

Abstract
Effective treatment for pancreatic ductal adenocarcinoma (PDAC) is an urgent, unmet medical need. Targeting KRAS, the oncogene that is present in >95% of PDAC, is a heavily pursued strategy, but remains unsuccessful in the clinic. Therefore, targeting key effector cascades of KRAS oncoprotein, particularly the mitogenic RAF-MEK-ERK pathway represents the next best strategy. However, RAF or MEK inhibitors have failed to show clinical efficacy in PDAC. Several studies have shown that cancer cells treated with RAF or MEK inhibitors adopt multiple mechanisms to re-activate ERK signaling. Therefore, development of ERK-specific inhibitors carries the promise to effectively abrogate this pathway. Ulixertinib (or BVD-523) is a first-in-class ERK-specific inhibitor that has demonstrated promising anti-tumor activity in a phase 1 clinical trial for advanced solid tumors with NRAS and BRAF mutations, providing a strong rationale to test this inhibitor in PDAC. In this study, we show that ulixertinib effectively inhibits in vitro growth of multiple PDAC lines and potentiates the cytotoxic effect of gemcitabine. Moreover, we found that PDAC cells treated with ulixertinib upregulates the parallel PI3K-AKT pathway through activating the HER/ErbB family proteins. Concurrent inhibition of PI3K or HER proteins synergizes with ulixertinib in suppressing PDAC cell growth in vitro and in vivo. Overall, our study provides the preclinical rationale for testing combinations of ulixertinib with chemotherapy or PI3K and HER inhibitors in PDAC patients.
Keywords: Pancreatic cancer, KRAS, ERK, Ulixertinib
INTRODUCTION
The prognosis for patients with pancreatic ductal adenocarcinoma (PDAC) is dismal, with a 5-year survival rate lower than 8%(1). Surgical resection is possible for only 10–15% of patients diagnosed at early stage. Most patients are diagnosed at advanced stages, where combinatorial chemotherapies are the only option. However, treatment responses are typically short-lived and associated with significant side effects (2). Therefore, development of new and effective clinical approaches is a significant unmet medical need. PDAC is typified by a near universal (~95%) mutational activation of the KRAS oncogene. Mutant KRAS proteins drive the malignant phenotype of PDAC through activating various downstream effector cascades, with the mitogenic RAF-MEK-ERK pathway (also known as the mitogen-activated protein kinase, MAPK pathway) and pro-survival PI3K-AKT-mTOR pathways being the best described (3). Of these, the MAPK pathway is probably the most critical, evidenced by observation in genetic mouse models that expression of activated BRAFV600E, but not PI3KH1047R, in cooperation with p53 loss, drives formation of PDAC that histologically mimics KRASG12D-mutant mouse PDAC models (or KPC mice)(4). However, pharmacologic inhibition of RAF and MEK have shown disappointing results in clinical trials for PDAC, in contrast to the relative success in BRAF-mutant melanoma, largely due to emergence of multiple escape mechanisms that reactivate the ERK kinases (5, 6). Therefore, recent attention has shifted to targeting the ERK kinases as a novel approach to suppress the MAPK pathway (7, 8).
A recent report showed that SCH772984, an ERK inhibitor, is effective in suppressing xenograft growth of PDAC cells, partly through degradation of c-Myc and induction of senescence-like phenotype (7), providing the first preclinical evidence in support of targeting ERK in PDAC. However, clinical development of SCH772984 is halted. Ulixertinib (or BVD-523) is a reversible, potent, ATP-competitive kinase inhibitor targeting ERK1 and ERK2 in the sub-nanomolar range, that has shown promising preclinical efficacy, and has clinical activity in NRAS- and BRAF-mutant melanomas in a phase Ib/IIa clinical trial (NCT01781429) (9, 10). This result, though early-staged, provides a strong rationale for testing ulixertinib in KRAS-mutant cancers such as PDAC. In this study, we showed that ulixertinib is very potent in suppressing growth of PDAC cells in vitro, but to a lesser degree, in vivo. However, ulixertinib greatly potentiates the cytotoxic effect of gemcitabine. Surprisingly, we found that ulixertinib upregulates phosphorylation of ERK1/2, MEK and the parallel PI3K-AKT pathways, partly through engagement of the HER/ErbB family proteins. We hypothesize that these pathways may enable PDAC cells to tolerate ERK inhibition. Supporting this notion, we showed that pan-HER and PI3K inhibitors synergized with ulixertinib in curbing PDAC growth in vitro and in vivo. Overall, our findings could have wide therapeutic implications for guiding ERK inhibitor-based therapeutic combinations in the treatment of PDAC.
MATERIAL AND METHODS
Cell lines.
All PDAC cells were purchased from ATCC, which performs authentication on its own cell lines. Pa01c, Pa02c, Pa03c and Pa14c were kind gifts from Dr. Channing Der at UNC-CH and were whole-exome sequenced(11). HEK T/tH cells were a kind gift from Dr. Christopher Counter and previously described(12). KPC2 cells were a kind gift from Dr. David DeNardo (Washington University, MO), whole-exome sequenced and published (13). All cells were cultured in DMEM or RPMI plus 10% FBS/1% Pen-Strep at 5% CO2 in 37°C incubators. Mycoplasma testing was performed every 6 months using MycoSEQ Detection kit (Applied Biosystems). All cell lines were used for fewer than 6 months after receipt or resuscitation from cryopreservation.
Drugs and reagents.
Gemcitabine was purchased from the Siteman Cancer Center Pharmacy. Ulixertinib was provided by BioMed Valley under MTA. GDC-0944, SCH772984, afatinib and neratinib were purchased from Selleckchem. GDC-0941 was purchased from Apexbio LLC and the structure was published (14).
Tumor section, immunohistochemistry (IHC) and Immunofluorescence (IF).
Tumors were formalin-fixed, paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E). IHC and IF staining were performed using the following antibodies: p-ERK, p-AKT, Ki-67, cleaved caspase-3, pan-keratin antibodies (CST). Unless otherwise specified, all quantification was performed under Nikon confocal fluorescence microscopy using NIS Element software as described (15). Briefly, red or green fluorescence-labelled cells per field were automatically detected, quantified by the software and exported in Excel format. Ten 200X or 400X power fields were analysed per tumor independently by two individuals (HJ and DZ), and data were presented as mean ± SEM of collated data. IHC sections was interpreted independently by HJ, MX, KHL and representative data agreeable to all members were presented.
Soft agar assay.
5,000 to 10,000 cells were suspended in Noble agar, seeded in triplicates in 24 well plates as indicated. Colony numbers from each well were counted manually under dissection microscopy after 3–4 weeks.
In vitro cell viability assay and calculation of combination indices.
5,000 to 10,000 cells/well were plated in triplicates in 96 well plates one day prior to addition of the inhibitors at the indicated final concentrations. After 5 or 7 days of culture, viability assay was measured using Resazurin colorimetric analysis as described (15). For drug interaction studies, cells were cultured in triplicates in the presence of six fixed-ratio concentrations for 72 hours followed by Alamar Blue viability assay. Combination indices were calculated using Compusyn software as described (16). All experiments were done at least three times in triplicates and one set of data most representative of the overall data was presented.
RPPA.
HPNE-KRASG12D and MIA Paca-2 cell lysates were prepared using pre-made lysis buffer provided by the RPPA core at MD Anderson Cancer Center. Samples were probed with antibodies by tyramide-based signal amplification approach and visualized by DAB colorimetric reaction. Slides were scanned on a flatbed scanner to produce 16-bit tif image. Spots from tif images were identified and the density was quantified by Array-Pro Analyzer. All the data points were normalized for protein loading and transformed to linear value, designated as “Normalized Linear”. “Normalized Linear” values were transformed to Log2 values, and median-centered for analysis.
Immunoblotting.
Cells were harvested in RIPA lysis buffer (25mM Tris pH7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton-X) with phosphatase and protease inhibitors. 30–50μg of lysates were resolved in the SDS-PAGE gels, transferred to PVDF membranes, blocked, probed with primary antibodies followed by HRP-conjugated secondary antibodies. The following antibodies were used: p-ERK1/2 (T202/Y204), total ERK1/2, p-MEK1/2 (S217/S221), total MEK1/2, p-AKT(S473), total AKT, p-EGFR (T1068), total EGFR, p-HER2 (Y1221/1222), total HER2, p-HER3 (Y1289), total HER3, p-RSK (S380), total RSK (all from CST), tubulin (Santa Cruz).
Caspase 3/7 reporter assay.
Caspase 3/7 reporter kit was purchased from Promega. All cells were plated in triplicates in 96 well plates, treated for 24 hours (for MIA Paca-2) or 48 hours (for HPNE-KRASG12D) and assayed according to the manufacturer’s protocol, and analyzed with a Synergy H4 Hybrid Multi-Mode Microplate Reader.
Xenograft tumorigenesis assay.
All animal experiments were conducted in accordance with IACUC protocol (#20130191 and #20160142) and per ethical principles of Declaration of Helsinki. Briefly, 5 million of MIA Paca-2 or HPNE-KRASG12D cells were inoculated into the flanks of 8- to 12-week-old nude female mice. Treatments were started by oral gavage when tumors reached ~100mm3 in volume (ulixertinib 100mg/kg twice daily, afatinib 12.5mg/kg daily, GDC-0941 50mg/kg twice daily). Mice were euthanized when vehicle-treated tumors reached maximum volumes. Experiment on MIA Paca-2 was performed twice, showing similar results.
Pharmacokinetic studies.
Concentrations of ulixertinib and gemcitabine in mouse plasma, collected two hours after the last dose of treatment, were stored at −80°C and measured using HPLC MS/MS (Shimadzu, Prominence model) as described (9).
Statistical Analyses.
All results, when applicable, were expressed as the mean ± SEM. Statistical analyses were performed using the Prism 6 software. Unpaired student’s two-tailed t-tests were used to compare two groups when appropriate. For multiple groups, one-way ANOVA analysis with Tukey’s post-test were used. P values <0.05 were considered as statistically significant.
RESULTS
Ulixertinib Has Promising Efficacy in PDAC Cells In Vitro
First, we tested the effect of ulixertinib as single agent in nine conventional PDAC and four patient-derived cell lines (PDCLs) grown as monolayer cultures. All these cell lines harbor an oncogenic KRAS mutation, except for BxPc-3, which has a BRAFV600E mutation, and Hs766T which has neither KRAS nor BRAF mutations (11, 17). We observed concentration-dependent inhibition of cell viability in all PDAC lines tested after 5 days of culture in 2D condition. We did not observe a correlation between the IC50 values and the genotype or KRAS mutant isoform of each cell line. The effect of ulixertinib was more prominent after 7 days, as shown by a further decline in IC50 in most cell lines tested (Fig. 1A, 1B), indicating that a continual dosing may be needed for optimal therapeutic effect in future clinical trials. Notably, ulixertinib also exhibited dose-dependent suppression in Hs766T cells, indicating a reliance of this cell line on the MAPK pathway. To clearly delineate the effect of ulixertinib in KRAS-driven cell growth, we utilized the murine KPC2 (derived from the PDAC of a p48-Cre/p53flox/WT/LSL-KRASG12D mouse) cells and an immortalized human pancreatic ductal cell line (HPNE) engineered to express KRASG12D (HPNE-KRASG12D, from ATCC). Supporting the paradigm of ERK being a key signal transducer downstream on KRAS, ulixertinib also dose-dependently suppressed the growth of these lines (Fig. 1A). As anchorage-independent (AI) growth is a hallmark of KRAS-induced oncogenic transformation, we also tested the effect of ulixertinib in AI growth. As expected, ulixertinib showed potent, dose-dependent suppression on all PDAC lines tested (Fig. 1C), including the KPC2, HPNE-KRASG12D and a human embryonic kidney cell line immortalized with hTERT, SV40 antigens and transformed with KRASG12V (HEK T/tH KRASG12V)(12). Overall, our results showed that ulixertinib as single agent has promising efficacy on PDAC cells of various genetic backgrounds in vitro.
Figure 1. Ulixertinib has promising single-agent efficacy in PDAC cells in vitro.
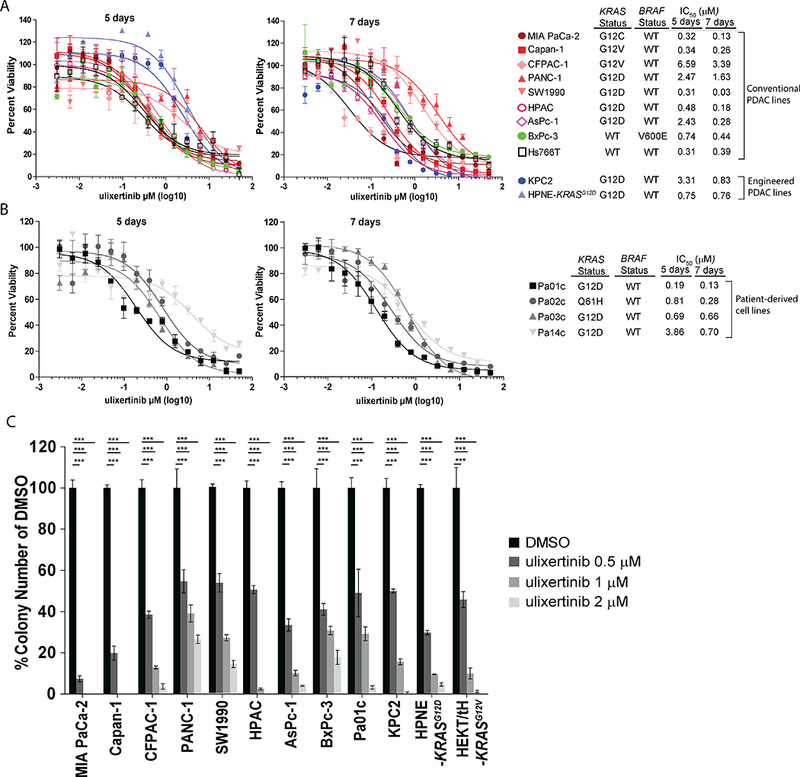
A, B, Alamar blue assays showing dose-dependent effect of ulixertinib on the viability of the indicated PDAC lines after 5 or 7 days of treatment. IC50 values were calculated using GraphPad software. C, Quantification of soft agar colonies of the indicated PDAC lines seeded in different concentrations of ulixertinib with reference to DMSO. All experiments were done at least three times in triplicates, and one set of data was presented. Data represents mean ± SEM. (*p<0.05, **p<0.01, ***p<0.001).
Ulixertinib Synergizes with Gemcitabine In Vitro And In Vivo
Recognizing that kinase inhibitor monotherapies have not been successful in PDAC, we tested whether ulixertinib may potentiate the effect of gemcitabine, a chemotherapeutic agent commonly used in PDAC treatment. We chose to focus on gemcitabine for two reasons: first, PDAC cells can upregulate ERK1/2 phosphorylation as a potential survival mechanism when challenged with gemcitabine (18), thereby providing a good rationale for testing ulixertinib in combination with gemcitabine. Second, 48% of patients treated with ulixertinib in a clinical study develop diarrhea (10), a side effect less commonly associated with gemcitabine-based chemotherapy regimens. We found that ulixertinib greatly potentiates the dose-dependent suppressive effect of gemcitabine on four selected PDAC lines cultured over 5 days (Suppl. Fig. 1A). We chose CFPAC-1 and MIA Paca-2 cells because they have the highest and one of the lowest IC50 values, respectively (Fig. 1A), as well as KPC2 and HPNE-KRASG12D because the malignant growth and hyperactivation of the MAPK pathway of these cell lines are driven by oncogenic KRAS. To better understand if the interaction between ulixertinib and gemcitabine was additive or synergistic, we quantified the drug effect interactions (by combination indices, or CI) between ulixertinib and gemcitabine using the widely adopted Chou-Talalay method (Compusyn software) (16). Of all four cell lines tested, ulixertinib synergizes with gemcitabine with CI of less than 0.7 (Fig. 2A). Mechanistically, ulixertinib at 2μM, which completely suppressed ERK catalytic activity, and ulixertinib plus gemcitabine induced more significant apoptosis in MIA Paca-2 cells as assayed by caspase 3/7 reporter activity (Fig. 2B left). In HPNE-KRASG12D cells, ulixertinib significantly augmented the pro-apoptotic effect of gemcitabine, although gemcitabine alone was more potent in inducing apoptosis compared to DMSO (Fig. 2B right). We next chose MIA Paca-2 and HPNE-KRASG12D cell lines for all subsequent in vivo studies because the MIA Paca-2 cells are KRAS-mutated, are highly aggressive, and readily form colonies in soft agar and tumors in mice (17); and the HPNE-KRASG12D are a genetically-defined model, which allows studies of KRASG12D-associated MAPK pathway in the absence of other confounding genetic changes (19). In MIA Paca-2 subcutaneous xenograft model, ulixertinib or gemcitabine alone were effective in suppressing tumor growth, but combined ulixertinib and gemcitabine was significantly more potent (Fig. 2C and 2D). Similar results were seen in HPNE-KRASG12D subcutaneous xenograft model (Fig. 2E, 2F). Importantly, mice treated with ulixertinib alone or in combination with gemcitabine showed no signs of systemic toxicity based on appearance and body weight (Suppl. Fig. 1B). In the MIA Paca-2 experiments, analyses of mouse plasma two hours after the last dose of treatment, before the mice were euthanized, showed ulixertinib concentrations to be statistically similar in the absence or presence of gemcitabine (17.7 ± 5.29 μg/mL vs 11.7 ± 2.4 μg /mL, p=0.54, Suppl. Fig. 1C). Similarly, plasma concentrations of gemcitabine were not statistically different between mice treated without or with ulixertinib (205 ± 29 ng/mL vs 302 ± 54ng/mL, p=0.093, Suppl. Fig. 1D). Therefore, we conclude that ulixertinib did not affect the plasma concentration of gemcitabine. Histologic analyses showed extensive areas of necrosis in harvested MIA Paca-2 tumors treated with both ulixertinib and gemcitabine, as opposed to tumors treated with vehicle or single agents (Fig. 2G). By quantitative immunofluorescence, combo-treated MIA Paca-2 tumors showed significantly more apoptotic cells (Fig. 2G middle panels, 2H), and lower abundance of proliferating Ki-67+ cells (Fig. 2G lower panels, 2I). Overall, these results support combining ulixertinib with gemcitabine-based chemotherapy regimen in clinical trials for PDAC patients.
Figure 2. Ulixertinib synergizes with gemcitabine in vitro and in vivo.
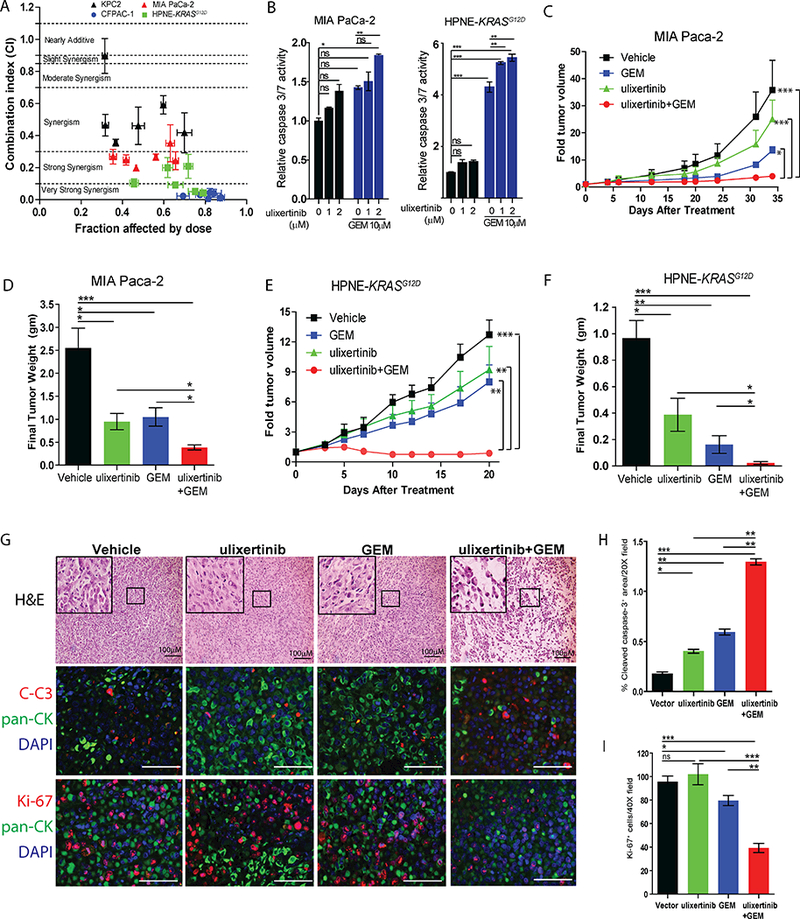
A, Combination indices (CI) between gemcitabine (GEM) and ulixertinib on four PDAC cells calculated using Compusyn software. Cells were cultured in six fixed-ratio concentrations (all in μM) of ulixertinib: gemcitabine (10:10, 5:5, 2.5:2.5, 1.25:1.25; 0.625:0.625, 0.31:0.31) for 5 days and viability masured by Alamar Blue assay. B, Caspase 3/7 reporter assay showing the effect of ulixertinib in combination with gemcitabine in MIA Paca-2 (24 hours treatment) and HPNE-KRASG12D cells (48 hours treatment). One of three sets of experiments, each done in triplicates, was presented. C, Serial measurements of relative tumor volume (to initial) and D, final weight of MIA Paca-2 tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=10/group, Fig. 2C and 2D represent two independent experiments). E, Serial measurements of relative tumor volume (to initial) and F, final weight of HPNE-KRASG12D tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=8/group). G H&E staining showing the effect of drug treatments on MIA Paca-2 tumors (upper panels); and representative immunofluorescence pictures and quantification of H, cleaved caspase-3+ area per 20X field and I, dual CK+ and Ki-67+ cells per 40X fields of the indicated MIA Paca-2 tumors (mid and lower panels). Eight to ten tumors were analyzed per group. Data represents mean ± SEM. (*p<0.05, **p<0.01, ***p<0.001, scale bars = 100μM).
Pharmacologic ERK Inhibition Increased ERK, MEK and AKT Phosphorylation
We next interrogated the effect of ulixertinib on the MAPK signaling by Western blots. To our surprise, we noted paradoxical, increased levels of phosphorylated ERK1/2, MEK1/2 and AKT in all PDAC lines treated with ulixertinib overnight (Fig. 3A, densitometric analyses in Suppl. Fig. 2A), although ulixertinib did dose-dependently suppress the phosphorylation (p-) of RSK (p-RSK), a canonical substrate of ERK. To ensure this phenomenon is not unique to ulixertinib, we used two other published ERK1/2 inhibitors, SCH772984 and GDC-0994 on MIA Paca-2 and HPNE-KRASG12D cells (20, 21). Intriguingly, we observed similar but less dramatic increase in p-ERK1/2 and p-MEK1/2 levels with GDC-0994 at equimolar concentrations (1μM and 2μM) with ulixertinib, and both agents suppressed p-RSK to similar extents (Fig. 3B, Suppl. Fig. 2B). On the contrary, SCH772984 was more potent in suppressing p-RSK at these concentrations but did not increase p-ERK1/2. It has been speculated that in addition to inhibiting ERK1/2 catalytic function, SCH772984 may additionally stabilize the conformation of ERK1/2 that prevents activation by MEK(20). Interestingly, SCH772984 treatment also potently increased p-MEK1/2 levels. These results suggest that each of the three ERK inhibitors has distinct mechanisms of action and PDAC cells respond to these inhibitors by activating MEK1/2, and in the case of ulixertinib and GDC-0994, ERK1/2. To further confirm target inhibition of ulixertinib, we treated MIA Paca-2 and HPAC cells engineered to stably express firefly luciferase driven by a serum-response element (SRE), and Renilla as internal control. Activation of SRE is driven by Elk-1, another substrate known to be activated by ERK (22). Overnight treatment of these two reporter lines with different MEK (trametinib and selumetinib) or ERK (ulixertinib and GDC-0994) inhibitors significantly suppressed SRE-reporter activity (Fig. 3C), further confirming that ulixertinib and GDC-0994 at 1μM and 2μM can effectively block the kinase activity of ERK1/2. We next performed time course analysis to evaluate the rapidity of action of ulixertinib. We observed noticeable increased levels of p-ERK1/2, p-MEK1/2, p-AKT, and decreased level of p-RSK as early as 5 minutes after drug treatment (Fig. 3D, densitometric analysis in Suppl. Fig. 2C). These changes appeared to start plateauing after 30 minutes and reach a maximum with overnight (16 hours) treatment. These striking results indicate that PDAC cells can rapidly rewire their kinome network following ERK inhibition, which we hypothesize may help PDAC cells adapt and survive ERK inhibition. To gain a more comprehensive view of the adaptive changes, we performed reverse-phase protein array analyses on HPNE-KRASG12D and MIA Paca-2 cells treated with DMSO or ulixertinib for 16 hours. We chose 16 hours of treatment for analyses because this is the time point when maximum suppression of p-RSK was seen (Fig 3D), and when we reasoned a stable adaptive balance has been reached. Consistent with Western blots, ulixertinib treatment resulted in more than a two-fold increase of p-ERK1/2 and p-AKT levels (Fig. 3E and 3F, MIA Paca-2 results in Suppl. Fig. 2D). Importantly, p-Rb and total c-Myc levels were significantly suppressed, as independently shown by another report (7), further validating the on-target effects of ulixertinib and the robustness of our results. Overall, our data demonstrated that ulixertinib treatment potently suppresses ERK kinase activity, while simultaneously resulting in rapid kinome changes that lead to increased phosphorylation of ERK, MEK, and AKT.
Figure 3. Pharmacologic ERK inhibition increased ERK, MEK and AKT phosphorylation.
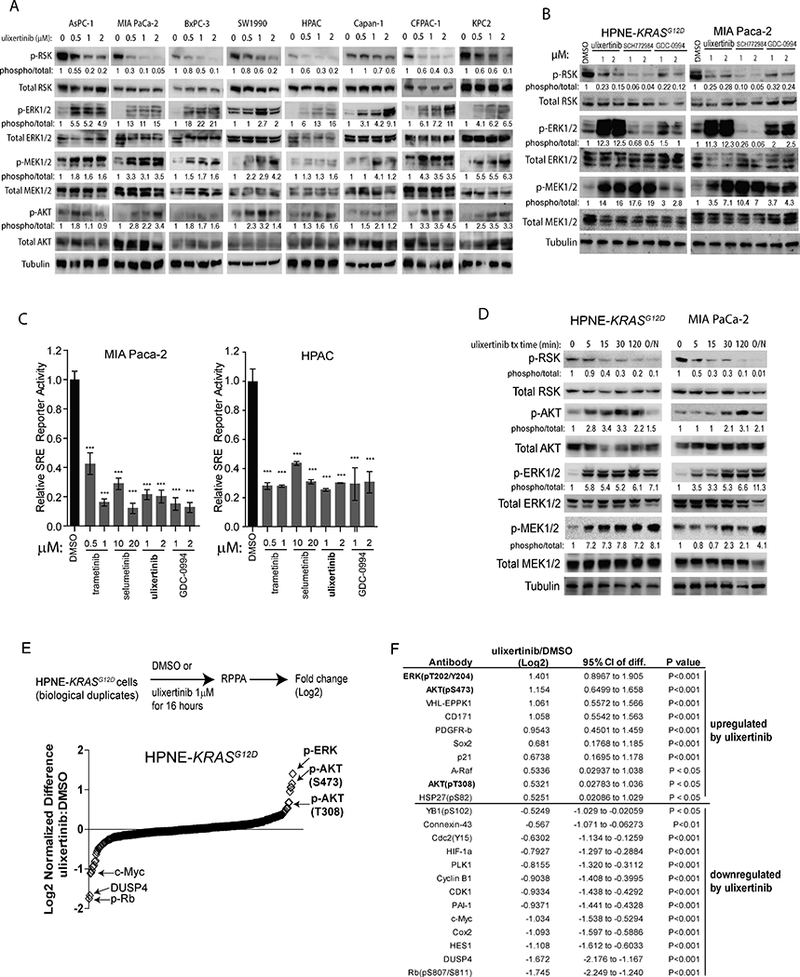
A, Western blots showing the effect of ulixertinib at different concentrations on the indicated PDAC lines after overnight (~16 hours) treatment. B, Western blots showing the effect of three different ERK inhibitors on MIA Paca-2 and HPNE-KRASG12D cells following overnight treatment. The indicated numbers represented relative densitometric intensity of each band measured by ImageJ software. C, SRE-driven luciferase reporter assay of MIA Paca-2 and HPAC cells showing the effect of MEK or ERK inhibitors on SRE activity levels as normalized to internal Renilla, after overnight treatment. One of three sets of experiments, each done in triplicates, was presented. D, Western blots showing time course changes in the intensity of the indicated proteins, measured by densitometry, in two different PDAC lines, following ulixertinib treatment. E, Graphical representation of RPPA data showing the quantitative changes in ulixertinib/DMSO ratio across all tested markers from HPNE-KRASG12D cells treated with ulixertinib 1μM or DMSO for 16 hours in biological duplicates. F, List of RPPA markers with intensity changes more than one-fold and meeting p<0.05 after ulixertinib treatment, compared to DMSO.
Pan-HER Inhibition Potentiates the Anti-Tumor Effect of Ulixertinib
Because most of our tested PDAC cells harbor oncogenic KRAS protein, which already chronically activates the MAPK and PI3K pathways, we hypothesized that the additional increase in p-ERK1/2, p-MEK and p-AKT levels induced by ulixertinib must, in part, be driven by signaling events independent of KRAS. We hypothesized that the HER/ErbB family members are the most likely candidates, based on several reasons (Fig. 4A). First, overexpression of EGFR (HER1), HER2 and HER3 protein is very common in PDAC (23). Second, genetic mouse models showed that EGFR is required for KRAS-driven PDAC formation and progression (24, 25). Third, erlotinib, an FDA-approved EGFR inhibitor, in combination with gemcitabine modestly improved the survival of PDAC patients (26). To this end, we tested the phosphorylation status of EGFR, HER2 and HER3 proteins by Western blots following overnight ulixertinib treatment in PDAC lines. Indeed, we observed various degree of dose-dependent increase of p-EGFR, p-HER2 and p-HER3 levels following ulixertinib treatment in all PDAC lines tested (Fig. 4B, densitometric analysis in Suppl. Fig. 3A). Importantly, each PDAC line differed in the degree of changes in phosphorylation and the HER family member involved. These results imply that different PDAC line may engage multiple HER family members upon ulixertinib treatment, and that all HER family members may need to be targeted to maximally abrogate kinome rewiring. Supporting our hypothesis, co-treatment with afatinib, a pan-HER1/2/3 inhibitor that is FDA-approved for treatment of EGFR-mutant non-small cell lung cancer (27), completely abrogated induction of p-AKT and p-EGFR in four ulixertinib-treated PDAC cell lines (Fig. 4C, densitometric analysis in Suppl. Fig. 3B). However, p-ERK1/2 was attenuated but not completely blocked, suggesting that ulixertinib-induced p-ERK1/2 may occur through other HER-independent mechanisms. Co-treatment ulixertinib and afatinib was also more effective in suppressing p-RSK (Fig. 4C), suggesting that HER signaling may partly drive p-RSK at baseline. Similar results were seen using another pan-HER inhibitor neratinib (Suppl. Fig. 3C). Next, we found that combined ulixertinib and afatinib treatment was significantly more effective in suppressing AI growth of various PDAC lines than either alone (Fig. 4D). We again used Chou-Talalay method to characterize the drug effect interaction between ulixertinib and afatinib. Of the six different paired concentrations tested, most CI values fell within the ranges of synergism (<0.7, Fig. 4E). Mechanistically, ulixertinib plus afatinib was more effective in inducing apoptosis, compared to either agent alone, in MIA Paca-2 cells (Fig. 4F). Supporting these findings, ulixertinib plus afatinib was significantly more effective in suppressing growth of established MIA Paca-2 (Fig. 4G, 4H) and HPNE-KRASG12D (Fig. 4I, 4J) xenografts in nude mice, without incurring any obvious toxicities based on appearance and weight of treated mice (Suppl Fig. 3D). Histologic analyses of MIA Paca-2 tumors showed more patches of necrotic areas in combo-treated tumors (Fig. 4K, top panels). Consistent with our results in Western blots, ulixertinib treatment markedly increased p-ERK and p-AKT staining by immunohistochemistry (IHC), which were attenuated by concurrent treatment with afatinib (Fig. 4K, middle and lower panels). Quantitative immunofluorescence showed significantly higher degree of apoptosis (cleaved caspase-3+), and modestly lower number of proliferating (Ki-67+) neoplastic cells in combo-treated tumors (Suppl. Fig. 3E and 3F). Overall, these results showed HER family proteins to be involved in kinome rewiring following ulixertinib treatment, and that concurrent inhibition of ERK1/2 and HER family proteins is a promising therapeutic strategy in PDAC.
Figure 4. Pan-HER inhibition potentiates the anti-tumor effect of ulixertinib.
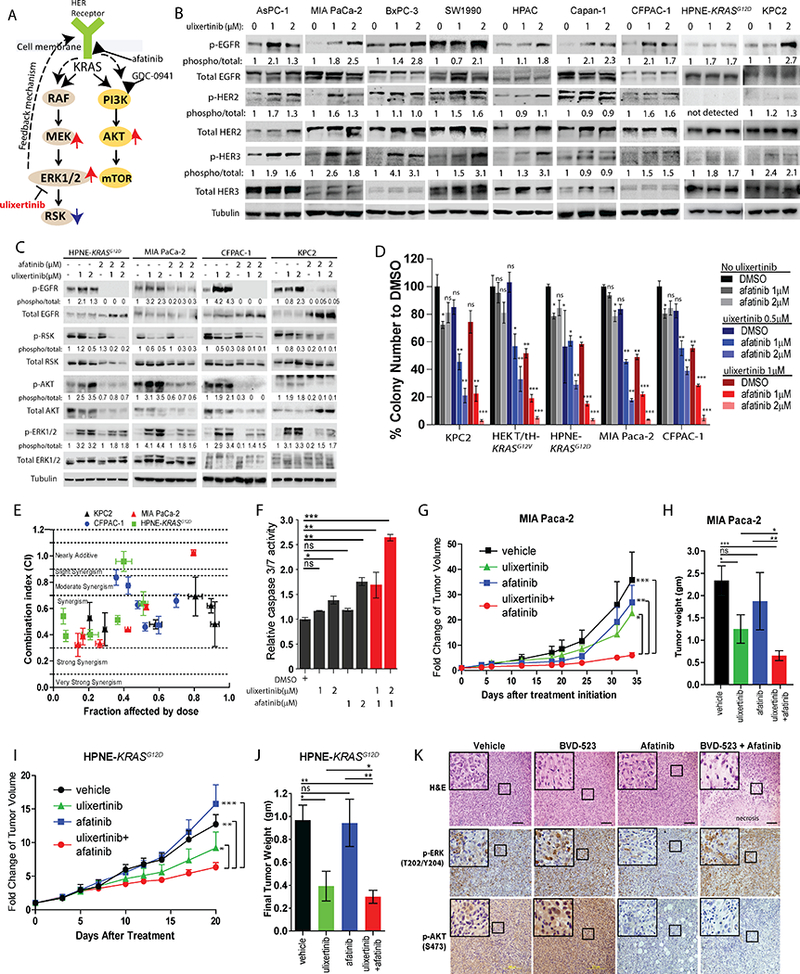
A, Hypothesized adaptive mechanisms to ulixertinib and proposed combinatorial strategies with afatinib or GDC-0941. B, Western blots showing changes in phospho-EGFR, phospho-HER2 and phospho-HER3 levels in various PDAC lines after overnight treatment with ulixertinib. C, Western blots showing the overnight treatment effect of ulixertinib, afatinib or both on the indicated proteins across the four PDAC lines. D, Relative quantification of soft agar colonies formed by PDAC lines treated as indicated. One of three sets of experiments, each done in triplicates, was presented. E, Median effect analyses of ulixertinib plus afatinib in four indicated PDAC lines as represented by combination indices at six different fixed-ratio concentrations (all in μM) of ulixertinib: afatinib (10:5, 5:2.5, 2.5:1.25, 1.25:0.625; 0.625:0.31, 0.31:0.15). One of three sets of experiments, each done in triplicates, was presented. F, Caspase 3/7 reporter assay showing the pro-apoptotic effect of ulixertinib and/or afatinib in MIA Paca-2 cells. G, Serial measurements of relative tumor volume (to initial) and H, final weight of MIA Paca-2 tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=10/group). I, Serial measurements of relative tumor volume (to initial) and J, final weight of HPNE-KRASG12D tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=8/group). K, H&E (upper panels), p-ERK and p-AKT IHC (middle and lower panels) staining showing the effect of drug treatments on MIA Paca-2 tumors. (*p<0.05, **p<0.01, ***p<0.001, scale bars = 100μM).
Combined ERK and PI3K Inhibition Is Synergistic in Inhibiting PDAC Growth
Because ulixertinib treatment led to increased p-AKT, another well-established pro-survival event driven by mutant KRAS(3), we theorized that concurrent inhibition of PI3K, the kinase that phosphorylates AKT, will potentiate the effect of ulixertinib. Indeed, co-treatment of different PDAC lines with GDC-0941, an orally available pan-PI3K inhibitor now being actively tested in clinical trials (14), potently blocked basal and ulixertinib-induced p-AKT levels, but had no effect on p-ERK or p-MEK (Fig. 5A, densitometric analysis in Suppl. Fig. 4A). Combined ulixertinib and GDC-0941 was significantly more effective in blocking AI growth of multiple PDAC lines in vitro (Fig. 5B). Analyses of CI values showed ulixertinib to be synergistic with GDC-0941 in curbing the growth of PDAC lines (Fig. 5C). Combined ulixertinib and GDC-0941 also induced significantly more apoptosis, as measured by caspase 3/7 reporter assay, in MIA Paca-2 cells (Fig. 5D). These results are consistent with a drug screen by Hayes et al. showing PI3K/AKT inhibitors to be synergistic with SCH772984 in inhibiting PDAC cell growth (7). Notably, GDC-0941 plus ulixertinib was significantly more effective in inhibiting the growth of MIA Paca-2 (Fig. 5E, 5F) and HPNE-KRASG12D xenografts in mice (Fig 5G, 5H), as measured by relative tumor volume and final tumor weight. Importantly, the tested doses of ulixertinib and GDC-0941 were well-tolerated as mice treated with this combination had normal appearance and body weight during the treatment period (Suppl. Fig. 4B). The anti-tumor effect of this combination appeared to be driven predominantly through induction of apoptosis, as we did not observe significant difference in the number of Ki-67+ neoplastic cells in tumors treated across four arms (Suppl. Fig. 4C, 4D). Histologic analyses showed GDC-0941 and ulixertinib co-treatment markedly disrupted the compact tumor architecture typical of MIA Paca-2 tumors (Fig. 5I, top panels). Immunohistochemical analyses showed almost complete abrogation of p-AKT staining in GDC-0941-treated tumors, whereas p-ERK staining was unaffected (Fig. 5I, middle and lower panels). Overall, these results support ulixertinib plus GDC-0941 as another promising therapeutic combination worthy of consideration for future clinical studies.
Figure 5. Concurrent Inhibition of PI3K potentiates the anti-tumor effect of ulixertinib.
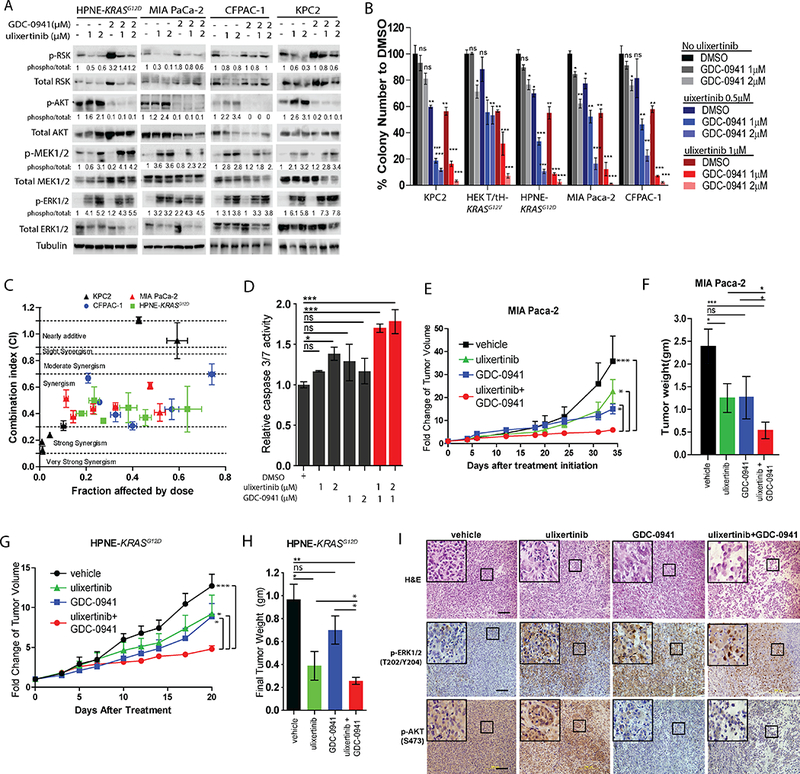
A, Western blots showing the effect of ulixertinib, GDC-0941 or both on p-RSK, p-MEK, p-ERK and p-AKT levels in four PDAC lines after overnight treatment. B, Relative quantification of soft agar colonies formed by PDAC lines treated as indicated. Experiments were done two to three times in triplicates, and one set of data was presented. C, Median effect analyses of afatinib plus GDC-0941 in four PDAC lines as represented by CI at six different fixed-ratio concentrations (all in μM) of ulixertinib: GDC-0941 (10:5, 5:2.5, 2.5:1.25, 1.25:0.625; 0.625:0.31, 0.31:0.15). One of three sets of experiments, each done in triplicates, is presented. D, Caspase 3/7 reporter assay showing the pro-apoptotic effect of ulixertinib and/or GDC-0941 in MIA Paca-2 cells. One of three sets of experiments each done in triplicates is presented. E, Serial measurements of relative tumor volume (to initial) and F, final weight of MIA Paca-2 tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=10/group). G, Serial measurements of relative tumor volumes (to initial) and H, final weight of HPNE-KRASG12D tumors grown subcutaneously in nude mice treated as indicated when tumors reached ~100mm3 (N=8/group). I, H&E (upper panels), p-ERK and p-AKT IHC (middle and lower panels) staining showing the effect of drug treatments on MIA Paca-2 tumors. (*p<0.05, **p<0.01, ***p<0.001, scale bars = 100μM).
DISCUSSION
Molecularly-targeted and immune-based therapies remain largely unsuccessful in PDAC. As a cancer type that is universally driven by oncogenic KRAS, targeting KRAS and/or its key effector pathways continues to be the most heavily pursued strategy with the highest likelihood to eventually improve patient outcome. The MAPK cascade is an established therapeutic target in PDAC (4, 28), as supported by decades of preclinical evidence. However, clinical success of RAF and MEK inhibitors in BRAF-mutant melanomas has so far failed to be recapitulated in PDAC. For example, sorafenib did not improve the effect of chemotherapy in patients with advanced or metastatic PDAC (29, 30). Use of the MEK inhibitor pimasertib also did not improve the effect of gemcitabine in treatment-naïve PDAC patients (31). These challenges strongly suggest context-dependent differences in the nature and scope of MAPK signaling circuitries driven by oncogenic RAF versus RAS. For instance, NRAS-mutant melanoma cells readily circumvent pharmacologic BRAF inhibition by engaging CRAF to reactivate ERK (32–34). These important observations strongly support targeting ERK as a promising therapeutic approach for RAS-driven cancers (35). Indeed, ulixertinib is the first ERK inhibitor to have shown treatment response in NRAS mutant melanomas in an early phase clinical trial (10), providing strong rationale for testing its efficacy in PDAC.
Ulixertinib is a highly potent, selective, reversible, ATP-competitive ERK 1/2 inhibitor with promising efficacy in BRAF- and RAS-mutant melanoma models (9). Notably, ulixertinib recently demonstrated promising anti-tumor activity in patients with various tumor types and mutations in either BRAF or NRAS, including patients that had progressed on BRAF and/or MEK inhibitors (10). In this study, we have demonstrated that ulixertinib potently suppresses the growth of KRAS and BRAF-mutant PDAC cells in vitro and synergizes with gemcitabine in attenuating PDAC growth in vitro and in vivo. However, we did not observe dramatic regression of MIA Paca-2 tumors treated with gemcitabine and ulixertinib, indicating a need to develop more potent combinatorial regimen. Addition of another cytotoxic agent such as nab-paclitaxel or targeted agents such as pan-HER inhibitors and PI3K inhibitors, as we proposed, are reasonable options. With the advent of immunotherapy, combinations of MAPK pathway inhibitors with immune checkpoint blockade are already being reported (36), and the utility of ERK inhibitor in this direction will surely need to be explored. Nonetheless, our results provide scientific rationale for the testing of ulixertinib in combination with gemcitabine and nab-paclitaxel in clinical trial, which is now being conducted (NCT02608229).
Strikingly, although ulixertinib potently suppresses downstream events including RSK phosphorylation and SRE reporter activity, treatment with ulixertinib increased levels of phosphorylated ERK, MEK, and AKT proteins in both KRAS and BRAF-mutant PDAC lines. We showed that ulixertinib-dependent AKT phosphorylation can be completely blocked by pharmacologic inhibition of the HER family members, which are themselves phosphorylated following ulixertinib treatment. Overexpression of HER family members is very common in PDAC (37–39), and contribute to various malignant phenotypes of PDAC. Studies in genetic mouse models showed that EGFR is required for KRAS-induced pancreatic tumorigenesis (24, 25). In another genetic mouse model with inducible KRAS expression, MEK1 inhibition results in sustained activation of HER1/2 and the PI3K-AKT-mTOR pathway (40), and MEK inhibitor trametinib synergizes with the HER1/2 inhibitor lapatinib in suppressing growth of patient-derived PDAC tumors (41). These observations demonstrated that PDAC cells are readily equipped with malleable and redundant mechanisms to uphold MAPK signaling, further substantiating the critical role of this pathway in cancer growth and the need to devise combinatorial strategies to effectively and durably block this pathway. Our proposed combination of ulixertinib plus pan-HER inhibitor represents one promising strategy. Meanwhile, further mechanistic work is needed to delineate how these HER family members are activated following ERK inhibition.
Although we showed that pan-HER inhibitors could completely block ulixertinib-associated AKT phosphorylation, phosphorylation of ERK and MEK were only partially attenuated. The ERK and MEK kinases are subject to multiple negative feedback mechanisms (42). Interestingly, our unbiased RPPA assay showed significant reduction in the protein level of DUSP4 (dual specificity phosphatase 4). The DUSP family members serve as negative regulators that help modulate the magnitude and duration of MAPK signaling in a spatio-temporal manner, mainly by dephosphorylating ERK and MEK (43). Genomic loss of DUSP4 occurs during PDAC progression in human samples, and re-expression of DUSP4 in PDAC lines suppresses PDAC growth and metastasis through inhibition of ERK (44). Therefore, it is plausible that abrogating the mechanism that downregulates DUSP4, thereby sustaining its expression, will block ERK phosphorylation to a greater extent. Because only DUSP4 protein was tested in our RPPA panel, whether other DUSP family members were affected is unclear and should be delineated.
In addition to the MAPK cascade, the parallel PI3K-AKT-mTOR cascade is another critical pathway in PDAC growth and should be co-targeted (12, 45–48). Unfortunately, combined use of MEK (selumetinib) and AKT (MK-2206) inhibitors did not improve overall survival compared to standard chemotherapy in gemcitabine-resistant PDAC patients (49). However, this study was limited by excessive clinical toxicities from combined selumetinib and MK-2206, which resulted in treatment interruptions and dose reductions, as well as a lack of pharmacodynamic analysis to inform on-target effects. Although our data supports combining ulixertinib with PI3K inhibitors, and that mice treated with this combination showed no signs of toxicity during the treatment period, toxicities remain a concern in the clinic, especially when these agents are commonly administered at maximum-tolerated doses over an extended period. It will be critical to include pharmacodynamic markers to inform on-target effects in future clinical trial design. Incorporation of cytotoxic agents with non-overlapping side effects may also lower the required dose of each agent, and hence overall toxicities, and should be explored in preclinical studies.
In summary, our study is the first to demonstrate the anti-tumor effects of ulixertinib in preclinical PDAC models in combination with chemotherapy as well as targeted agents directed at two escape pathways, which PDAC cells may utilize circumvent pharmacologic inhibition of ERK. Further work is needed to elucidate how these mechanisms can be overcome to improve treatment response.
Supplementary Material
ACKNOWLEDGMENTS
K.H.Lim is supported by 2016 Pancreatic Cancer Action Network-AACR Career Development Award, supported by Laurie MacCaskill, WUSTL Pancreatic Cancer SPORE (NIH/NCI grant 1P50CA196510–01A1), Siteman Cancer Center and The Foundation for Barnes-Jewish Hospital, Concern Foundation Conquer Cancer Now Award, the Washington University Institute of Clinical and Translational Sciences grant (UL1 TR000448 from the NCATS/NIH). A.Wang-Gillam is supported WUSTL Pancreatic Cancer SPORE (NIH/NCI grant 1P50CA196510–01A1), Siteman Cancer Center and The Foundation for Barnes-Jewish Hospital. Q.Li is supported by the National Natural Science Foundation of China (grant 81401735). We thank the WUSTL Digestive Disease Research Core Center (grant P30DK052574 from the NIDDK) for assistance in tissue sections and staining. The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
Footnotes
The authors have declared no conflicts of interest
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Teague A, Lim KH, Wang-Gillam A. Advanced pancreatic adenocarcinoma: a review of current treatment strategies and developing therapies. Ther Adv Med Oncol. 2015;7(2):68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers (Basel). 2016;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer discovery. 2012;2(8):685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nature medicine. 2013;19(11):1401–9. [DOI] [PubMed] [Google Scholar]
- 6.Salama AK, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(16):4326–34. [DOI] [PubMed] [Google Scholar]
- 7.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer cell. 2016;29(1):75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nissan MH, Rosen N, Solit DB. ERK pathway inhibitors: how low should we go? Cancer discovery. 2013;3(7):719–21. [DOI] [PubMed] [Google Scholar]
- 9.Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ, et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (ulixertinib). Molecular cancer therapeutics. 2017. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer discovery. 2018;8(2):184–95. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8(5):381–92. [DOI] [PubMed] [Google Scholar]
- 13.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–32. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D, Li L, Jiang H, Knolhoff BL, Lockhart AC, Wang-Gillam A, et al. Constitutive IRAK4 Activation Underlies Poor Prognosis and Chemoresistance in Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017;23(7):1748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. [DOI] [PubMed] [Google Scholar]
- 17.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39(4):425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyabayashi K, Ijichi H, Mohri D, Tada M, Yamamoto K, Asaoka Y, et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res. 2013;73(7):2221–34. [DOI] [PubMed] [Google Scholar]
- 19.Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007;67(5):2098–106. [DOI] [PubMed] [Google Scholar]
- 20.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer discovery. 2013;3(7):742–50. [DOI] [PubMed] [Google Scholar]
- 21.Blake JF, Burkard M, Chan J, Chen H, Chou KJ, Diaz D, et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-y l)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J Med Chem. 2016;59(12):5650–60. [DOI] [PubMed] [Google Scholar]
- 22.Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, et al. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14(5):951–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ioannou N, Seddon AM, Dalgleish A, Mackintosh D, Modjtahedi H. Expression pattern and targeting of HER family members and IGF-IR in pancreatic cancer. Front Biosci (Landmark Ed). 2012;17:2698–724. [DOI] [PubMed] [Google Scholar]
- 24.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22(3):304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6. [DOI] [PubMed] [Google Scholar]
- 27.Kohler J Second-Line Treatment of NSCLC-The Pan-ErbB Inhibitor Afatinib in Times of Shifting Paradigms. Front Med (Lausanne). 2017;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. [DOI] [PubMed] [Google Scholar]
- 29.Krege S, Rexer H, vom Dorp F, de Geeter P, Klotz T, Retz M, et al. Prospective randomized double-blind multicentre phase II study comparing gemcitabine and cisplatin plus sorafenib chemotherapy with gemcitabine and cisplatin plus placebo in locally advanced and/or metastasized urothelial cancer: SUSE (AUO-AB 31/05). BJU Int. 2014;113(3):429–36. [DOI] [PubMed] [Google Scholar]
- 30.Cascinu S, Berardi R, Sobrero A, Bidoli P, Labianca R, Siena S, et al. Sorafenib does not improve efficacy of chemotherapy in advanced pancreatic cancer: A GISCAD randomized phase II study. Dig Liver Dis. 2014;46(2):182–6. [DOI] [PubMed] [Google Scholar]
- 31.Van Cutsem E, Hidalgo M, Bazin I, Canon J-L, Poddubskaya E, Manojlovic N, et al. Phase II randomized trial of MEK inhibitor pimasertib or placebo combined with gemcitabine in the first-line treatment of metastatic pancreatic cancer. Journal of Clinical Oncology. 2015;33(3_suppl):344-. [Google Scholar]
- 32.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5. [DOI] [PubMed] [Google Scholar]
- 34.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan MB, Der CJ, Wang-Gillam A, Cox AD. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1(3):183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity. 2016;44(3):609–21. [DOI] [PubMed] [Google Scholar]
- 37.Lemoine NR, Lobresco M, Leung H, Barton C, Hughes CM, Prigent SA, et al. The erbB-3 gene in human pancreatic cancer. J Pathol. 1992;168(3):269–73. [DOI] [PubMed] [Google Scholar]
- 38.Lemoine NR, Hughes CM, Barton CM, Poulsom R, Jeffery RE, Kloppel G, et al. The epidermal growth factor receptor in human pancreatic cancer. J Pathol. 1992;166(1):7–12. [DOI] [PubMed] [Google Scholar]
- 39.Kimura K, Sawada T, Komatsu M, Inoue M, Muguruma K, Nishihara T, et al. Antitumor effect of trastuzumab for pancreatic cancer with high HER-2 expression and enhancement of effect by combined therapy with gemcitabine. Clin Cancer Res. 2006;12(16):4925–32. [DOI] [PubMed] [Google Scholar]
- 40.Pettazzoni P, Viale A, Shah P, Carugo A, Ying H, Wang H, et al. Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res. 2015;75(6):1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, et al. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013;15(2):143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73(23):4397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280(2):489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hijiya N, Tsukamoto Y, Nakada C, Tung Nguyen L, Kai T, Matsuura K, et al. Genomic Loss of DUSP4 Contributes to the Progression of Intraepithelial Neoplasm of Pancreas to Invasive Carcinoma. Cancer Res. 2016;76(9):2612–25. [DOI] [PubMed] [Google Scholar]
- 45.Baer R, Cintas C, Dufresne M, Cassant-Sourdy S, Schonhuber N, Planque L, et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev. 2014;28(23):2621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21(4):739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holt SV, Logie A, Davies BR, Alferez D, Runswick S, Fenton S, et al. Enhanced apoptosis and tumor growth suppression elicited by combination of MEK (selumetinib) and mTOR kinase inhibitors (AZD8055). Cancer Res. 2012;72(7):1804–13. [DOI] [PubMed] [Google Scholar]
- 48.Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, et al. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PLoS One. 2012;7(8):e44146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017;3(4):516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.