

Abstract
Background:
Severe toxicity is experienced by a substantial minority of patients receiving fluoropyrimidine-based chemotherapy, with approximately 20% of these severe toxicities attributable to polymorphisms in the DPYD gene. The DPYD codes for the enzyme dihydropyrimidine dehydrogenase (DPD) important in the metabolism of fluoropyrimidine-based chemotherapy. We questioned whether prospective DPYD mutation analysis in all patients commencing such therapy would prove more cost-effective than reactive testing of patients experiencing severe toxicity.
Methods:
All patients experiencing severe toxicity from fluoropyrimidine-based chemotherapy for colorectal cancer in an Irish private hospital over a 3-year period were tested for 4 DPYD polymorphisms previously associated with toxicity. The costs associated with an index admission for toxicity in DPD-deficient patients were examined. A cost analysis was undertaken comparing the anticipated cost of implementing screening for DPYD mutations versus current usual care. One-way sensitivity analysis was conducted on known input variables. An alternative scenario analysis from the perspective of the Irish health-care payer (responsible for public hospitals) was also performed.
Results:
Of 134 patients commencing first-line fluoropyrimidine chemotherapy over 3 years, 30 (23%) patients developed grade 3/4 toxicity. Of these, 17% revealed heterozygote DPYD mutations. The cost of hospitalization for the DPYD-mutated patients was €232 061, while prospectively testing all 134 patients would have cost €23 718. Prospective testing would result in cost savings across all scenarios.
Conclusions:
The cost of hospital admission for severe chemotherapy-related toxicity is significantly higher than the cost of prospective DPYD testing of each patient commencing fluoropyrimidine chemotherapy.
Keywords: DPYD, fluoropyrimidine, colorectal cancer, cost-effectiveness, pharmacogenomics
Introduction
Fluoropyrimidine chemotherapy drugs such as 5-fluorouracil (5FU) and the oral 5FU prodrugs are widely used as both monotherapies and combination chemotherapy regime in the treatment of a wide variety of cancers. Potential toxicities associated with this class of chemotherapy include emesis, diarrhea, mucositis, alopecia, myelosuppression, palmar-plantar erythrodysesthesia, and cardiac toxicity. These adverse reactions may be severe and rarely fatal. They often compromise optimal patient treatment due to delays in drug administration or discontinuation of therapy before completion of a planned treatment.
The DPYD gene encodes for the enzyme dihydropyrimidine dehydrogenase (DPD), which functions as the rate-limiting step in the metabolism of fluoropyrimidine chemotherapies1,2; greater than 80% of 5FU is metabolized by DPD, and factors such as age, race, comorbidities, and concomitant therapies also influence metabolism (Figure 1). Over 50 polymorphisms of DPYD have been described, with certain alleles associated with reduced functionality of the enzyme leading to decreased metabolism of 5FU and more treatment-based toxicities.3-8
Figure 1.
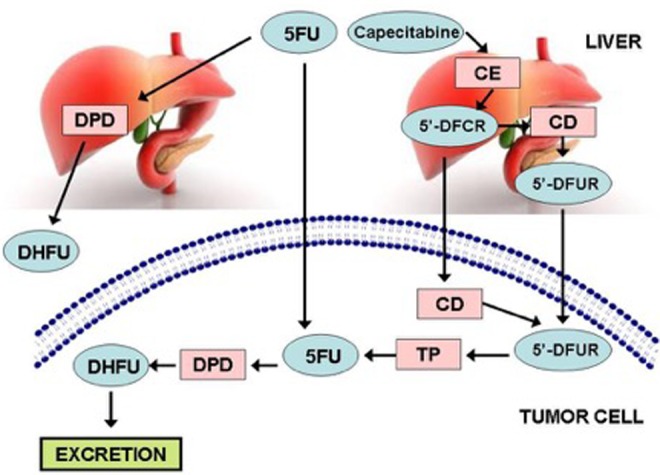
5-Fluorouracil and capecitabine metabolism in liver and tumor cells. CD indicates cytidine deaminase; CE, carboxyl esterase; 5′-DFCR, 5′-deoxy-5-fluorocytidine; 5′-DFUR, 5′-deoxy-5-fluorouridine; DHFU, dihydro-5-fluorouracil; DPD, dihydropyrimidine dehydrogenase; 5FU, fluorouracil; TP, thymidine phosphorylase.
Prospective testing for DPYD mutations is not routinely carried out due to concerns over the cost-effectiveness of upfront testing and the absence of clear guidelines for dose reductions in patients found to be DPD deficient on prospective testing.9 In addition, the absence of a mutation does not guarantee freedom from severe toxicity. Nonetheless, the potential advantage of prospectively identifying DPYD mutations is that careful monitoring and dose escalation may allow DPD-deficient patients to safely receive fluoropyrimidine chemotherapy.6-8
We became concerned by a number of patients treated at our institution (a large Irish private hospital) suffering prolonged hospitalizations, having to curtail or abandon adjuvant chemotherapy due to fluoropyrimidine toxicity.10 In this study, we followed current practice of reactive testing for DPYD polymorphisms in patients experiencing severe toxicity. We questioned whether prospective testing of all patients treated during this period would reduce the economic and medical toxicity of fluoropyrimidine-based treatment. We examined the costs associated with an index admission for fluoropyrimidine toxicity in DPD-deficient patients. A cost analysis was undertaken comparing the anticipated cost and outcomes of implementing screening for DPYD mutations as routine care versus current practice.
Routine prospective screening can potentially benefit our practice through the identification of those patients who are at increased risk of toxicity. Subsequent individualization of affected patients’ chemotherapy management may reduce the risk of adverse outcomes.
Methods
Patient Population
Patients commencing chemotherapy for colorectal cancer (CRC) at our institution over a 3-year period between January 1, 2010, and December 31, 2012, who developed severe (grade 3/4) toxicity were reactively tested for DPYD mutations. The type and durations of toxicity were recorded using the National Cancer Institute Common Toxicity Criteria for Adverse Events version 4.0. Following informed consent, EDTA blood samples were procured from each patient and genotyped for 4 DPYD mutations associated with fluoropyrimidine toxicity at St Thomas’ Hospital, London. The specific genotypes tested for were 1905+1G>A (DPYD*2A), 2846A>T, 1601G>A (DPYD*4), and 1679T>G (DPYD*13). From June 2011, quantitative polymerase chain reaction targeting 4 specific DPYD variants associated with fluoropyrimidine toxicity was adopted as the testing method. Prior to this, testing was performed by sequencing exons 13, 14, and 22 (including 4 DPYD variants targeted by the later method).
Economic Analysis
Cost analysis comparing the impact of systematic screening for DPYD mutation with routine care was performed, consisting of testing for DPYD mutation in the event of severe toxicity following commencement of chemotherapy. All costs are in form of Euro (€) at 2012 values.
The cost of routine DPYD mutation testing if it had been applied prospectively in all patients commencing on fluoropyrimidine therapy for CRC in this time frame was calculated. Cost per test (€177) was obtained from internal hospital data and then compared with the cost of the index admission with grade 3/4 toxicity for the patients identified retrospectively as having DPYD mutations. For patients with multiple admissions, the cost of the key admission, which led to DPYD mutation testing only, was assessed.
Costs associated with the study are based on microcosting methods unless otherwise stated. The study was conducted in a private hospital, enabling analysis of costs of care incurred by individual patients. Discounting was not applied as only costs associated with the index admission were evaluated. All costs associated with treatment of severe toxicity at the index admission were analyzed. Mean patient costs for an admission for severe toxicity are detailed in Table 1. Administrative and overhead costs were excluded as they were considered to be equivalent in both comparison groups. The primary analysis was conducted from an Irish private hospital perspective.
Table 1.
Mean Patient Costs for Index Admission With Severe Toxicity.
| Category | Mean (€) | Standard Deviation |
|---|---|---|
| Accommodation fees | 25 981 | 14 893 |
| Consumables | 114 | 242 |
| Paramedical | 4063 | 4229 |
| Pathology | 12 017 | 11 570 |
| Pathology send outs | 99 | 102 |
| Pharmacy | 2470 | 1759 |
| Procedures | 480 | 295 |
| Radiology | 1041 | 578 |
| Theater/ward packs | 244 | 141 |
Sensitivity Analysis
One-way sensitivity analysis was conducted on all known input variables. Variations of ±50% were applied due to the low numbers involved in the study. An alternative scenario analysis, from the perspective of the Irish health-care payer (responsible for public hospitals) based on diagnosis-related group costs, is also presented. Cost was based on an average length of stay of 31.8 days for admission due to sequelae of treatment. Budget impact analysis of potential costs and outcomes associated with implementing DPYD screening at a national level was also conducted. Analysis was informed by national incidence, treatment, and mortality data.
Results
A total of 134 patients were commenced on first-line fluoropyrimidine-based chemotherapy for CRC over the 3-year period, 66 in the adjuvant setting and 68 with metastatic disease. In all, 30 patients (23%) developed grade 3/4 toxicity during chemotherapy and therefore tested for DPYD mutations. Of these, 5 (17% of those tested, 4.5% of total population) revealed heterozygote DPYD mutations. Of the 4 deleterious DPYD variants tested for, 2 were identified among our group of patients with severe toxicity (DPYD*2A and *4). The DPYD genotype, toxicity type, and duration of hospitalization are summarized in Table 2.
Table 2.
Characteristics of Patients Diagnosed With DPYD Mutations.
| Pt | Gender | Regimen | Cycle of Toxicity | Type of Toxicity | Mutation | Status | Length of Admission (Days) |
|---|---|---|---|---|---|---|---|
| 1 | Female | FLOX | Post C1D15 | GI | *4 | Heterozygous | 64 |
| 2 | Male | mFolfox6 | Post C4 | GI and hematologic | *2A | Heterozygous | 37 |
| 3 | Female | Xelox | Post C2* | GI | Compound *2A & *4 | Heterozygous | 26 |
| 4 | Male | mFolfox6 | Post C4 | GI | *4 | Heterozygous | 17 |
| 5 | Male | mFolfox6 | Post C1 | GI and hematologic | *2A | Heterozygous | 15 |
Abbreviations: C, cycle; D, day; GI, gastrointestinal; Pt, patient.
aPatient 3 had received previous capecitabine therapy with neoadjuvant radiation, requiring dose reductions for toxicity.
The total cost related to hospitalization with toxicity for these 5 patients was €232 061, an average of €46 412 per case. At €177 per test, the cost to prospectively test all 134 patients would have been €23 718. As seen in Table 3, if 60% of patients identified with a DPYD mutation were prevented from experiencing a severe toxicity resulting in hospitalization, approximately €120 000 in additional cost would have been avoided over a 3-year period.
Table 3.
Costs of Systematic Screening Versus Usual Care of 134 Patients Commenced on First-Line Fluoropyrimidine-Based Chemotherapy (2010-2012) and 1-Way Sensitivity Analysis.
| Systematic Screening (€) | Usual Care (€) | |
|---|---|---|
| Cost of DPYD screening | 23 718 | 5310 |
| Cost of severe toxicity-related admission to hospital | 92 824a | 232 061 |
| Total cost of care | 116 542 | 232 371 |
| Incremental cost of systematic screening versus usual care | −120 829 | |
| Effectiveness of DPYD screening and altered chemotherapy protocols (lower limit = 30% success; upper limit = 90% success) | −51 210 | −190 447 |
| Cost of hospital care (±95% confidence interval) | −37 265 | −204 392 |
| Irish health-care payer scenario | −54 074 | |
aBased on the assumption that routine screening for DPYD mutations and revised chemotherapy protocol has a 60% success rate in preventing severe toxicity-related admissions.
Sensitivity Analysis
A wide variation was applied to help address uncertainty surrounding the intervention. Variations of ±50% were evaluated during sensitivity analysis (Table 3). If only 30% of the patients identified with DPYD mutations were successfully prevented from experiencing severe toxicity, the proposed update in practice would still result in a cost saving. All scenarios evaluated were in favor of routine DPD screening, including an analysis from an Irish public hospital scenario.
Budget Impact Analysis
Based on a 2009 report, the average incidence of early-stage (stage I-III) CRC in the Republic of Ireland is 1484 patients per year11. Of these patients, 40% receive chemotherapy. Based on estimates from the primary analysis presented in this article, the budgetary impact of implementing DYPD screening on a routine basis is €105 000 per year; however, savings of approximately €630 000 could be achieved annually through the prevention of unexpected hospital admissions for severe toxicity from fluoropyrimidine.
Discussion
The novel finding of our study is that cost of admissions for severe chemotherapy-related toxicity with reactive DPYD testing is higher than the cost of prospectively testing each new patient commencing fluoropyrimidine chemotherapy.
Using a panel of 4 mutations associated with fluoropyrimidine toxicity, we report a prevalence of heterozygous DPYD mutation in this Irish population with CRC of 4.5%. DPYD mutations were found to be present in a sizeable minority (17%) of patients developing grade 3/4 toxicities with fluoropyrimidine therapy. Previous studies have described deleterious mutations in DPYD in up to 12% to 25% of patients developing grade 3/4 toxicities with fluoropyrimidine-based chemotherapy,12-15 although curiously one study found the dominant *2A polymorphism in as low as 2.2% of patients with severe toxicity.16
To date, there have been some proponents of routine testing for DPD deficiency prior to starting treatment17; however, these remain the minority, the perceived wisdom being that preemptive screening is not cost-effective. This consensus was challenged by a nonrandomized study in patients receiving fluoropyrimidine-based chemotherapy for CRC, demonstrating that prospective screening for DPD deficiency (genotype ± phenotype) could be a cost-effective strategy.18 Our study supports this finding by showing that routine prospective DPYD mutation testing in the Irish population would be associated with significant cost savings.
Patients reactively diagnosed with DPYD mutations following admissions with severe toxicity were unlikely to resume therapy at reduced doses, potentially compromising curative outcomes.19 Prospective identification of patients with a DPYD mutation, coupled with dose reduction from therapy initiation, may protect patients, improve quality of life, and avoid severe and potentially fatal chemotherapy-related toxicity.
Although the practice of pharmacogenomic drug dosing is in its infancy, the Clinical Pharmacogenetics Implementation Consortium (CPIC) has published guidelines containing dosing recommendations for fluoropyrimidines based on DPYD genotype.20 They recommend a minimum of a 50% reduction in the initiation dose in patients heterozygous for the nonfunctional alleles *2a, *13, and 2846A>T. Notably, the consortium currently recommend that the *4 variant be categorized as “normal” activity, in part based on functional analyses of enzymatic activity in transfected cell lines which suggested supranormal activity.21,22 This is, however, contradictory to our study: the *4 allele was identified as a heterozygote variant in 2 of our patients with severe toxicity and a compound heterozygote in combination with the *2A variant in another. This variant has been found to be previously associated with decreased enzyme activity in functional analyses of human donor peripheral blood mononuclear cells, as well as severe toxicity in patients receiving fluoropyrimidines.6,23-25 We suggest that initial dose reduction for this variant should continue to be considered, while we recognize that further study is required to produce more definitive pharmacogenomic-based dosing guidelines.
By definition, pharmacogenomic dosing will vary between individuals, with initial dose reduction needing adjustment on a patient-by-patient basis. Currently, the CPIC guidelines do not report dosing recommendations for all variants of DPYD, due to weak or conflicting data on the effect these alleles have on DPD activity.
There is understandable concern that patients may have dose reductions performed for identified mutations which may not have resulted in increased toxicity, resulting in patients receiving reduced doses of potentially curative chemotherapy without achieving a gain from toxicity avoidance. Additional information regarding the increased toxicity associated with various polymorphisms is required to optimize pharmacogenomic dosing. Currently, it is reasonable to reduce doses appropriately for the better characterized polymorphisms listed above and avoid testing for polymorphisms of as yet undetermined significance. Similarly, where such polymorphisms are detected, one may recommend increased education and vigilance for toxicity in such patients, without utilizing dose reductions from the start of therapy.
One way to clearly establish predictable drug exposure to minimize undue toxicity while maximizing therapeutic exposure is to conduct continuous pharmacokinetic monitoring, as has been utilized in a French study.26-28 Although this is an interesting concept, it is an excessively labor-intensive and time-consuming approach rendering it impractical in clinical practice.
Our study suggests that while routine testing is economically viable, further research and clear guidance on dose reduction are needed. DPYD testing has the potential to avoid premature cessation of potentially curative therapy for patients with deficiencies.
We acknowledge that this was a small single-center-based study and that results may be biased by the fact that 2 of the hospitalized cases required prolonged treatment in an intensive care unit. Nonetheless, proposed routine screening of patients remained cost beneficial in all sensitivity analysis conducted, including one encompassing an approximate 50% reduction in costs. As with the vast majority of clinical-based studies, the diverse nature of health-care systems across jurisdictions must be considered when analyzing results; however, it must be noted that the costs of inpatient care for complications due to both medical and surgical therapy were among the fastest rising costs of hospital stays in US health care in 2010.29
Acknowledgments
The authors would like to thank our patients for participating in this study.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Andrew Kenny  http://orcid.org/0000-0001-5059-8520
http://orcid.org/0000-0001-5059-8520
References
- 1. Heggie GD, Sommadossi JP, Cross DS, et al. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987;47(8):2203–2206. [PubMed] [Google Scholar]
- 2. Deenen MJ, Cats A, Beijnen JH, et al. Part 2: pharmacogenetic variability in drug transport and phase I anticancer drug metabolism. Oncologist. 2011;16(6):820–834. doi:10.1634/theoncologist.2010-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maekawa K, Saeki M, Saito Y, et al. Genetic variations and haplotype structures of the DPYD gene encoding dihydropyrimidine dehydrogenase in Japanese and their ethnic differences. J Hum Genet. 2007;52(10):804–819. doi:10.1007/s10038-007-0186-6. [DOI] [PubMed] [Google Scholar]
- 4. Deenen MJ, Tol J, Burylo AM, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17(10):3455–3468. doi:10.1158/1078-0432.CCR-10-2209. [DOI] [PubMed] [Google Scholar]
- 5. Johnson MR, Wang K, Diasio RB. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin Cancer Res. 2002;8(3):768–774. [PubMed] [Google Scholar]
- 6. Loganayagam A, Arenas Hernandez M, Corrigan A, et al. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer. 2013;108(12):2505–2515. doi:10.1038/bjc.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosmarin D, Palles C, Pagnamenta A, et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut. 2015;64(1):111–120. doi:10.1136/gutjnl-2013-306571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosmarin D, Palles C, Church D, et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: investigation in the QUASAR2 study, systematic review, and meta-analysis. J Clin Oncol. 2014;32(10):1031–1039. doi:10.1200/JCO.2013.51.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Opdam FL, Swen JJ, Wessels JAM, et al. SNPs and haplotypes in DPYD and outcome of capecitabine—letter. Clin Cancer Res. 2011;17(17):5833; author reply 5835-5836. doi:10.1158/1078-0432.CCR-11-1208. [DOI] [PubMed] [Google Scholar]
- 10. van Staveren MC, Jan Guchelaar H, van Kuilenburg ABP, et al. Evaluation of predictive tests for screening for dihydropyrimidine dehydrogenase deficiency. Pharmacogenomics J. 2013;13(5):389–395. doi:10.1038/tpj.2013.25. [DOI] [PubMed] [Google Scholar]
- 11. National Cancer Registry of Ireland. http://www.ncri.ie/sites/ncri/files/pubs/ColorectalCancerIncidenceMortalityTreatmentandSurvivalinIreland1994-2010.pdf. Accessed September 19, 2018.
- 12. Saif MW. Dihydropyrimidine dehydrogenase gene (DPYD) polymorphism among Caucasian and non-Caucasian patients with 5-FU- and capecitabine-related toxicity using full sequencing of DPYD. Cancer Genomics Proteomics. 2013;10(2):89–92. [PubMed] [Google Scholar]
- 13. Van Kuilenburg AB, Meinsma R, Zoetekouw L, et al. High prevalence of the IVS14 + 1G>A mutation in the dihydropyrimidine dehydrogenase gene of patients with severe 5-fluorouracil-associated toxicity. Pharmacogenetics. 2002;12(7):555–558. [DOI] [PubMed] [Google Scholar]
- 14. Raida M, Schwabe W, Häusler P, et al. Prevalence of a common point mutation in the dihydropyrimidine dehydrogenase (DPD) gene within the 5′-splice donor site of intron 14 in patients with severe 5-fluorouracil (5-FU)-related toxicity compared with controls. Clin Cancer Res. 2001;7(9):2832–2839. [PubMed] [Google Scholar]
- 15. Loganayagam A, Arenas-Hernandez M, Fairbanks L, et al. The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer Chemother Pharmacol. 2010;65(2):403–406. doi:10.1007/s00280-009-1147-x. [DOI] [PubMed] [Google Scholar]
- 16. Magné N, Etienne-Grimaldi MC, Cals L, et al. Dihydropyrimidine dehydrogenase activity and the IVS14+1G>A mutation in patients developing 5FU-related toxicity. Br J Clin Pharmacol. 2007;64(2):237–240. doi:10.1111/j.1365-2125.2007.02869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Del Re M, Di Paolo A, van Schaik RH, et al. Dihydropyrimidine dehydrogenase polymorphisms and fluoropyrimidine toxicity: ready for routine clinical application within personalized medicine? EPMA J. 2010;1(3):495–502. doi:10.1007/s13167-010-0041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Traoré S, Boisdron-Celle M, Hunault G, et al. DPD deficiency: medicoeconomic evaluation of pretreatment screening of 5-FU toxicity. J Clin Oncol. 2012;30(suppl 4):410–410. doi:10.1200/jco.2012.30.4_suppl.410. [Google Scholar]
- 19. United States Food and Drug Administration label information on capecitabine. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020896s037lbl.pdf. Accessed September 19, 2018.
- 20. Caudle KE, Thorn CF, Klein TE, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin Pharmacol Ther. 2013;94(6):640–645. doi:10.1038/clpt.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. CPIC dosing guideline for fluorouracil and DPYD. http://www.pharmgkb.org/guideline/PA166122686. Accessed September 19, 2018.
- 22. Offer SM, Fossum CC, Wegner NJ, et al. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74(9):2545–2554. doi:10.1158/0008-5472.CAN-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lazar A, Mau-Holzmann UA, Kolb H, et al. Multiple organ failure due to 5-fluorouracil chemotherapy in a patient with a rare dihydropyrimidine dehydrogenase gene variant. Onkologie. 2004;27(6):559–562. doi:10.1159/000081338. [DOI] [PubMed] [Google Scholar]
- 24. Collie-Duguid ESR, Etienne MC, Milano G, et al. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics. 2000;10:217–223. [DOI] [PubMed] [Google Scholar]
- 25. Seck K. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin Cancer Res. 2005;11(16):5886–5892. doi:10.1158/1078-0432.CCR-04-1784. [DOI] [PubMed] [Google Scholar]
- 26. Gamelin E, Delva R, Jacob J, et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(13):2099–2105. doi:10.1200/JCO.2007.13.3934. [DOI] [PubMed] [Google Scholar]
- 27. Boisdron-Celle M, Capitain O, Metges JP, et al. Severe fluoropyrimidines toxicities: a simple and effective way to avoid them. Screen effectively for DPD deficiencies. Ann Oncol. 2012;23(suppl 4):iv5–iv18, abstract O-0019.22774231 [Google Scholar]
- 28. Boisdron-Celle M, Capitain O, Faroux R, et al. Prevention of 5-FU-induced toxicities using pretherapeutic DPD deficiency screening: medical and economic assessment of a multiparametric approach. J Clin Oncol. 2013;31(suppl 4):351–351. doi:10.1200/jco.2013.31.4_suppl.351. 23233706 [Google Scholar]
- 29. Pfuntner A, Wier LM, Steiner C. Costs for hospital stays in the United States, 2010. HCUP Statistical Brief #146. 2013. Rockville, MD: Agency for Healthcare Research and Quality; Available at: http://www.hcup-us.ahrq.gov/reports/statbriefs/sb146.pdf [PubMed] [Google Scholar]