

Abstract
Oncogenic transformation of hematopoietic stem cells by chimeric fusion kinases causing constitutive activation of FGFR1 leads to a stem cell leukemia/lymphoma (SCLL) syndrome, accompanied by widespread dysregulation of gene activity. We now show that FGFR1 activation is associated with upregulation of MYC and pharmacological suppression of FGFR1 activation leads to downregulation of MYC and suppression of MYC target genes. Luciferase reporter assays demonstrate FGFR1 can directly regulate MYC expression and this effect is enhanced in the presence of chimeric FGFR1 kinases. In SCLL cells, a truncated form of FGFR1 is generated by granzyme B cleavage of the chimeric kinases, producing a nucleus-restricted derivative that can bind MYC regulatory regions. Mutation of the granzyme B cleavage site prevents relocation to the nucleus but does not suppress MYC activation, suggesting additional mechanisms of MYC activation in the presence of cytoplasm-restricted chimeric kinases. We show one of these mechanisms involves activating cytoplasmic STAT5, which upregulates MYC independent of the truncated FGFR1 kinase. Targeting MYC function using shRNA knockdown and 10054-F8 in SCLL cells leads to inhibition of cell proliferation and synergizes with the BGJ398 FGFR1 inhibitor, suggesting a combination therapy that could be used in the treatment of SCLL.
Introduction
Constitutive activation of FGFR1 kinase in hematopoietic stem cells following chromosome translocations, leads to the development of an atypical myeloproliferative disease that frequently progresses to AML and often either a B- or T-cell lymphoma in the same patients depending on the fusion partner gene.1 Available evidence suggests, however, that the constitutive activation of FGFR1 kinase in hematopoietic cells is, in itself, perhaps not sufficient to cause malignant transformation but rather expands the stem cell pool,2 increasing the pool of potential tumor precursor cells. FGFR1 fusion kinases, expressed exogenously in murine stem cells, lead to the development of an SCLL phenotype closely resembling the human disease.3-5 Acquisition of additional genetic changes are thought to be required for fully transformed phenotypes. During the genetic analyses of leukemic cell types associated with the development of human and mouse SCLL, we demonstrated increased expression of the MYC oncogene whether the outcome was AML, T- or B-cell lymphoma,4,6-8 suggesting that the constitutive expression of FGFR1 may directly influence MYC gene expression, which has profound consequences for hematopoiesis.
The MYC gene family members have similar functions but operate in a cell dependent context.9 MYC plays a central role in promoting early myeloid and lymphoid development with expression levels increasing as cells progress to multipotent progenitors.10 The carefully orchestrated role of MYC in hematopoiesis is disrupted during leukemogenesis, usually though over expression. MYC, as a dimer with MAX, is a transcription factor that promotes both upregulation and down regulation of a wide range of target genes, presumably contributing to tumor development and progression.9 It is unclear, however, whether dysregulation of MYC alone can initiate leukemogenesis11 and, as with overexpression of chimeric FGFR1 kinases, additional genetic factors may be required. Here we investigated the interrelationship between expression of MYC and chimeric FGFR1 kinases during SCLL development and demonstrate that FGFR1 directly activates MYC, leading to upregulation of its target genes. Further, we demonstrated that while a truncated form of FGFR1 is responsible for MYC activation, interactions between the full-length fusion kinases and cytoplasm proteins, specifically STAT5, also contribute to the upregulation of MYC and the oncogenic process.
Materials and methods
Reagents
Drugs used in these studies were; BGJ398, 10058-F4 and SH-4-54 (Selleckchem, Houston, TX) and Ponatinib (AP24534) from Ariad pharmaceuticals.
Cell and Molecular analyses
Cell cycle and apoptosis analysis was performed as previously described.12 Quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blotting assays were carried out as described previously.4 Detection of FGFR1 fusion kinase and its variants, used several different anti-FGFR1 antibodies designed against the human FGFR1 sequence; #9740 (Cell signaling, Inc) was raised against a recombinant protein specific to the FGFR1 carboxy terminus, ab76464 (Abcam) was raised against a synthetic peptide corresponding to aa 800 to the C-terminus and ab58516 (Abcam) was raised against a synthetic non-phosphopeptide derived from human FGFR1 around the phosphorylation site of tyrosine 654 (D-Y-YP-K-K). An anti-Myc tag antibody (#05-724, EMD Millipore) was also used to detect exogenous Myc-tagged BCR-FGFR1 fusion protein. ChIP-qPCR was performed with an FGFR1 antibody against total FGFR1 (#9740, Cell signaling) and truncated FGFR1 (ab58516, Abcam), as described previously.13,14 Mutation or truncation of BCR-FGFR1 was generated using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs).
Luciferase reporter assay
HEK293T cells were seeded at 105 cells/500 μl in each well of a 24-well plate and cultured overnight. 400 ng of the HBM-Luc plasmid (Addgene #35155) which carries the ~2KB upstream of the MYC transcription initiation site15 was co-transfected into HEK293T cells with the previously described chimeric FGFR1 kinases3-5, 8 driven by the MSCV promoter, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Cells were harvested 48 h after transfection and analyzed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). Renilla luciferase was used to normalize for transfection efficiency, and the ratio of firefly/Renilla luciferase activities defined relative promoter activity.
Virus production and transduction
Retrovirus was produced by transfecting the Phoenix Amphotropic cell line with retroviral expression vectors pMIG3, and lentivirus was produced by transfecting 293FT cells with pLKO.1, pLP1, pLP2 and pVSVG vectors using lipofectamine 2000 (Invitrogen, Carlsbad, CA). Virus was harvested and target cells were infected with RetroNectin reagent (Clontech, Mountain View, CA). After 2 days, transduced cells were challenged with 1 μg/ml puromycin to generate stable cell lines.
Protein subcellular localization assay
Cytoplasmic and nuclear components were isolated using the NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific). For confocal microscopy, the cytoplasmic and nuclear FGFR1 were first probed with an anti-Myc tag antibody (#2272, Cell Signaling), then visualized with APC goat anti-rabbit IgG secondary antibody (Product # A-10931, Invitrogen). Nuclei were stained with DAPI.
RNA-Seq and analysis
Preparation of RNA and sequence analysis was performed as described previously.16 Data deposited in GEO database accession number GSE110457.
Results
MYC overexpression occurs in FGFR1-driven leukemia/lymphoma in mouse models
Analysis of MYC expression in primary T-cell lymphomas and myeloid leukemias arising in various SCLL mouse models we have developed3,4,17 shows both mRNA and protein levels were consistently upregulated (Figure 1). Through our development of mouse models for leukemogenesis driven by various chimeric FGFR1 kinases, we previously isolated a series of cell lines that expressed different chimeric FGFR1 fusion genes; ZNF112 expresses ZMYM2-FGFR1,3 BBC1/2 express BCR-FGFR117 and CEP2A and CEP5A express CNTRL-FGFR1.4 To determine whether MYC expression levels correlate with FGFR1 fusion kinase expression in human cells, we included the human KG1 myeloid leukemia cell line, which expresses a chimeric FGFR1OP2-FGFR1 gene. MYC protein levels in all of these cell lines is also remarkably increased (Figure 1C) compared with healthy human peripheral mononuclear cells (PBMC). Taken together, these results indicate that high expression of MYC is associated with constitutive expression of chimeric FGFR1 kinases.
Figure 1. Overexpression of MYC in cells transformed with chimeric FGFR1 kinases in mouse models.
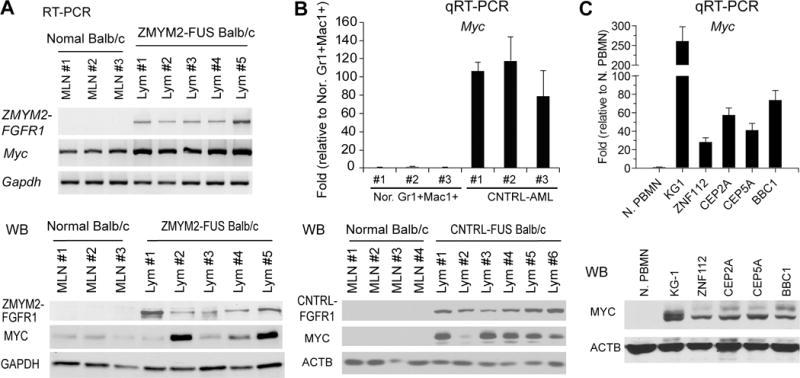
(A) RT-PCR (above) and western blot (below) analysis for T-lymphomas from five independently developed founders from the ZMYM2-FGFR1 mouse model, shows upregulation of MYC expression compared with cells from normal mesenteric lymph nodes (MLN). In the CNTRL-FGFR1 model (B), enhanced MYC expression levels (above) and protein levels (below), were seen in AML cells compared with normal Mac1+Gr1+ myeloid cells obtained from spleen of bone marrow as well as MLN from normal BALB/c mice. MYC was also upregulated (C) in a series of human and murine cell lines transformed with the various FGFR1 fusion kinases (see text).
MYC is overexpressed in human CD34+ derived leukemia/lymphoma induced by FGFR1 fusion kinases
Because human leukemia/lymphomas associated with FGFR1 rearrangement is rare, we have developed several humanized mouse models engrafted with the FGFR1 fusion kinase-transduced human CD34+ progenitor cells in immunocompromised mice as described previously.4,7,8 CD34+ progenitor cells transduced with either ZMYM2-FGFR1,7 CNTRL-FGFR1,4 or BCR-FGFR1,8 developed acute myeloid leukemia (AML) with a human CD45+CD13+CD34+/CD38+ immunophenotype. Quantitative RT-PCR analysis showed that MYC is highly transcribed in cells from mouse spleens from all three models compared with either control pMIG3 transduced mouse splenocytes or healthy human PBMC (Figure 2). Furthermore, PBMC from one patient with the CNTRL-FGFR1 rearrangement and two patients with the ZMYM2-FGFR1 rearrangement also showed higher expression of MYC compared with healthy human PBMC (Figure 2B). Consistently, MYC protein levels were also highly expressed in these human leukemic cells compared with their normal counterparts (Figure 2). Thus, upregulation of MYC is also present in human cell leukemias carrying chimeric FGFR1 kinases.
Figure 2. Upregulation of MYC in human cell-derived SCLL models.
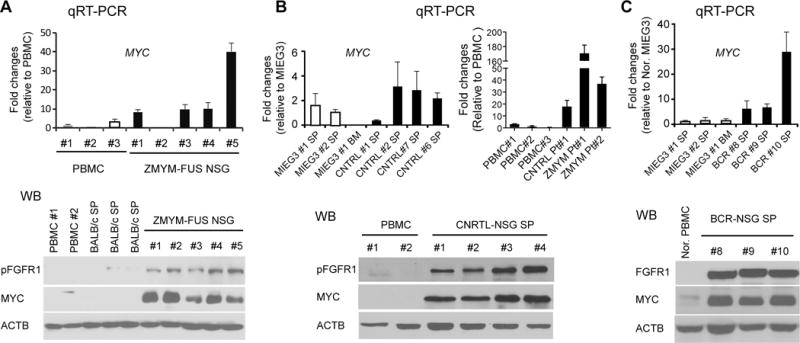
Quantitative RT-PCR analysis (A) of MYC mRNA expression in PBMC cells from mice that developed AML following transformation of human CD34+ cord blood cells with ZMYM-FGFR1 shows upregulation of MYC. Analysis of spleen cells from NSG mice transplanted with human CD34+ cells transformed with CNTRL-FGFR1 (B, left) also shows upregulation of MYC compared with empty vector control transfected spleen (SP) and bone marrow (BM) cells (MIEG3). MYC is also upregulated in primary bone marrow cells from an SCLL patient (B, right) with CNTRL-FGFR1 and two patients with ZMYM2-FGFR1. Upregulation of MYC is also seen in spleen cells in three representative NSG founder mice from the human cell model of BCR-FGFR1 disease (C). In all three models, upregulation of MYC protein levels further confirmed the mRNA expression data (lower panel in each case).
MYC expression levels correlate with activated FGFR1 expression levels
When starved, FGFR1-expressing H520 human lung cancer cells were treated with either mouse or human FGF1 or FGF2 ligands, increased FGFR1 protein levels were seen (Figure 3A), which correlated with increased MYC protein levels. Stably overexpressing FGFR1 in mouse NIH-3T3 and BaF3 cells, or human M07e leukemia cells, resulted in increased MYC expression (Figure 3A) either directly or as a consequence of FGF-induced increased proliferation. Conversely, SCLL cell lines (KG1, BBC1/2 and CEP2A) treated with FGFR1 inhibitors (either BGJ398 or ponatinib), showed reduced MYC expression (Figure 3B). As a result, impaired cell cycle progression was demonstrated by upregulation of p21 and the proportion of cells in S/G2/M was reduced (Supplementary Figure S1). After withdrawal of ponatinib, FGFR1 and MYC levels returned to normal (Figure 3B), suggesting that MYC expression in SCLL cells may be regulated by both FGFR1 fusion kinases and ligand activated, wild-type FGFR1. When HEK293 cells were co-transfected with a MYC promoter-luciferase reporter construct HBM-Luc15 and four different fusion kinase constructs, increased luciferase activity was seen (Figure 3C) compared with wild type FGFR1.
Figure 3. MYC expression correlates with FGFR1 and chimeric FGFR1 expression levels.
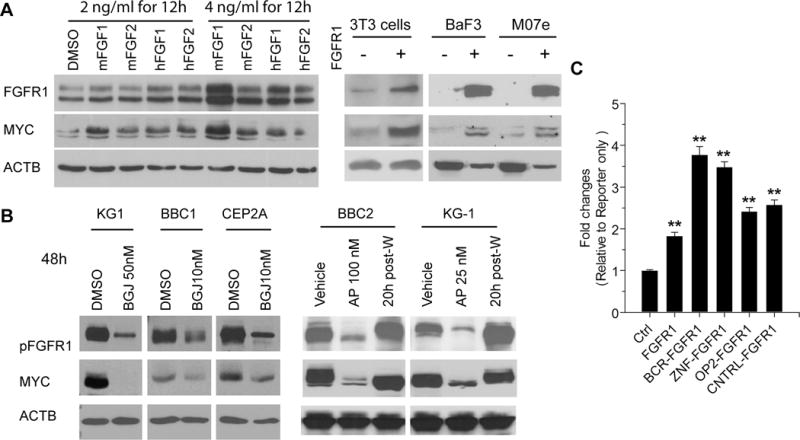
A520 cells, which overexpress FGFR1, were treated with mouse and human FGF1 and FGF2, at two concentrations for 12 hours. In each case, upregulation of FGFR1 is seen, which correlated with increased MYC expression (A, left). Forced expression of wild type FGFR1 in mouse 3T3 or BaF3 cells or human M07e cells leads to increased MYC levels (A, right). Treatment of human (KG1) and mouse (BBC1 and CEP2A) leukemia cells with the BGJ398 FGFR kinase inhibitor for 48 hours at the IC50 dose leads to downregulation of MYC (B, left). Treatment of BBC2 and KG1 cells with the ponatinib (AP) FGFR1 inhibitor shows similar down regulation of activated FGFR1 and MYC (B, right). When this drug was washed out (post-W) and the cells re-assayed 20 hours later, FGFR1 and MYC levels recover proportionately. Luciferase assays (C) using a MYC promoter construct shows that co-transfection with wild type FGFR1 leads to increased luciferase activity which are further increased in cells co-transfected with four different FGFR1 chimeric kinases compared with cells co-transfected with the pcDNA3.1 control (Ctrl). ** = p < 0.001.
FGFR1 fusion kinases can be cleaved by granzyme B to generate a truncated, nuclear FGFR1
In breast cancer cells, wild-type FGFR1 can be cleaved by granzyme B leading to nuclear translocation of the truncated kinase.18,19 To detect whether the truncated FGFR1 is present in fusion kinase disease model, different anti-FGFR1 and anti-Myc tag antibodies were evaluated for their ability to distinguish between the nuclear and cytoplasmic forms of FGFR1 (Supplementary Figure 2). Only FGFR1 antibody ab58516, designed against the region containing the Y654 phosphorylatable site showed specificity for the truncated nuclear FGFR1. Using antibodies described in the materials and methods, ZMYM2-FGFR1 (present in ZNF112) was exclusively located in the nucleus, the 65kD FGFR1OP2-FGFR1 (present in KG1 cells) was present in nucleus and cytoplasm, and BCR-FGFR1 is predominantly located in the cytoplasm (Figure 4A). The cytoplasmic/nuclear location of the BCR-FGFR1 chimeric kinase was also seen in the recently derived, BCRF8C murine BCR-FGFR1-expressing myeloid cell line.16 In all three cases, the fusion kinase is cleaved and the truncated form is present exclusively in the nucleus. When the granzyme B site, which is retained in all chimeric FGFR1 transcripts (Figure 4B), was mutated (D432N) and this BCR-FGFR1 derivative was stably expressed in 3T3 cells, the truncated 55kD FGFR1 was lost and nuclear localization was absent (Figure 4B), and the mutated fusion kinase was retained in the cytoplasm. Immunofluorescence analysis (Figure 4C) using a MYC-tag that was present in all 3 constructs confirmed this distribution pattern. In primary SCLL models (Supplementary Figure S2), consistent with the in vitro studies, nFGFR1 levels were relatively low for BCR-FGFR1 and relatively high for ZMYM2-FGFR1.
Figure 4. Cleavage and subcellular localization of the chimeric FGFR1 kinases.
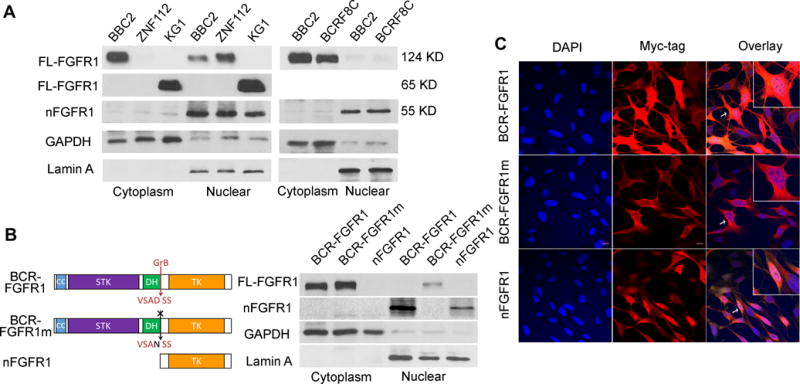
Cell fractionation studies using SCLL cell lines (A) show the relative locations of the various chimeric kinases between the cytoplasm and nucleus. The relatively pure isolation of both cell compartments is shown by the presence of cytoplasmic (GAPDH) and nuclear (LaminA) markers. In the three examples, the 55 kD truncated derivative (nFGFR1) can be seen exclusively in the nucleus. Similarly, the presence of the nuclear truncated form is seen in the AML-derived BCRF8C cell line compared with murine BBC2, both expressing BCR-FGFR1. Schematic representation of BCR-FGFR1 showing the coiled coil domain (CC), the serine/threonine kinase domain (STK), the Dbl homology domain (DH) and the FGFR1 tyrosine kinase domain (TK). The arrow indicates granzyme B cleavage site (GrB) at Asp432 of FGFR1 (B, left). The absence of the truncated nuclear variant in transfected 3T3 cells following mutation of the GrB cleavage site (BCR-FGFR1m) is seen compared with the parent chimeric kinase (B, right). The presence of the exogenous truncated form of FGFR1 is shown exclusively in the nucleus. Immunofluorescence analysis of the three variant kinases further demonstrated the cytoplasmic location of the full length BCR-FGFR1 chimeric kinases as well as exclusive nuclear presence of the truncated form in the NIH 3T3 cells (C). Insets represent the cell indicated by the arrow at higher magnification.
Both cytoplasm and nuclear FGFR1 kinases lead to upregulation of MYC
Overexpression of both BCR-FGFR1 and nFGFR1 led to upregulation of MYC (Figure 5A) in HEK293, NIH3T3 and BaF3 model cell systems. Since the mutant BCR-FGFR1 protein does not enter the nucleus, activation of MYC in this case must be indirect. STAT proteins, which are activated by chimeric FGFR1 kinases21,26, can also activate MYC20. Activated STAT5 levels are high in cells expressing chimeric kinases and are dependent on FGFR1 activation (Supplementary Figure S3A). Inhibition of SCLL cell lines with the SH-4-54 STAT inhibitor reduces viability in a dose-dependent manner (Supplementary Figure S3B). When BaF3 cells expressing the mutated (D432N) BCR-FGFR1 were treated with the SH-4-54 STAT inhibitor (Figure 5B), MYC protein levels were inhibited in a dose dependent manner and mRNA levels were reduced (Supplementary figure S3C). Similarly, treatment of ZNF112 cells also shows reduced Myc expression (Supplementary figure S3D). Independently, knockdown of STAT5, using two different shRNAs also leads to reduced Myc expression (Figure 5B, Supplementary figure S3C).
Figure 5. Dual effect of FGFR1 fusion kinase activation of MYC expression.
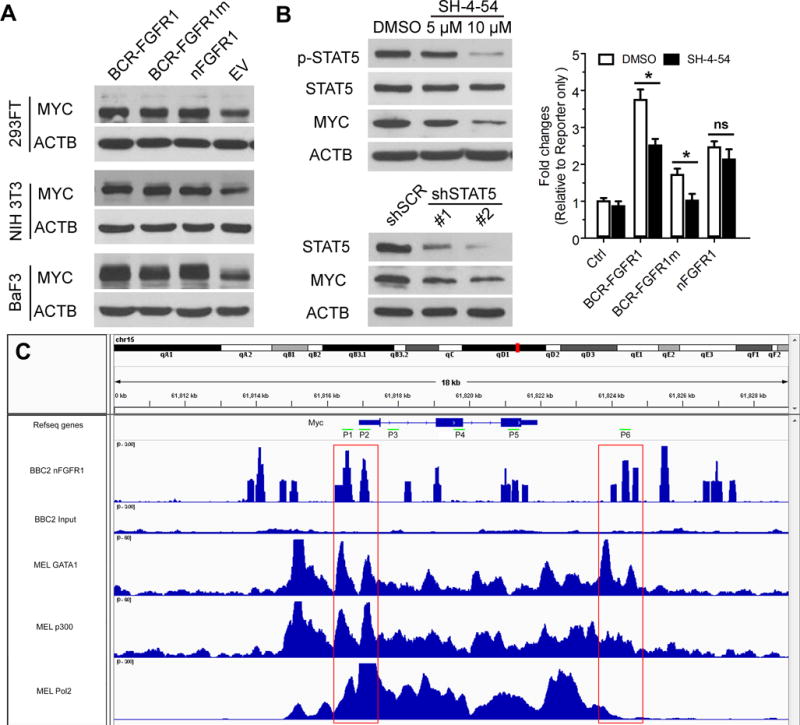
(A) Introduction of chimeric kinase (BCR-FGFR1), truncated kinase (nFGFR1) or Asp432 mutant chimeric kinase (BCR-FGFR1m) (A) shows an increase in MYC expression in three model cell systems compared with cells expressing the empty vector (EV). When STAT5 activation is suppressed using SH-4-54 for 24 hours, there is a dosage dependent inhibition of STAT5 phosphorylation (B, above), which is correlated with down regulation of MYC. ShRNA knockdown of STAT5 (B, below) shows a similar relationship between MYC and STAT5. Luciferase reporter assays (B, right) show that when cells transfected with the normal or mutant chimeric kinases are treated with the STAT5 inhibitor (SH-4-54), there is a significant reduction in MYC activation. In contrast, there is no significant difference in MYC activation in cells transfected with the truncated FGFR1 kinase (nFGFR1). A ChIP-seq scan for the Myc locus (C) identifies the binding sites for nFGFR1 which overlap (red boxes) with the Gata1 and p300 transcription factors as well as Pol2 (data from mouse erythroid leukemia cell analysis; GEO accession numbers GSM912907, GSM912893 and GSM912895 respectively). * p<0.05; ns, not significant.
Using luciferase reporter assays, where the MYC promoter vector was cotransfected into HEK293 cells with the wild type or mutant FGFR1 kinases in the presence and absence of the STAT5 inhibitor, MYC activation was significantly prevented (Figure 5B) but there is no significant difference in MYC activation in the presence of the nuclear FGFR1 truncated kinase. These data support the suggestion that the cytoplasm-restricted fusion kinase activates proteins such as STAT5 which contributes to MYC activation in these cells, whereas the truncated FGFR1 kinase in the nucleus has a direct effect on MYC activation. From a genome wide ChIP-Seq study to identify nFGFR1 binding sites, a scan of the Myc locus demonstrated specific binding sites (Figure 5C). This data was used to define six locations (P1-P6) to perform ChIP-qPCR analysis, which confirmed nFGFR1 binding and demonstrated that only the truncated nuclear kinase showed occupancy on these regulatory sequences (Supplementary Figure 4). An extensive comparison of the nFGFR1 occupancy pattern with the TFBS ChIP-seq signal from ENCODE21 revealed that the nFGFR1 peak regions showed overlap with Gata1 and p300 binding sites as well as Pol2, indicating its involvement in transcription regulation.
Inhibition of the FGFR1 chimeric kinase decreases expression of MYC and its direct targets
Since MYC was down-regulated following FGFR1 inhibition, we investigated whether MYC target genes were also affected. RNASeq data from a single run from three different SCLL cell models (BBC2, ZNF112 and BCRF8C) expressing different FGFR1 chimeric kinases, which had been treated with BGJ398 was analysed. Hierarchical clustering of the 131 genes known to be direct MYC targets (systematic GSEA name M6506) showed their consistent downregulation following FGFR1 inhibition (Supplementary Figure S5 and Supplementary Table 1). To confirm these results, we performed an independent analysis of our RNASeq data using the Gene Set Enrichment Analysis algorithm, where the genes reported22 as upregulated in breast cancer cells overexpressing MYC (systematic GSEA name M2711), were also downregulated by BJG398 treatment in FGFR1 overexpressing cells (Figure 6A-B). Analysis of several randomly selected downregulated genes in the different SCLL cell lines using real-time RT-PCR analysis confirmed the RNASeq results (Figure 6C). Although RNASeq data was not available for human KG1 cells, a qPCR analysis of a series of randomly selected MYC target genes from Figure 6B were also shown to be downregulated following FGFR1 inhibition (Figure 6C).
Figure 6. Activation of MYC by FGFR1 kinases leads to upregulation of MYC target genes.
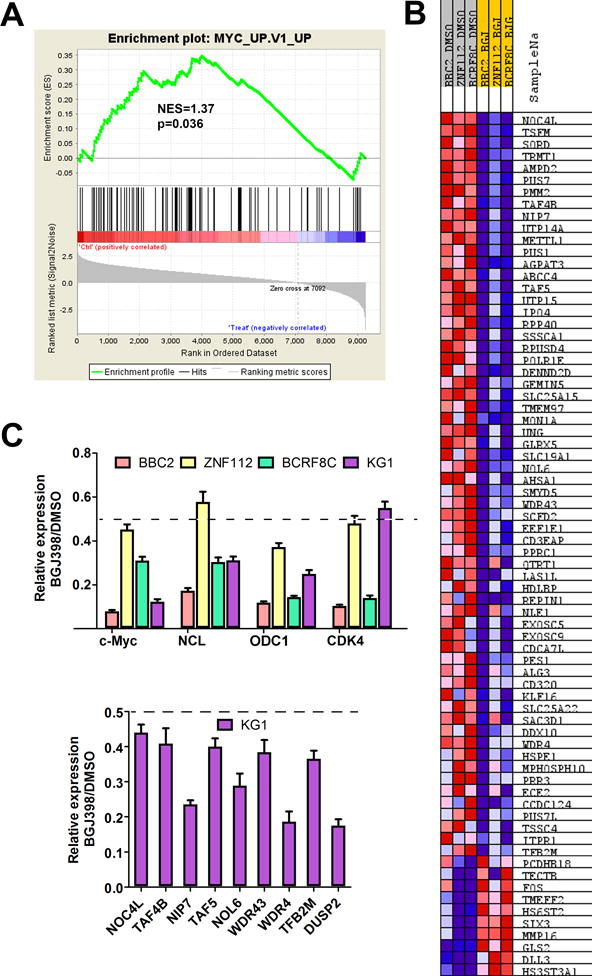
GSEA analysis of RNASeq data from BBC2, ZNF112 and BCRF8C following treatment with the BGJ398 FGFR1 inhibitor shows statistically significant (A) downregulation of MYC target genes. The majority of genes up-regulated upon MYC overexpression show reduced expression following BGJ398 treatment. The expression levels of MYC and three selected targets of MYC (NCL, ODC1 and CDK4), as determined by qPCR, shows down regulation (55-85%) following BGJ398 treatment in all 4 cell lines expressing the chimeric kinases (C, above). Focused analysis of a wider selection of MYC target genes in the human KG1 cell line (for which RNASeq data is not available) also shows consistent down regulation (C, below). 50% reduction in expression levels is shown by the broken lines in each case.
Suppression of MYC function in SCLL suppresses cell growth and induces apoptosis
The consistent activation of MYC in SCLL suggested a potential for treatment of this disease. MYC function requires interaction with MAX, and 10058-F4 disrupts this interaction.23 Using three different SCLL cell lines, 10058-F4 treatment effectively reduced cell viability in a dose dependent manner and caused cell cycle arrest and an increase in apoptosis (Figure 7A). More importantly, our observations indicate chimeric FGFR1 kinases can activate MYC both indirectly through activation of cell signaling pathways and directly by genomic occupancy of the truncated nuclear form. Since this difference could lead to different drug responses, BaF3 cell stably expressing the mutated (D432N) BCR-FGFR1 and the truncated nFGFR1 were treated with FGFR1 inhibitor BGJ398 and MYC inhibitor 10058-F4. Cells expressing nFGFR1 did not respond to BGJ398, while both derivatives responded to 10058-F4 (Supplementary Figure 6A). Using a combination of FGFR1/MYC inhibitors in the effect on cell viability and apoptosis was enhanced in SCLL cell lines compared with either drug alone (Figure 7B). Isobologram analysis demonstrated a synergistic effect at different combinations of drug concentration (Supplementary Figure 6). Consistent with the pharmacological studies, shRNA knockdown of MYC in KG1 cells also suppressed cell cycle progression and enhanced apoptosis (Figure 7C).
Figure 7. Synergistic effect of targeting FGFR1 and MYC function.
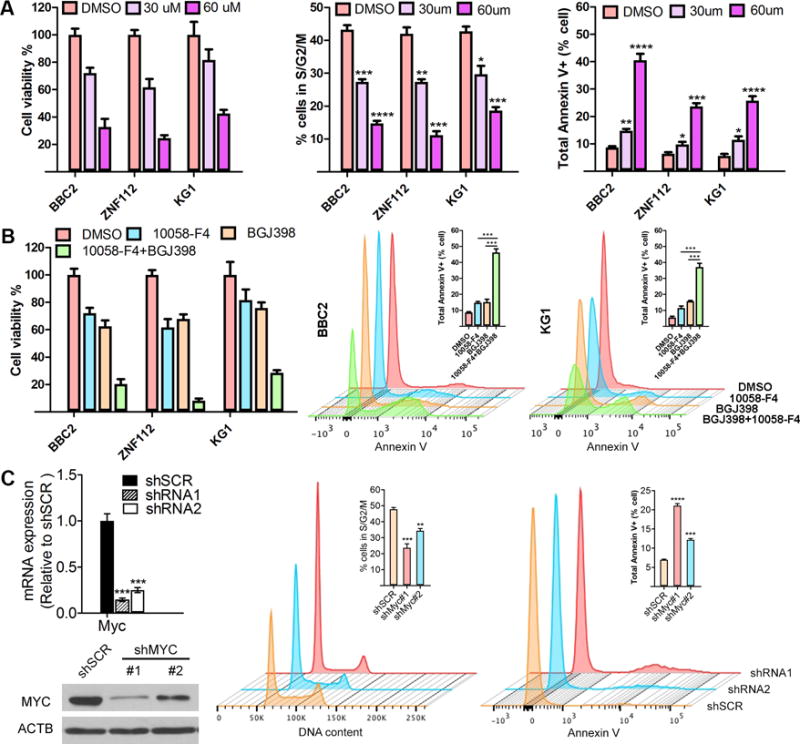
(A) Treatment of BBC2, ZNF112 and KG1 cells for 48 hours with the 10054-F4 MYC/MAX inhibitor shows a dose dependent reduction in cell viability. Cell cycle analysis using flow cytometry demonstrates a G0/G1 arrest following treatment with the MYC inhibitor and flow cytometric analysis using Annexin V antibodies shows a dose-dependent increase in apoptosis as a result of suppression of MYC function. A combined treatment with FGFR1 inhibitor BGJ398 and MYC inhibitor 10058-F4 in BBC2 and KG1 cells shows enhanced suppression of cell viability and significantly increased levels of apoptosis, compared with the single inhibitor alone (B). * p<0.05, ** p<0.001, *** p<0.0001, **** p<0.00001. When Myc is knocked down using 2 different shRNAs in KG1 cells, protein levels are reduced proportionately (C, left). As a result, the proportion of cells in S/G2/M is significantly reduced compared with a scrambled control and the proportion of apoptotic cells positive for Annexin-V is significantly increased (C, right).
Discussion
The molecular etiology of SCLL is complex, with a diverse reorganization of gene expression patterns promoting stemness and suppressing differentiation,4,16 supplemented by extensive changes in the micro RNA expression profile.12 The constitutive, ligand-independent activation of FGFR1 kinases in SCLL suggests two possible roles in regulation of molecular function of genes and proteins depending on intracellular location. A cytoplasmic location, for example, potentially phosphoactivates other cytoplasmic proteins and a nuclear location can either activate nuclear proteins or bind DNA and function as a transcription factor or co-factor. In addition, the specific partner genes in the chimeric kinases can potentially superimpose added complexity based on their intracellular location and the specific pattern of protein-protein interactions. Designing rational therapeutic strategies to treat this almost inevitably fatal disease, therefore, depends on identifying the more consistent changes that occur during its development. We now show that upregulation of MYC, as a direct consequence of FGFR1 activation is a consistent observation in SCLL, which highlights the broad influence of the chimeric kinases on dysregulation of gene expression profiles, since the large number of genes regulated by MYC also become dysregulated.
It has been demonstrated that the different chimeric kinases can have specific effects on the SCLL phenotype1 which in turn, may depend on the different subcellular localization of the fusion kinases. Here we show that the processing of the chimeric kinases by Granzyme B leads to a truncated FGFR1 derivative which, unlike the fusion kinases, can transcriptionally activate MYC and presumably other FGFR1 target genes. Activation of MYC, however, can occur in the absence of the truncated kinase through activation of alternative transcription factors such as STAT5, which are activated by FGFR1 in the cytoplasm and promote increased MYC expression levels. It has been shown that this regulation of MYC is due to STAT5 binding to a super enhancer upstream of the MYC promoter.24 Higher levels of MYC activation, therefore, are due to a combination of direct and indirect activation. By analogy, therefore, activation of other cytoplasmic proteins may also contribute to disease progression depending on the nature of the specific rearrangements. Indeed, it is known that FGFR1 can activate cytoplasmic proteins such as PLCΥ and SRC,24-26 which also contribute to the activation of genes through promoter engagement. While we have focused on STAT5 as an intermediate activator of MYC, STAT3 is also activated by FGFR1 in SCLL21,27-30 and STAT3 has also been shown to drive MYC expression.31 In addition, in T-lymphomas associated with SCLL, upregulation of Notch1 also leads to upregulation of MYC.6 It seems likely, therefore, that the high levels of MYC activation associated with expression of the fusion kinases is due to compound effects related to fusion kinase actions both in the cytoplasm and the nucleus cooperating to enhance their oncogenic capacity.
One of the consistent observations in both primary SCLL, and animal models of this variant disease, is a more aggressive development of the disease in the presence of the BCR-FGFR1 chimeric kinase. The BCR component carries a serine/threonine kinase, which may cause activation of other proteins that contribute to development of a more aggressive disease progression independent of the FGFR1 tyrosine kinase. This suggestion is supported by the demonstration that the full length BCR-FGFR1 kinase is found predominantly in the cytoplasm, implying activation of different cytoplasmic proteins in addition to the effects of the truncated form of the tyrosine kinase in the nucleus.
In summary, despite the suggestion that MYC can independently drive leukemogenesis in animal models,32 in SCLL its role is clearly dependent on the ability of FGFR1 kinase to regulate its activity. Since FGFR1 activates other proteins independently of MYC, the development of SCLL seems dependent on the functions of FGFR1 kinase. In this case, targeting FGFR1 appears to be the most rational approach for treating this disease,25,26 although the genomic study of mouse models is identifying downstream mechanisms driving SCLL. These observations are suggesting combination therapies that may prove more effective4,33 and, in particular, may address the issue of resistance to FGFR1 inhibitors resulting from single drug regimens.33
Supplementary Material
Acknowledgments
Grant support: This work was supported by grant CA076167 from the National Institutes of Health
Footnotes
Conflict of Interest statement: The authors declare no conflicts of interest associated with this work.
References
- 1.Jackson CC, Medeiros LJ, Miranda RN. 8p11 myeloproliferative syndrome: a review. Hum Pathol. 2010;41:461–476. doi: 10.1016/j.humpath.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Roumiantsev S, Krause DS, Neumann CA, Dimitri CA, Asiedu F, Cross NC, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5:287–298. doi: 10.1016/s1535-6108(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 3.Ren M, Li X, Cowell JK. Genetic fingerprinting of the development and progression of T-cell lymphoma in a murine model of atypical myeloproliferative disorder initiated by the ZNF198-fibroblast growth factor receptor-1 chimeric tyrosine kinase. Blood. 2009;114:1576–1584. doi: 10.1182/blood-2009-03-212704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren M, Qin H, Kitamura E, Cowell JK. Dysregulated signaling pathways in the development of CNTRL-FGFR1-induced myeloid and lymphoid malignancies associated with FGFR1 in human and mouse models. Blood. 2013;122:1007–1016. doi: 10.1182/blood-2013-03-489823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin H, Wu Q, Cowell JK, Ren M. FGFR1OP2-FGFR1 induced myeloid leukemia and T-cell lymphoma in a mouse model. Haematologica. 2016;101:e91–94. doi: 10.3324/haematol.2015.137695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren M, Cowell JK. Constitutive Notch pathway activation in murine ZMYM2-FGFR1-induced T-cell lymphomas associated with atypical myeloproliferative disease. Blood. 2011;117:6837–6847. doi: 10.1182/blood-2010-07-295725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren M, Qin H, Wu Q, Savage NM, George TI, Cowell JK. Development of ZMYM2-FGFR1 driven AML in human CD34+ cells in immunocompromised mice. Int J Cancer. 2016;139:836–840. doi: 10.1002/ijc.30100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowell JK, Qin H, Chang CS, Kitamura E, Ren M. A model of BCR-FGFR1 driven human AML in immunocompromised mice. Br J Haematol. 2016;175:542–545. doi: 10.1111/bjh.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer. 2015;15:593–607. doi: 10.1038/nrc3984. [DOI] [PubMed] [Google Scholar]
- 10.Delgado MD, Leon J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1:605–616. doi: 10.1177/1947601910377495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu T, Chong Y, Qin H, Kitamura E, Chang CS, Silva J, et al. The miR-17/92 cluster is involved in the molecular etiology of the SCLL syndrome driven by the BCR-FGFR1 chimeric kinase. Oncogene. 2017 doi: 10.1038/s41388-017-0091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu T, Pi W, Zhu X, Yu M, Ha H, Shi H, et al. Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucleic Acids Res. 2017;45:4479–4492. doi: 10.1093/nar/gkx055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghoshal P, Teng Y, Lesoon LA, Cowell JK. HIF1A induces expression of the WASF3 metastasis-associated gene under hypoxic conditions. Int J Cancer. 2012;131:E905–915. doi: 10.1002/ijc.27631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Facchini LM, Chen S, Marhin WW, Lear JN, Penn LZ. The Myc negative autoregulation mechanism requires Myc-Max association and involves the c-myc P2 minimal promoter. Mol Cell Biol. 1997;17:100–114. doi: 10.1128/mcb.17.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva J, Chang CS, Hu T, Qin H, Kitamura E, Hawthorn L, et al. Gene expression profiling during progression of FGFR1 driven AML in a mouse model of Stem Cell Leukemia Lymphoma syndrome. BMC Cancer. 2017 doi: 10.1016/j.ygeno.2018.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Ren M, Tidwell JA, Sharma S, Cowell JK. Acute progression of BCR-FGFR1 induced murine B-lympho/myeloproliferative disorder suggests involvement of lineages at the pro-B cell stage. PLoS One. 2012;7:e38265. doi: 10.1371/journal.pone.0038265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loeb CR, Harris JL, Craik CS. Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J Biol Chem. 2006;281:28326–28335. doi: 10.1074/jbc.M604544200. [DOI] [PubMed] [Google Scholar]
- 19.Chioni AM, Grose R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J Cell Biol. 2012;197:801–817. doi: 10.1083/jcb.201108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez JV, Frank DA. Genome-wide analysis of STAT target genes: elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol Ther. 2004;3:1045–1050. doi: 10.4161/cbt.3.11.1172. [DOI] [PubMed] [Google Scholar]
- 21.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baumann H, Kunapuli P, Tracy E, Cowell JK. The oncogenic fusion protein-tyrosine kinase ZNF198/fibroblast growth factor receptor-1 has signaling function comparable with interleukin-6 cytokine receptors. J Biol Chem. 2003;278:16198–16208. doi: 10.1074/jbc.M300018200. [DOI] [PubMed] [Google Scholar]
- 22.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse DJ, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 23.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 24.Pinz S, Unser S, Rascle A. Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC Mol Biol. 2016;17:10. doi: 10.1186/s12867-016-0063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren M, Qin H, Ren R, Tidwell J, Cowell JK. Src activation plays an important key role in lymphomagenesis induced by FGFR1 fusion kinases. Cancer Res. 2011;71:7312–7322. doi: 10.1158/0008-5472.CAN-11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia. 2013;27:32–40. doi: 10.1038/leu.2012.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Q, Bhole A, Qin H, Karp J, Malek S, Cowell JK, et al. SCLLTargeting FGFR1 to suppress leukemogenesis in syndromic and de novo AML in murine models. Oncotarget. 2016;7:10. doi: 10.18632/oncotarget.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath C, Cross NC. Critical role of STAT5 activation in transformation mediated by ZNF198-FGFR1. J Biol Chem. 2004;279:6666–6673. doi: 10.1074/jbc.M308743200. [DOI] [PubMed] [Google Scholar]
- 28.Smedley D, Demiroglu A, Abdul-Rauf M, Heath C, Cooper C, Shipley J, et al. ZNF198-FGFR1 transforms Ba/F3 cells to growth factor independence and results in high level tyrosine phosphorylation of STATS 1 and 5. Neoplasia. 1999;1:349–355. doi: 10.1038/sj.neo.7900035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu TL, Goss VL, Reeves C, Popova L, Nardone J, Macneill J, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108:4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- 30.Guasch G, Ollendorff V, Borg JP, Birnbaum D, Pebusque MJ. 8p12 stem cell myeloproliferative disorder: the FOP-fibroblast growth factor receptor 1 fusion protein of the t(6;8) translocation induces cell survival mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt/mTOR pathways. Mol Cell Biol. 2001;21:8129–8142. doi: 10.1128/MCB.21.23.8129-8142.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001;98:7319–7324. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo H, Li Q, O'Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood. 2005;106:2452–2461. doi: 10.1182/blood-2005-02-0734. [DOI] [PubMed] [Google Scholar]
- 33.Cowell JK, Qin H, Hu T, Wu Q, Bhole A, Ren M. Mutation in the FGFR1 tyrosine kinase domain or inactivation of PTEN is associated with acquired resistance to FGFR inhibitors in FGFR1-driven leukemia/lymphomas. Int J Cancer. 2017;141:1822–1829. doi: 10.1002/ijc.30848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.