG azide, Berry & Associates) dissolved in PBS and diluted in OptiMEM I with no phenol-red (Gibco). Cells were irradiated on ice with 365 nM UV for 10 minutes using a CL-1000 crosslinker (UVP). Prolonged UVA irradiation should be avoided as it might decompose the azide moiety. Cells were lysed using RNeasy lysis buffer. Proteins were degraded by proteinase K (NEB) and RNA was purified using RNeasy midi kit (Qiagen).
Viral RNA enrichment.
Total RNA was mixed with an array of 50 biotinylated DNA oligos, 20 nucleotides-long each (IDT) designed to capture the ZIKV genomic RNA and was maintained at 37 °C for 6 hours rotating in the following hybridization buffer: 500 mM NaCl, 0.7% SDS, 33 mM Tris-Cl pH 7, 0.7 mM EDTA, 10% Formamide. Hybridization and wash conditions were adapted from21. At the end of incubation Dynabeads MyOne Streptavidin C1(Invitrogen) were added and the RNA was incubated for additional 1 hour at 37 C. Beads were captured on a magnet and were washed 5 times with 2x SSC buffer containing 0.5% SDS. RNA was released from beads by degrading the DNA-probes with 0.1 units/μl Turbo DNase (Invitrogen) at 37 °C for 30 minutes. RNA was cleaned by RNA Clean & Concentrator (Zymo Research).
Crosslink pulldown.
RNA was fragmented to an average size of 100 nucleotides using RNase III (Ambion) and was cleaned by RNA clean & concentrator (Zymo Research). Copper-free Click reaction was carried at 37 °C for 90 minutes in the presence of 150 μM Click-IT Biotin DIBO Alkyne (Life technologies) and 0.5 units/μl Superase-In (Invitrogen). Reaction was terminated by RNA Clean & Concentrator (Zymo Research). Biotinylated RNA was pulldown using Dynabeads MyOne Streptavidin C1(Invitrogen) at the following reaction conditions: 100 mM Tris-Cl pH 7.5, 10 mM EDTA, 1 M NaCl, 0.1% Tween-20, 0.5 unit/μl Superase-In. Beads were captured on a magnet and were washed 5 times with 100 mM Tris-HCl pH 7.5, 10 mM EDTA, 3.5M NaCl, 0.1% Tween-20. RNA was eluted by adding 95% Formamide, 10 mM EDTA solution and incubating at 65oC for 5 minutes. To avoid enrichment of small RNA chimeric reads that cannot be double-aligned to the reference ZIKV genome / Human transcriptome, RNA was size fractionated on 10% TBE-Urea gel and fragments corresponding to a size of 100-200 nucleotides were eluted overnight at 4 °C in 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 250 mM NaCl, 0.1% SDS. RNA was concentrated using RNA Clean & Concentrator (Zymo Research).
Proximity ligation and crosslink reversal.
At this stage, the RNA sample was divided in two. One half was used for proximity ligation and then crosslink reversal (i.e. COMRADES sample), while in the other half, crosslink reversal was done before proximity ligation (i.e. control sample). We included an additional control containing an equimolar concentration (albeit a non-similar composition) of non-psoralen treated, non-crosslinked enriched RNA (i.e., Non-crosslinked sample). Before proximity ligation, the RNA was heated to 85 °C for 2 minutes and was cooldown rapidly on ice. Proximity ligation was done under the following conditions: 1 unit/μl RNA ligase 1 (New England Biolabs), 1x RNA ligase buffer, 50 mM ATP, 1 unit/μl Superase-in (Invitrogen), final volume: 200 μl. Reaction was incubated for 16 hours at 16 °C and was terminated by cleaning with RNA Clean & Concentrator (Zymo Research). Crosslink reversal was done by irradiating the RNA on ice with 2.5 KJ/m2 UVC.
Sequencing library preparation.
Library preparation was done as described in22 with the following modifications: 6N unique molecular identifiers were added to the 5' end of the 3' sequencing adapter; primers and adapters concentrations were lowered to match the low RNA input; Agencourt RNAClean XP beads (Beckman Coulter) were used for clean-up and size separation; pre-adenylated 5' and 3' adapters were used and all ligation reactions were carried without ATP to reduce ligation artefacts. All libraries and controls went through 13 PCR cycles using KAPA HiFi HotStart Ready Mix (KAPA Biosystems). PCR products were size-selected on a 1.8% agarose gel before loading on a HiSeq 1500 sequencer (Illumina).
Cell culture.
JEG-3 placental trophoblasts (ATCC) and Hela cells (ATCC) were cultured in Minimum Essential Medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, GlutaMAX, non-essential amino acids and penicillin-streptomycin. Vero cells (Sigma-Aldrich) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, GlutaMAX and penicillin-streptomycin. All cell lines were cultured in a humidified CO2 incubator at 37 °C and were regularly examined to exclude mycoplasma contamination.
Virus inoculation.
ZIKV isolate PE243 was originated from Recife, Brazil in 2015. The virus was propagated in Vero cells and titer was determined by measuring the 50% Tissue Culture Infective Dose (TCID50) in JEG-3 cells. For measurements of virus replication, JEG-3 or Hela cells were inoculated with ZIKV at MOI: 0.1 TCID50/cell for three hours, after which cells were washed 3 times with PBS and supplemented with fresh growth medium. 24 hours post inoculation medium was removed, cells were washed 3 times with PBS and RNA was extracted using RNeasy kit (Qiagen). Virus copy number was determined using a TaqMan real-time PCR Assay (Primerdesign) and was normalized to GAPDH and ribosomal RNA. All virus work was handled in a containment level 2 facility registered with the HSE under COSHH.
Replicon assay.
ZIKV wildtype and 5' CS - 3' CS double mutated replicons were described previously23. Replicon RNA was synthesized in vitro using MEGAscript T7 Transcription Kit (Ambion). Replicon RNA was Capped using the ScriptCap m7G Capping System (Cellscript) and transfected to Hela cells using the TransIT-mRNA Transfection Kit (Mirus). Replicon levels were analyzed after 24-48 hours using a microplate luminometer (Promega) and normalized to baseline luminescence values measured at 6 hours post transfection.
MIR21 knockout.
JEG-3 cells were transfected with CAS9-gRNA riboprotein complexes using Lipofectamine RNAiMAX (Life technologies) according to the Alt-R CRISPR-Cas9 user guide (IDT). MIR21 knockout clone1 was generated using the following guide RNA: 5'-TCATGGCAACACCAGTCGATGGG-3' and contains a homozygous deletion at the positions 59841310-59841326 on chromosome 17 (GRCh38/hg38 Assembly). MIR21 knockout clone2 was generated using a mixture of two guide RNAs: 5'-ATGTCAGACAGCCCATCGACTGG-3', 5'-CTACCATCGTGACATCTCCATGG-3', and contain a homozygous deletion at positions 59841249-59841321 on chromosome 17. MIR21 knockout and control clones were validated by sanger sequencing and by TaqMan Advanced miRNA Assay targeting the mature miR-21 (Life Technologies).
miR-21 inhibition.
Hela cells were transfected with inhibitors targeting human miR-21 or non-targeting control A (Power inhibitors, Exiqon) at a final concentration of 25 nM using Lipofectamine RNAiMAX. 6 hours post transfection medium was replaced and cells were inoculated with ZIKV or re-transfected with ZIKV replicons as described above. miR-21 inhibition was validated using a psiCHECK-2 reporter (Promega) carrying a fully complementary miR-21 site at the 3' UTR of a Renilla luciferase reporter along with a Firefly reporter to normalise transfection efficiency. The miR-21 psiCHECK-2 reporter was deposited in addgene (plasmid number 114206). Luminescence was assessed using the Dual-reporter assay (Promega) and normalized to control psiCHECK-2 without the miR-21 binding site.
Gel-based Reverse Transcription Stalling (RTS) assay.
RTS assay was performed as previously described24 using a Cy5-labeled primer targeting the human 5.8S ribosomal RNA: 5'-Cy5-AAGCGACGCTCAGACAGG-3'.
Dot blot analysis.
50 ng crosslinked RNA, or the indicated amount of 50 nt-long biotinylated standards were spotted on to a Biodyne B Nylon Membrane (Life technologies) and dried by baking at 80 °C for 10 minutes. Biotinylated RNA was detected using the chemiluminescent nucleic acid detection module Kit (Life technologies) and visualized using ChemiDoc MP Imaging System (Biorad).
Purification of human AGO2 loaded with miR-21.
Human AGO2 homogeneously loaded with miR-21 was prepared according to a published protocol25. Human AGO2 was expressed in Sf9 cells using the Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific). Sf9 cells were lysed and human AGO2 was purified by Ni-NTA affinity chromatography using a His tag. Human AGO2 was loaded with synthetic 5'-phosphorylated miR-21 (IDT), and the His tag was removed using Tobacco etch virus protease. Human AGO2 loaded with miR-21 was captured using an antisense oligonucleotide (IDT), eluted, and purified by size exclusion chromatography on an ÄKTA FPLC (GE Healthcare Life Science). Protein concentration was measured using absorption at 280 nM with extinction coefficients obtained from the protparam tool (www.expasy.org) and from the ribotask oligocalculator (www.ribotask.com).
Target RNA labelling.
Synthetic RNA oligonucleotides (IDT) were radiolabelled at the 5'-end using gamma 32P ATP (Perkin Elmer) and T4 polynucleotide kinase (NEB), and purified by denaturing polyacrylamide gel and ethanol precipitation. RNA concentration was determined from absorption at 260 nM using extinction coefficients calculated with the ribotask oligocalculator (www.ribotask.com).
Electrophoretic mobility shift assay (EMSA).
Binding reactions were prepared in reaction buffer (28 mM Tris pH 8.0, 20 mM KCl, 80 mM KOAc, 1.6 mM Mg(OAc)2, 0.5 mM TCEP, 0.004% NP-40, 0.01 g/l baker’s yeast tRNA) with a final volume of 20 μl, and a final concentration of the labeled RNAs of 10 nM and of the non-labeled RNA or AGO2-miR-21 of 100 nM. Reactions were incubated for 10 minutes at room temperature and analyzed on a 15% acrylamide native gel in 0.5x TBE.
Kd measurements.
Binding experiments were conducted according to the protocol published in25. AGO2-miR-21 (0-200 nM) was incubated with 0.1 nM radiolabaled target in reaction buffer (28 mM Tris pH 8.0, 20 mM KCl, 80 mM KOAc, 1.6 mM Mg(OAc)2, 0.5 mM TCEP, 0.004% NP-40) with a total volume of 25 μl for 45 minutes at room temperature. Filter-binding was performed using a dot-blot apparatus (GE Healthcare Life Sciences) with Protran nitrocellulose membrane (Amersham, GE Healthcare Life Sciences) and Hybond N+ nylon membrane (Amersham, GE Healthcare Life Sciences). Samples were applied with vacuum and washed with 50 μl wash buffer (30 mM Tris pH 8.0, 100 mM KOAc, 2 mM Mg(OAc)2, 0.5 mM TCEP). After air drying, the membrane strips were used to expose phosphor screens (GE Healthcare Life Sciences) for visualization. Screens were imaged on a Typhoon phosphorimager (GE Healthcare Life Science) and signals were quantified with ImageQuant (GE Healthcare Life Sciences). Dissociation constants were calculated by fitting the data to a single site binding equation:
F = fraction target RNA bound, Bmax = maximal number of binding sites, [AGO2] = total concentration of the AGO2-miR-21 complex, and KD = calculated dissociation constant, using Prism (GraphPad Software). For weakly binding RNAs Bmax was constrained to ≤1.
Data analysis and statistical testing
Processing and visualization of sequencing data.
Sequencing data were pre-processed to combine FASTQ files of two sequencing lanes (cat) and to remove adapters (cutadapt). Paired end reads were merged by paired-end read merger (pear). UMIs were collapsed by collapse.py (T.D. Domenico, https://github.com/tdido). Chimeric reads were called and annotated with the hyb package26, using the command:
Hyb uses bowtie227 in local mapping mode to map reads to a transcriptome database and to identify chimeras, and it annotates the chimeras with RNA base-pairing information generated by hybrid-min28. The transcriptome database used by hyb, “hOH7_and_Zika”, consists of human spliced mRNAs and noncoding RNAs described in4, and the genome sequence of the Zika virus (Zika virus isolate ZIKV/H.sapiens/Brazil/PE243/2015, complete genome). To evaluate the folding energy of chimeric reads, we used hybrid-min28 with default settings. We then randomly reassigned (shuffled) pairs of fragments found in chimeric reads, and repeated the folding energy analysis. The folding energies of experimentally identified and shuffled chimeras were compared by Wilcoxon test.
Virus interaction heatmaps were plotted using Java Treeview29, such that color intensity represents the coverage of chimeric reads at every pair of positions. The first read of each pair is plotted along the X axis, and the second read along the Y axis. As a result, chimeras found in the 5'-3' orientation are shown above the diagonal, and chimeras in the 3'-5' orientation are below the diagonal. Viewpoint histograms were plotted with gnuplot, and arc plots were plotted with R-chie30.
For every pair of positions (i, j) along the virus genome we calculated the COMRADES score, Cij: the number of chimeric reads that, when analyzed with the program hybrid-min with default settings, indicated base-pairing between positions i and j. We used COMRADES scores to calculate per-base Shannon entropy for each nucleotide position along the virus. Shannon entropy of position i is defined as:
where n is the length of the genome (10,807 nt); and P(Cij) is:
High entropy indicates flexible positions that may form multiple alternative base-pairs, whereas low entropy indicates positions that always pair with the same nucleotide partner. We visualized RNA structures using VARNA31, where the colour scale represents the COMRADES score for each base pair.
RNA structure prediction.
For RNA structure predictions, we collected all potential base pairs with a non-zero Cij value, assembled sets of adjacent base pairs into uninterrupted stem structures, and calculated the base-pairing score of each stem as the sum of Cij values of individual base pairs. We then ranked these stem elements by their scores. In a preliminary analysis, we folded the 10,807 nt virus genome in a set of 50 overlapping 1,000 nt fragments, using the hybrid-ss-min program28. Each fragment was folded using a set of 250 top-ranked in vivo probed stem elements as folding constraints. Based on this preliminary analysis, we identified high-scoring stem-loop structures that were reproducibly predicted across multiple fragments, and we defined new fragment boundaries to prevent the disruption of these reproducible structural elements. As a result, we obtained fragment sizes that vary in size, but are approximately 1,000 nt long each.
We then performed full folding analysis using the following fragment boundaries: 5'UTR: 1-107, F1: 108-1275, F2: 1276-2287, F3: 2288-3323, F4: 3324-4521, F5: 4522-5551, F6: 5552-6810, F7: 6811-7757, F8: 7758-8755, F9: 8756-9543, F10: 9544-10379, 3'UTR: 10380-10807.
For each fragment, we assembled a set of folding constraints that represented the 75 top-scoring stem elements within that fragment, we randomly shuffled this set of constraints 1,000 times, and we used the shuffled constraints for folding prediction by hybrid-ss-min. The resulting individual structures typically incorporate 25%-40% of these constraints. We recorded the folding energy of each structure, as predicted by hybrid-ss-min, and we used the sum of Cij values to calculate an overall score for each structure. To assemble the top-scoring full-genome structure shown in Supplementary Fig. 4a, we assembled the top-scoring structures for each coding sequence fragment (F1-F10), and the previously proposed structures of the 5' and 3' UTRs. An additional analysis of folding within and between the 5' and 3' UTRs is shown in Fig. 2a-c and Supplementary Fig. 4c.
We also repeated the folding analysis with shuffled sets of 50-250 top-scoring constraints per fragment. This yielded similar results, but we found that either reducing or increasing the numbers of constraints tended to reduce the number of high-scoring structures.
To explore the sets of alternative structures, we computed pairwise distances between structures as the number of positions with discordant base-pairing. This resulted in a 1,000 × 1,000 matrix of distances, which we then represented on a two-dimensional surface using multidimensional scaling (using the R function cmdscale). Multidimensional scaling, also known as Principal Coordinate Analysis32, maps multidimensional objects (in this case, RNA structures) to a set of points on a plane, such that the distances between RNA structures are well-approximated by Euclidean distances between points, by minimization of a stress function:
The statistical significance of host-virus RNA-RNA interactions was calculated using DESeq233, by comparing counts of chimeric reads from 3 COMRADES and 3 control datasets.
Discovery of ZIKV miR-21 interaction with an independent analysis pipeline.
Alignment.
The first read of each pair was processed using UMI-tools34 to extract the 6 nucleotide unique molecular identifier (UMI) at the start of the read. Processed reads were aligned using the STAR aligner35, reporting all reads in their original order (--outSAMtype BAM Unsorted --outSAMunmapped Within); only reporting unique alignments (--outFilterMultimapNmax 1); and reporting alignments to individual segments of chimeric reads (--chimOutType WithinBAM --chimSegmentMin 20 --chimScoreJunctionNonGTAG 0 --chimMainSegmentMultNmax 1). The reference consisted of the hg38 build of the human genome, combined with the genome sequence of the PE243 strain of the Zika virus. Each read of the pair was aligned separately to avoid preferencing alignment to the same genomic locus.
For each library, the pair of BAM files were collated and pair information was fixed using samtools36. PCR duplicates were removed on the basis of their UMIs, using UMI-tools in paired mode.
Detecting significant interactions.
We considered the "interaction space" between the human and Zika genomes, consisting of pairs of 1 kbp bins (one on each genome). For each replicate library in each condition (COMRADES and control), we counted the number of read pairs with one read in each bin using diffHic37. This yielded a count matrix that was normalized using the trimmed mean-of-M-values method38 to correct for composition biases, under the assumption that most read pairs mapping across the Zika and human genomes was caused by non-specific ligation. We then applied the quasi-likelihood framework in edgeR39 with 2 residual degrees of freedom for dispersion estimation to test for significant differences between the read pair counts for COMRADES and control. This was performed using an additive design matrix that blocked on the batch to reflect the paired-sample design of the experiment.
Robust empirical Bayes shrinkage40 was also used to stabilise the dispersion estimates in the presence of limited replication. Bin pairs were aggregated into clusters based on whether they overlapped the same human gene
Test statistics were combined for each gene-Zika interaction using Simes' method41 prior to applying the Benjamini-Hochberg method. Interactions that were significantly enriched in COMRADES over the control were defined at a false discovery rate threshold of 5%.
Data availability.
All sequencing data sets have been deposited in ArrayExpress under accession number: E-MTAB-6427. Base-pairing prediction, structure prediction and clustering data are available as Supplementary Data files. Additional data that support the findings of this study are available from the corresponding authors upon request. A step-by-step protocol is available as a Supplementary protocol and an open resource in Protocol Exchange41.
Code availability.
The RNA structure prediction pipeline can be downloaded from: https://github.com/gkudla/comrades.
Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
The authors thank A. Kohl, Centre for Virus Research, University of Glasgow and L.J. Pena and R.F. França, Fiocruz Recife, Pernambuco, Brazil for providing the PE243 ZIKV RNA used to generate the virus stock. We thank Y. Galanty and F.M. Martínez for assisting with CRISPR-Cas9 knockouts; T.D. Domenico and W. Matsushima for collapsing U.M.Is; G. Sanguinetti for suggesting the RNA folding strategy; S. Moss for assisting with risk assessments; M.S. Diamond, T. Sweeney, A. Firth, D. Jordan, A. Zeisel and members of E.A.M. group for their comments and C. Flandoli for illustrations. This work was supported by Cancer Research UK (C13474/A18583, C6946/A14492) and the Wellcome Trust (104640/Z/14/Z, 092096/Z/10/Z) to E.A.M. O.Z. was supported by the Human Frontier Science Program (HFSP, LT000558/2015), the European Molecular Biology Organization (EMBO, ALTF1622-2014), and the Blavatnik Family Foundation postdoctoral fellowship. G.K. and M.G. were supported by Wellcome Trust grant 207507 and UK Medical Research Council. A.T.L.L. and J.C.M. were supported by core funding from Cancer Research UK (award no. 17197 to JCM). J.C.M was also supported by core funding from EMBL. I.G. and L.W.M. were supported by the Wellcome Trust Senior Fellowship in Basic Biomedical Science to I.G. (207498/Z/17/Z). I.J.M., L.F.G. and J.S.-G. were supported by grants R01GM104475 and R01GM115649 from NIGMS. C.K.K was supported by City University of Hong Kong Projects 9610363 and 7200520, Croucher Foundation Project 9500030 and Hong Kong RGC Projects 9048103 and 9054020. C.-F.Q. was supported by the NSFC Excellent Young Scientist Fund 81522025 and the Newton Advanced Fellowship from the Academy of Medical Sciences, UK.
Footnotes
Competing interests
The authors declare no competing interests
References
- 1.Lu Z et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aw JGA et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 62, 603–617 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Sharma E, Sterne-Weiler T, O’Hanlon D & Blencowe BJ Global Mapping of Human RNA-RNA Interactions. Mol. Cell 62, 618–626 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Helwak A, Kudla G, Dudnakova T & Tollervey D Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramani V, Qiu R & Shendure J High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol 33, 980–984 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugimoto Y et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Z-Y et al. Viral RNA switch mediates the dynamic control of flavivirus replicase recruitment by genome cyclization. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez DE, Lodeiro MF, Ludueña SJ, Pietrasanta LI & Gamarnik AV Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol 79, 6631–6643 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friebe P & Harris E Interplay of RNA elements in the dengue virus 5’ and 3' ends required for viral RNA replication. J. Virol 84, 6103–6118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filomatori CV et al. A 5’ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 20, 2238–2249 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirakitikulr N, Kohlway A, Lindenbach BD & Pyle AM The Coding Region of the HCV Genome Contains a Network of Regulatory RNA Structures. Mol. Cell 62, 111–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watts JM et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hahn CS et al. Conserved elements in the 3’ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol 198, 33–41 (1987). [DOI] [PubMed] [Google Scholar]
- 14.Manzano M et al. Identification of Cis-Acting Elements in the 3′-Untranslated Region of the Dengue Virus Type 2 RNA That Modulate Translation and Replication. J. Biol. Chem 286, 22521–22534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z-Y et al. Novel cis-acting element within the capsid-coding region enhances flavivirus viral-RNA replication by regulating genome cyclization. J. Virol 87, 6804–6818 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akiyama BM et al. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354, 1148–1152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouskin S, Zubradt M, Washietl S, Kellis M & Weissman JS Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding Y et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Spitale RC et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo YE & Steitz JA Virus meets host microRNA: the destroyer, the booster, the hijacker. Mol. Cell. Biol 34, 3780–3787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu C, Qu K, Zhong FL, Artandi SE & Chang HY Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44, 667–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwok CK, Marsico G, Sahakyan AB, Chambers VS & Balasubramanian S rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 13, 841–844 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Liu Z-Y et al. Characterization of cis-acting RNA elements of Zika virus by using a self-splicing ribozyme-dependent infectious clone. J. Virol 91, e00484–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwok CK, Ding Y, Tang Y, Assmann SM & Bevilacqua PC Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun 4, 2971 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Schirle NT, Sheu-Gruttadauria J & MacRae IJ Structural basis for microRNA targeting. Science 346, 608–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Travis AJ, Moody J, Helwak A, Tollervey D & Kudla G Hyb: a bioinformatics pipeline for the analysis of CLASH (crosslinking, ligation and sequencing of hybrids) data. Methods 65, 263–273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markham NR & Zuker M UNAFold in Bioinformatics: Structure, Function and Applications (ed. Keith JM) 3–31 (Humana Press, 2008). [Google Scholar]
- 29.Saldanha AJ Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Lai D, Proctor JR, Zhu JYA & Meyer IM R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 40, e95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darty K, Denise A & Ponty Y VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gower JC Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53, 325–338 (1966). [Google Scholar]
- 33.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lun ATL & Smyth GK diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics 16, 258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lun ATL, Chen Y & Smyth GK It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol. Biol. 1418, 391–416 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Phipson B, Lee S, Majewski IJ, Alexander WS & Smyth GK ROBUST HYPERPARAMETER ESTIMATION PROTECTS AGAINST HYPERVARIABLE GENES AND IMPROVES POWER TO DETECT DIFFERENTIAL EXPRESSION. Ann. Appl. Stat 10, 946–963 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lun ATL & Smyth GK De novo detection of differentially bound regions for ChIP-seq data using peaks and windows: controlling error rates correctly. Nucleic Acids Res. 42, e95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziv O & Miska EA COMRADES: Crosslinking Of Matched RNAs And Deep Sequencing. Protocol Exchange. DOI: 10.1038/protex.2018.091 (2018). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data sets have been deposited in ArrayExpress under accession number: E-MTAB-6427. Base-pairing prediction, structure prediction and clustering data are available as Supplementary Data files. Additional data that support the findings of this study are available from the corresponding authors upon request. A step-by-step protocol is available as a Supplementary protocol and an open resource in Protocol Exchange41.