

Abstract
Liver cirrhosis is an independent risk factor for hepatocellular carcinoma (HCC). The mechanisms that contribute to HCC development in the cirrhotic microenvironment are unknown. We found that HCC grown in the highly stressed cirrhotic microenvironment undergoes autophagy switching from a protective state characterized by high macroautophagy with low chaperone-mediated autophagy (CMA) to an HCC-promoting state characterized by low macroautophagy with high CMA. This study examined how the stress response executes oncogenic cell programming through autophagy switching using hepatitis C virus cell culture. Protein kinase R–like endoplasmic reticulum kinase expression increased to high levels in hepatitis C virus culture. Protein kinase R–like endoplasmic reticulum kinase–dependent activation of nuclear factor erythroid 2–related factor (Nrf2) led to increased transcription of the cytoprotective genes: heat shock cognate 70 kDa protein and lysosome-associated membrane protein 2A (LAMP2A) and precipitated the induction of CMA. CMA selectively targeted beclin1 degradation, leading to accumulation of the autophagy flux protein p62 due to impaired autophagosome-endosome fusion. This impaired autophagosome-endosome fusion due to beclin1 degradation inhibited endocytosis and degradation of epidermal growth factor receptor. Silencing Nrf2 and LAMP2A reduced cell viability, suggesting that the stress response activates CMA as a compensatory mechanism of cell survival. We report a novel mechanism through which stress response triggers oncogenic Nrf2 signaling that promotes autophagy switching to favor cell survival.
Hepatitis C virus (HCV), a positive-stranded RNA virus that belongs to the Flaviviridae family, is the leading cause of liver cirrhosis and hepatocellular carcinoma (HCC) in the United States.1 A number of recently approved direct-acting antiviral agents have produced high rates of viral clearance and are expected to decrease the incidence of liver cirrhosis and HCC related to chronic HCV infection in the near future.2 Although the causal relationship between HCV infection and HCC is well documented, our understanding of the molecular mechanisms through which chronic HCV infection leads to liver cirrhosis and HCC is limited.3 The infection cycle of HCV involves a series of molecular events that include virus attachment and entry into hepatocytes through receptor-mediated endocytosis. A number of candidate proteins have been implicated as putative cellular receptors that facilitate virus attachment for HCV.4 During the entry process, the virus envelope fuses with the endosomal membrane, and the subsequent reduction in pH results in the release of the viral RNA genome. In infected hepatocytes, HCV uses the endoplasmic reticulum (ER) extensively to continue its translation and replication cycles during chronic infection.5 Large amounts of viral proteins and double-stranded RNA replicative intermediates accumulate in the ER of infected hepatocytes and generate a substantial amount of stress (termed ER stress).6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 ER stress activates several cellular transcription programs, known collectively as the unfolded protein response (UPR), to restore cellular homeostasis and improve the survival of infected hepatocytes. The UPR can be divided into three branches: protein kinase RNA-activated–like ER kinase (PERK), activating transcription factor-6α (ATF6α), and inositol-requiring enzyme-1α (IRE1α).6 The low-level accumulation of misfolded proteins in the ER is cleared by ubiquitin-proteasome degradation referred to as type I ER-associated protein degradation (ERAD). When type I ERAD is not sufficient, the ER initiates a second line of protein degradation through the induction of autophagy (type II ERAD).7
Autophagy is a diverse family of processes with three main subtypes: macroautophagy (hereafter termed autophagy), chaperone-mediated autophagy (CMA), and microautophagy. Autophagy involves the formation of a double-membrane autophagosome that sequesters misfolded proteins in the ER and then fuses with lysosome to form an autolysosome, where its contents are degraded.26, 27 CMA requires heat shock proteins, such as heat shock cognate 70 kDa (Hsc70), which bind proteins that contain KFERQ motifs for transport into lysosomes through lysosome-associated membrane protein 2A (LAMP2A) receptor.28, 29, 30 During microautophagy, cargo is directly engulfed by lysosomes. Dysregulation of ER stress and ERAD plays an important role in viral-induced metabolic alteration (lipid accumulation), inflammation, and immunity. We contend that these changes comprise the direct and indirect mechanisms implicated in HCC development.
We and others have reported that ER stress, UPR, and the autophagy response persist during chronic liver disease and liver cirrhosis, suggesting that viral-induced chronic ER stress and ERAD play a major role in HCC.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 Although ER-stress markers are elevated in HCC, HCC development in human and mouse models is associated with an impaired autophagy response. Impaired autophagy has been implicated in the pathogenesis of many human diseases, including various cancers, Parkinson disease, and Crohn disease. ER stress is a strong inducer of autophagy. However, in case of extreme stress, such as prolonged starvation, the other forms of autophagy compensate for impaired autophagy.28 The mechanism of autophagy regulation in the setting of a highly stressed ER during chronic HCV infection is unknown. Therefore, we are interested in understanding the mechanism through which the hepatic autophagy process functions as a prosurvival defense mechanism under high ER stress and sets the course of HCC development in the cirrhotic microenvironment.
In this study, we examined how macroautophagy and CMA compensate for each other to promote hepatocyte survival using a persistently infected HCV cell culture model. We demonstrate that persistent HCV infection resulted in the prolonged activation of the PERK axis of the UPR, leading to the activation of nuclear factor erythroid 2–related factor (Nrf2) signaling and its nuclear translocation. Nrf2 nuclear translocation induced Hsc70 and LAMP2A expression and subsequently activated CMA. CMA activation led to beclin1 degradation and inhibition of autophagy. Loss of beclin1 in infected cells resulted in impaired fusion of autophagosomes with endosomes and lysosomes, which impaired epidermal growth factor receptor (EGFR) degradation and led to the accumulation of p62. Our results provide a potential autophagy switching mechanism triggered during HCV infection that promotes improved cell survival under chronic ER stress.
Materials and Methods
Cell Culture, Antibodies, and Chemicals
Nontransformed primary human hepatocytes (PHHs) were supplied by XenoTech LLC, Kansas City, MO. A transformed hepatoma cell line (Huh-7.5) developed by the Charles M. Rice Laboratory (Rockefeller University, New York, NY) that was highly permissive for HCV infection was cultured using a protocol described in our previous publications.24, 25 PHHs were cultured in a six-well tissue culture plate using growth media supplied by the XenoTech LLC plus 10% human serum (Invitrogen, Brown Deer, WI). The Huh-7.5 cell line was cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 2 mmol/L l-glutamine, sodium pyruvate, nonessential amino acids, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 10% fetal bovine serum. Antibodies used in immunostaining, Western blotting, and immunoprecipitation experiments were obtained from the following commercial sources. The antibody to NS3 was purchased from Virogen Inc. (Boston, MA). The antibody to HCV core was purchased from Thermo Scientific (Waltham, MA). Antibodies to phosphorylated Nrf2 and LAMP2A were purchased from Abcam (Cambridge, MA). Antibodies to glucose-regulated protein 78 [binding immunoglobulin protein (BiP)], PERK, IRE1α, Beclin1, β-actin, autophagy-related 7 (ATG7), autophagy-related 16-like 1 (ATG16L1), Rab5, Rab7, VPS34, LAMP1, light chain 3B (LC3B), EGFR, phosphorylated EGFR, and Hsc70 were obtained from Cell Signaling (Beverly, MA). Antibodies to ATF6α, total Nrf2, p62, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant GAPDH (G2267) and epidermal growth factor (EGF) were purchased from Sigma-Aldrich (St. Louis, MO). Sulforaphane, Bafilomycin A1, Torin 1, and 3-methyladenine were obtained from Sigma-Aldrich. Ferrofluid-iron nanoparticles (EMG 508) were obtained from FerroTech (Boston, MA).
HCV Replication in Nontransformed (PHHs) and Transformed (Huh-7.5 Cells) Human Liver Cells
PHHs seeded in six-well plates were infected with JFH-ΔV3-Rluc virus (HCV genotype 2a) with a multiplicity of infection of 0.1, as described previously.18 Cell pellets of uninfected and HCV-infected PHHs were harvested at 3-day intervals and used for Western blot analysis. The success of HCV replication in the infected PHHs was assessed through Western blots for viral NS3. Huh-7.5 cells were infected with JFH-ΔV3-Rluc HCV as previously described.18 Infected Huh-7.5 cells were split every 3 days for the measurement of viral protein expression by immunostaining, Western blotting, and flow cytometry. The success of HCV replication in the infected Huh-7.5 cells was measured by Renilla luciferase activity. The efficiency of infection and spread of HCV in Huh-7.5 cell culture were examined by immunostaining and Western blotting for core protein.
Western Blotting and Immunohistochemical Staining
Infected cells were harvested at different time points and examined for the expression of viral proteins, ER stress, and autophagy markers by immunostaining and Western blotting. To monitor the effect of virus replication on cellular ER stress and autophagy, infected cells were immobilized on glass slides and then stained for HCV core or p62 by immunostaining using a standard protocol established in our laboratory.25 Cell lysates (approximately 20 μg of protein) were prepared from the infected culture at different time points and examined for expression of stress markers and autophagy by SDS-PAGE and Western blotting using a standard protocol established in the laboratory.25
Quantification of mRNA Levels by RT-qPCR
Infected Huh-7.5 cells were harvested at different time points, and total RNA was isolated using the RNeasy Mini kit (Qiagen, Germantown, MD). This purification protocol includes a DNase treatment step to obtain cellular RNA free of any genomic DNA. The first strand cDNA was synthesized from 1 μg of total RNA using an iScript Reverse Transcription Supermix containing Moloney murine leukemia virus reverse transcriptase, RNase inhibitor, dNTPs, and oligo (dt) random primers (Bio-Rad, Hercules, CA). A total of 100 ng of newly synthesized cDNA was amplified using iTaq Universal SYBR Green Supermix (Bio-Rad) with gene-specific sense and antisense primers using the standard protocol provided in the kit. The mRNA levels of PERK, IRE1α, ATF6α, Hsc70, LAMP2A, Beclin1, and GAPDH (internal control) were quantified using quantitative RT-PCR (RT-qPCR). Amplification, data acquisition, and analysis were performed on the CFX96 real-time instrument using CFX manager software version 3.0 (Bio-Rad). The expression level of each mRNA was compared with the GAPDH mRNA level using the comparative threshold cycle method. The nucleotide sequences of oligonucleotide primers for PERK mRNA (sense primer 5′-GGCTTGAAAGCAGTTAG-3′ and antisense primer 5′-GGACAGTTGCCTTACAGA-3′),31 IRE1α mRNA (sense primer 5′-GAAAAGGAATCCCTGGATGG-3′ and antisense primer 5′-TCAGAGGGCGTCTGGAGTC-3′),32 ATF6α mRNA (sense primer 5′-AACAAGACCACAAGACCA-3′ and antisense primer 5′-AGGAGGAACTGACGAACT-3′),33 Hsc70 mRNA (sense primer 5′-AAACGTCTGATTGGACGCAG-3′32 and antisense primer 5′-GCACGTTTCTTTCTGCTCCA-3′), and LAMP2A mRNA (sense primer 5′-GTCTCAAGCGCCATCATACT-3′ and antisense primer 5′-TCCAAGGAGTCTGTCTTAAGTAGC-3′) were derived from a previous report.34 Beclin1 mRNA (sense 5′-AGCTGCCGTTATACTGTTCTG-3′ and antisense 5′-ACTGCCTCCTGTGTCTTCAATCTT-3′) was also derived from a published report.35
Co-Immunoprecipitation
Infected Huh-7.5 cells were harvested on day 6 and day 21, and cell pellets were washed once with 10 mL of phosphate-buffered saline (PBS). Cell pellets were solubilized using 3 mL of ice-cold radioimmunoprecipitation assay buffer supplemented with a protease inhibitor cocktail (Roche, Basel, Switzerland) on a shaker for 30 minutes. Lysate was clarified by centrifugation at 10,700 × g for 10 minutes. One milligram of total cell lysate was incubated with antibodies against beclin1 (Cell Signaling) at a dilution of 1:1000 overnight at 4°C in a shaker. The next day, 20 μL of protein A/G plus-agarose beads (Santa Cruz Biotechnology) were added for 1 hour. The beads were washed three times with radioimmunoprecipitation assay buffer, and the bound protein-complexes were eluted in Laemmli buffer. Samples were boiled for 5 minutes, 20 μL of lysate were loaded on a 12% SDS-PAGE gel, and Western blotting was performed using a standard protocol.24
Measurement of CMA Activity in Vitro
Lysosomes were purified from infected Huh-7.5 cells after labeling with magnetic iron nanoparticles (EMG-508, Ferrotec) using a procedure described previously.36 Uninfected, serum starved, and infected Huh-7.5 cells cultured in a 15-cm tissue culture plate seeded at a density of 3 × 106 cells/mL. The next day, cells were incubated with magnetic iron nanoparticles (1:100 dilution) in culture media for 4 hours. Following this step, the media was aspirated, and cells were incubated with fresh media for an additional 20 hours. Cells were harvested by scraping the plate using disposable cell scraper, centrifuged at 20 × g for 3 minutes, and washed once with cold PBS in a tabletop centrifuge. The cell pellet was resuspended in 700 μL of subcellular fractionation buffer that contained 20 mmol/L HEPES potassium salt, 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L EDTA, 1 mmol/L EGTA, and 8 mmol/L dithiothreitol at pH 7.5. Cells were homogenized using a tight-fitting hand-held homogenizer (five strokes). The homogenates were transferred to an Eppendorf tube and spun at 1100 × g at 4°C for 10 minutes to remove debris (basically remove pellet and collect supernatants). The centrifugation step was then repeated with supernatants to remove all cellular debris. The supernatants were transferred to a clean tube and incubated in a magnetic rack for 1 hour at 4°C. The lysosome-bound magnetic beads were washed twice using 1 mL of subcellular fractionation buffer. After the last step of washing, the suspended iron-bound lysosomes were centrifuged at 13,200 × g to pellet the lysosomes. The lysosome pellets were resuspended in 100 μL of fractionation buffer, and the protein concentration was determined. The activity of CMA was tested using GAPDH uptake and degradation by the purified lysosomes as previously described.37 In brief, an aliquot of lysosomes (75 μg of protein) with or without protease inhibitors (catalog number 78,030, Thermo Scientific) was incubated with GAPDH (25 μg, catalog number G2267, Sigma-Aldrich) in an isotonic medium [10 mmol/L 3-(N-morpholino) propanesulfonic acid (MOPS), 0.25 sucrose at pH 7.3] for 20 minutes at 37°C. After this step, the contents were diluted in 0.5 mL of fractional buffer and lysosomes were recovered by centrifugation at 13,200 × g for 10 minutes at 4°C. Lysosomes were resuspended in lysis buffer, and the amount of GAPDH associated with the lysosomes was assessed by SDS-PAGE and immunoblotting. LAMP2A, LAMP1, Cathepsin-D, and β-actin can be used as controls during analysis. Quantification was performed by densitometric analysis of the immunoblots.
Electron Microscopy
Uninfected and infected Huh-7.5 cells on day 6 and day 21 were harvested using trypsin-EDTA. Cell pellets were washed with PBS and then suspended in 3% glutaraldehyde fixative (Sigma-Aldrich). Cell pellets were fixed in 1% osmium tetroxide and dehydrated with an ethyl alcohol series. Samples were infiltrated and embedded in eponate 12 resin and polymerized at 60°C for 24 hours. Thin sections (70 nm) of the samples were placed on copper grids. Cells were examined using a G2 F30 Tecnai transmission electron microscope at 200 kV. Cytoplasmic areas of 10 different cells were captured under the grid, and the number of autophagosomes was counted in uninfected, early-infeted, and late-infected Huh-7.5 cells.
Flow Analysis
HCV-infected Huh-7.5 cells were collected at different time points using trypsin-EDTA. Cells were washed with PBS, fixed for 10 minutes with 2% paraformaldehyde, then permeabilized for 15 minutes using ice-cold methanol. At each time point, the expression of p62 and HCV core was measured by flow analysis after staining. In brief, cells were divided into two equal aliquots. One aliquot of cells was incubated with a rabbit monoclonal antibody to p62 tagged with Alexa Fluor 488 (1:100) for 60 minutes. For detection of HCV core, cells were incubated with an HCV core-specific mouse monoclonal antibody (1:200) for 60 minutes. After three washes, cells were incubated with an Alexa Fluor 488–conjugated secondary antibody (1:50) for 60 minutes. After these steps, cells were washed three times with PBS and analyzed separately by flow analysis. The percentage of core-positive and p62-positive cells was analyzed by flow analysis.
siRNA Transfection
Persistently HCV-infected Huh-7.5 cells were cultured in six-well plates (up to 60% confluence in DMEM supplemented with 10% FBS media) without antibiotics. The next day, culture media was replaced with fresh DMEM with 2% FBS, and the cells were then transfected with siRNA to PERK (5′-CAAGAGGAAGACAUCCUGCTT-3′),38 IRE1α (5′-GGACGUGAGCGACAGAAUATT-3′),39 ATF6α (5′-GCAACCAAUUAUCAGUUUATT-3′),40 Nrf2 (siRNA1: 5′-GAAUGGUCCUAAAACACCA-3′; siRNA2: 5′-UGACAGAAGUUGACAAUUA-3′),41, 42 LAMP2A (5′-CUGCAAUCUGAUUGAUUAUU-3′),24 beclin1 (siRNA 1: 5′-UGGAAUGGAAUGAGAUUAATT-3′; siRNA 2: 5′-GCUCAGUAUCAGAGAGAAUTT-3′),43 or GAPDH (5′-GUAUGACAACAGCCUCAAGTT-3′) using lipofectamine (Life Technology, Grand Island, NY). Knockdown efficiency was analyzed by Western blot analysis.
Statistical Analysis
Western blot, immunostaining, and immunofluorescence images were quantified using the computer image analysis software package ImageJ version 1.52c (NIH, Bethesda, MD; http://imagej.nih.gov/ij).44, 45 All measurements were made at least in triplicate (n = 3). One-factor analysis of variance was performed using the GraphPad Prism software version 5.01 to compare means between groups. Data were tested and found to be normally distributed. In cases in which the overall P value obtained from the analysis of variance analysis was significant (P < 0.05), Dunnet's post hoc test was used to compare the control and experimental samples. To compare multiple groups, the Bonferroni correction was used to calculate a revised cutoff for statistical significance that produces a combined 5% type I error probability.
Results
Persistent HCV Replication Leads to Increased PERK Activation
To examine the long-term effect of HCV replication on ER stress, autophagy, and cell survival, an HCV infection model was developed using nonproliferative PHHs. Infected and uninfected PHHs were harvested every 3 days, and HCV replication in the infected culture was measured using Western blotting targeting the HCV core protein during 15 days. The highly permissive Huh-7.5 cell–based infection model was also used as an alternative method to investigate the effect of high-level HCV replication on cellular ER-stress response, autophagy, and cell survival. High-level replication of JFH-ΔV3-Rluc chimera virus could be seen in both nontransformed (PHHs) and transformed (Huh-7.5) cells (Supplemental Figure S1). It is now well established that viral infection–induced ER stress can lead to cell death or cell survival. HCV infection promotes cell survival because infected cultures can be maintained for 15 days in the PHH model and for >1 month in infected Huh-7.5 cells. The expression levels of GRP78 (alias BiP), which controls the activation of three UPR axes, were measured by Western blotting. BiP was induced in infected PHHs and Huh-7.5 cells. PERK, IRE1α, and ATF6α expression levels also increased because of HCV infection and remained elevated in the infected culture for 15 days in PHHs and for 21 days in Huh-7.5 cells (Figure 1, A and B). Activation of PERK, IRE1α, or ATF6α in uninfected PHHs and Huh-7.5 cells was not seen when cultured for similar time points (Supplemental Figure S2). These results suggest that HCV replication produces chronic ER stress. Total RNA was isolated from HCV-infected Huh-7.5 cells, and mRNA levels of PERK, ATF6α, and IRE1α in infected Huh-7.5 cells were measured by real-time RT-PCR for 15 days (Figure 1, C–E). The mRNA level of PERK was induced significantly more (>30-fold) on day 15 compared with IRE1α or ATF6α, suggesting that HCV-induced ER stress leads to preferential activation of the PERK axis. PERK is an integral component of the UPR that phosphorylates eukaryotic translation initiation factor-2α. In a previous publication, we found increased eukaryotic translation initiation factor-2α phosphorylation in liver biopsy specimens of patients with chronic HCV infection and in HCV-infected explant cirrhotic livers due to activation of the PERK axis.22
Figure 1.

Hepatitis C virus (HCV) infection activates endoplasmic reticulum (ER) stress response and induces expression of unfolded protein response (UPR) genes in infected primary human hepatocytes (PHHs) and Huh-7.5 cells. A: Cell lysates prepared from infected PHHs were examined for the expression of ER-stress chaperone, binding immunoglobulin protein (BiP), and expression levels of protein kinase RNA-activated–like endoplasmic reticulum kinase (PERK), activating transcription factor-6α (ATF6α), and inositol-requiring enzyme-1α (IRE1α) by Western blot analysis. B: Cell lysates of HCV-infected Huh-7.5 cells were prepared at different days, and the expression levels of PERK, IRE1α, and ATF6α were examined by Western blot analysis. C: mRNA levels of PERK in infected Huh-7.5 cells at different days measured by real-time quantitative RT-PCR (RT-qPCR). D: mRNA levels of IRE1α in infected Huh-7.5 cells at different days measured by real-time RT-qPCR. E: mRNA levels of ATF6α in infected Huh-7.5 cells at different days measured by real-time RT-qPCR. ∗∗P < 0.01, ∗∗∗P < 0.001 versus day 0.
Persistent HCV Infection Leads to Activation of Nrf2 Oncogenic Signaling
Nrf2 is a transcription factor in the cap‘n'collar subfamily that can be activated by PERK and oxidative stress.46, 47, 48, 49 The number of cells that express viral core protein increased in the infected Huh-7.5 cell cultures during 21 days by immunostaining (Figure 2A). Quantification of core-positive cells by ImageJ shows that the level of HCV replication and spread increased with time (Figure 2B). Predominant nuclear accumulation of Nrf2 was found in 100% of cells infected with HCV starting from day 9, suggesting that HCV replication promotes Nrf2 activation and nuclear translocation (Figure 2C). Nrf2 nuclear expression was quantified by ImageJ, thus confirming the time-dependent activation of Nrf2 signaling after HCV infection (Figure 2D). An siRNA-based silencing approach was used to determine which UPR branch was responsible for Nrf2 activation in HCV infection. At day 21, infected Huh-7.5 cells were transfected with siRNAs targeted to PERK, ATF6α, or IRE1α. After 72 hours, cell lysates were examined for Nrf2 protein expression by Western blot analysis. Silencing PERK resulted in decreased Nrf2 expression, suggesting that Nrf2 signaling is activated by the PERK axis in persistent HCV infection (Supplemental Figure S3A). Silencing IRE1α and ATF6α did not reduce the expression of Nrf2 (Supplemental Figure S3, B and C). In the nucleus, phosphorylated Nrf2 forms a heterodimer with its partner sMAF (v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog) and ATF4 to bind a consensus sequence known as the antioxidant responsive element (ARE).47 This interaction regulates the transcription of several gene families encoding enzymes involved in antioxidation, detoxification, heme, lipid, and sugar metabolism, nicotinamide adenine dinucleotide phosphate generation, and proteins involved in autophagy induction.50 Hsc70 and LAMP2A are vital to the uptake of cellular substrates by lysosomes for degradation during CMA. Because Nrf2 is a transcription factor, the possibility that Nrf2 induces Hsc70 and LAMP2A by directly binding to their promoter regions was examined. Two ARE (TGAnnnnGC) and three ARE-like (TGAnnnGC or TGAnnnnnGC) binding sites were identified in the LAMP2A promoter region. A similar search also revealed five ARE (TGAnnnnGC) and four ARE-like (TGAnnnGC or TGAnnnnnGC) sequences in the Hsc70 promoter (Figure 3A). It was examined whether Nrf2 activation induces expression of Hsc70 and LAMP2A using lysates prepared from HCV-infected PHHs by Western blotting (Figure 3B). These results indicate that expression of Hsc70 and LAMP2A was increased compared with uninfected PHHs at day 6 onward and remained elevated through 15 days. HCV infection resulted in increased Nrf2 expression and Nrf2 phosphorylation in the PHH model. The expression level of β-actin did not change, suggesting that HCV infection of PHHs induced CMA during infection. These results suggest that Nrf2 likely induces Hsc70 and LAMP2A directly. The increase in LAMP2A and Hsc70 expression levels in HCV-infected Huh-7.5 cells was verified by immunocytochemical staining. Compared with uninfected culture, the cytoplasmic expression of Hsc70 and LAMP2A was elevated from day 9 onward and remained high through day 21 (Figure 3, C and D). The staining intensity was quantified using ImageJ, which shows that persistent HCV infection induced expression of Hsc70 and LAMP2A (Figure 3, E and F). To determine whether this increase in expression of Hsc70 and LAMP2A occurs at the mRNA level, HCV-infected Huh-7.5 cells were collected at different time points, and mRNA levels of Hsc70 and LAMP2A were measured using real-time RT-PCR (Figure 3G). To determine whether silencing Nrf2 blocks the induction of LAMP2A and Hsc70 in HCV cell culture, infected cells at day 15 were treated with increasing concentrations of Nrf2 siRNA, and the expression levels of LAMP2A and Hsc70 were examined by Western blot analysis 48 hours after transfection. Nrf2 silencing decreased LAMP2A and Hsc70 expression (Figure 3H). To test whether Nrf2-induced CMA activation in late-stage HCV culture is a mechanism of cell survival, the viability of chronically infected cells was measured 21 days after infection after silencing LAMP2A and Nrf2 for 72 hours (Figure 3, I and J). LAMP2A silencing decreased cell viability by >90%, and Nrf2 silencing decreased cell viability by 70%. The amalgamation of these data suggest that Nrf2-induced CMA activation is required to improve cell survival during chronic HCV infection.
Figure 2.

Persistent hepatitis C virus (HCV) replication in Huh-7.5 cells induces nuclear factor erythroid 2–related factor (Nrf2) activation and nuclear translocation. A: Immunocytochemical staining of core protein in HCV-infected Huh-7.5 cells at different days after HCV infection. B: Quantification of core-positive infected Huh-7.5 cells. Three high-power fields (HPFs) were measured using ImageJ version 1.52c. C: Immunocytochemical staining of phosphorylated Nrf2 (pNrf2) in HCV-infected Huh-7.5 cells at different days after HCV infection. D: Quantification of nuclear Nrf2-positive infected Huh-7.5 cells; three high-power fields were measured using ImageJ. ∗∗∗∗P < 0.0001 versus day 0. Scale bars = 200 μm (A and C).
Figure 3.

Persistent hepatitis C virus (HCV) infection activates nuclear factor erythroid 2–related factor (Nrf2) that induces chaperone-mediated autophagy (CMA). A: The human lysosome-associated membrane protein 2A (LAMP2A) and heat shock cognate 70 kDa (Hsc70) promoter sequence were examined for the presence of antioxidant response element (ARE) consensus sequences (TGAnnnnGC) indicated by arrowheads and ARE-like sequences (TGAnnnGC or TGAnnnnnGC) shown by arrows. B: Western blot analysis shows expression levels of Nrf2, phosphorylated Nrf2 (pNrf2), Hsc70, and LAMP2A in infected primary human hepatocytes (PHHs) during 15 days. C: Expression of Hsc70 levels in HCV-infected Huh-7.5 cells by immunostaining. D: Expression of LAMP2A in HCV-infected Huh-7.5 cells by immunostaining of cytospin slides at different days. E: Quantification of cells expressing Hsc70 by ImageJ version 1.52c. F: Quantification of LAMP2A-positive cells by ImageJ. G:Hsc70 and LAMP2A mRNA levels in HCV-infected Huh-7.5 cells measured by real-time quantitative RT-PCR. H: Western blot shows expression of LAMP2A, Hsc70, and β-actin after siRNA-mediated silencing of Nrf2 in HCV-infected Huh-7.5 cells. I: MTT assay showing viability of HCV-infected Huh-7.5 cells with or without silencing LAMP2A. J: MTT assay showing viability of HCV-infected Huh-7.5 cells with or without Nrf2 silencing. ∗P < 0.05, ∗∗∗P < 0.001 versus day 0. Scale bars = 200 μm (C and D).
Stress-Induced Nrf2 Promotes Beclin1 Degradation by CMA
CMA is responsible for the degradation of cytosolic proteins that contain a consensus pentapeptide motif (KFERQ).51 Four KFERQ-like motifs were found in human beclin1, a protein critical for autophagy and autophagosome-endosome fusion (Figure 4A). Serum starvation is a well-established method for inducing CMA. Uninfected Huh-7.5 cells were cultured in serum-free media for 0, 2, 4, 6, and 24 hours, and beclin1 expression levels were examined by Western blot (Figure 4B). Beclin1 levels were decreased after 6 hours of serum starvation and remained low through 24 hours. Serum starvation also increased LAMP2A expression. It was then examined whether CMA induced during HCV infection degrades beclin1 in infected PHHs by using Western blotting (Figure 4C). Beclin1 expression was found to be decreased after day 9 in HCV-infected PHHs. Similar results were obtained in HCV infected Huh-7.5 cells (Figure 4D). It is possible that the decrease in beclin1 expression by Western blot analysis was due to decreased mRNA transcription. To eliminate this possibility, the effect of HCV infection on transcription of beclin1 was measured by real-time RT-PCR. The decreased protein expression of beclin1 was not due to mRNA transcription (Figure 4E). Silencing LAMP2A expression restored beclin1 expression in infected Huh-7.5 cells (Figure 4F). The levels of beclin1, LAMP2A, Hsc70, and Nrf2 were comparable to infected cultures that were transfected with scramble siRNA or siRNA targeted to GAPDH (Supplemental Figure S4). Interactions between beclin1 and the CMA machinery were examined using co-immunoprecipitation with antibodies against beclin1, LAMP2A, and Hsc70 that verified their interaction in uninfected, early-infected, and late-infected culture (Figure 4G). Uninfected Huh-7.5 cells cultured with sulforaphane, a known Nrf2 activator, showed Nrf2 nuclear stabilization (Figure 4H) and increased beclin1 degradation along with LAMP2A induction (Figure 4I). Lysosomes were isolated from uninfected Huh-7.5 cells, chronically HCV-infected Huh-7.5 cells, and serum-starved Huh-7.5 cells after ferrofluid labeling using a protocol described earlier.36 The purified lysosomes were incubated with recombinant GAPDH in the presence and absence of protease inhibitors for 20 minutes at 37°C using a protocol described earlier.37 The amount of GAPDH degradation in the presence and absence of protease inhibitors was examined by Western blotting (Figure 4J). The lysosomal uptake was calculated as the difference of GAPDH level with or without protease inhibitors (Figure 4K). These results suggest that CMA is induced in serum-starved cells and HCV-infected Huh-7.5 cells.
Figure 4.

Chaperone-mediated autophagy (CMA) targets beclin1 degradation in hepatitis C virus (HCV) infection. A: Schematic representation showing the presence of multiple pentapeptide motif (KFERQ-like), CMA motifs, and their location in beclin1 protein sequences. B: Western blot shows beclin1 and lysosome-associated membrane protein 2A (LAMP2A) expression in Huh-7.5 cells after serum starvation. C: Beclin1 expression in HCV-infected primary human hepatocytes (PHHs). D: Beclin1 expression in HCV-infected Huh-7.5 cells. E: Beclin1 mRNA levels in HCV-infected Huh-7.5 cells by real-time quantitative RT-PCR. F: Western blot analysis shows silencing LAMP2A expression restores beclin1 expression without altering HCV replication in infected Huh-7.5 cells. G: Co-immunoprecipitation shows interaction among beclin1, LAMP2A, and heat shock cognate 70 kDa (Hsc70) in uninfected and HCV-infected Huh-7.5 cells at 6 days and 21 days. H: Sulforaphane induces nuclear factor erythroid 2–related factor (Nrf2) activation and its nuclear translocation in uninfected Huh-7.5 cells. I: Western blot analysis demonstrating expression of LAMP2A and beclin1 in uninfected Huh-7.5 cells after treatment with increasing concentrations of sulforaphane. J: CMA functional assay showing lysosomal degradation of recombinant glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the presence and absence of protease inhibitors. K: Quantification of GAPDH bands in Western blot showing amount of GAPDH uptake and degradation during serum starvation and late HCV infection. ∗∗∗P < 0.001 versus day 0. Scale bars = 200 μm.
Persistently Infected HCV Culture Shows Impaired Autophagy Flux
Many previous publications, including those from our own laboratory, have shown that p62 is an autophagy flux protein and that its expression is decreased due to autophagy induction.23, 28 Because decreased autophagy flux has been observed in hepatocellular carcinoma due to p62 accumulation, it was examined whether beclin1 degradation during chronic HCV infection alters p62 levels. Huh-7.5 cells infected with HCV were harvested at 0, 3, 6, 9, 12, 17, 23, and 30 days, and their HCV core protein and p62 expression was quantified using flow cytometry. At each time point, cells were divided into two parts and stained for HCV core or p62. HCV core expression was compared with p62 expression at different time points during 30 days. Uninfected Huh-7.5 cells showed high p62 expression, whereas HCV infection resulted in a time-dependent decrease in p62 expression. On day 12, the number of cells expressing p62 had decreased 75% compared with the uninfected control. On days 17, 23, and 30, p62 expression was increased compared with the uninfected culture (Figure 5, A and B). The percentage of infected cells increased rapidly from day 3 to day 12 (10% to 83%) and remained constant afterward (Figure 5C). The percentage of HCV-infected cells decreased slightly (86% to 79%) between days 23 and 30 due to impaired autophagy. The results were confirmed through Western blotting (Figure 5D). The impaired autophagy flux was verified in HCV-infected cultures (day 30) with or without bafilomycin A1 treatment. Bafilomycin A1 treatment increased LC3B-I/II and p62 levels in infected Huh-7.5 cells (Figure 6A). In contrast, no further enhancement of LC3-II or p62 levels was observed in the HCV-infected culture, thereby confirming that autophagy flux was blocked in persistently infected Huh-7.5 cells. To verify that the loss of beclin1 is the reason for the impaired autophagy flux (accumulation of p62) in late-infected culture, levels of LC3B-I/II and p62 were examined after silencing or overexpression of beclin1 by transient transfection. Silencing beclin1 led to the accumulation of p62 and LC3B-I/II in uninfected Huh-7.5 cells (Figure 6B). On the other hand, overexpression of beclin1 decreased expression of p62 and LC3B-I/II in a dose-dependent manner (Figure 6C). The expression of HCV core did not change.
Figure 5.

Quantification of hepatitis C virus (HCV) core and autophagy flux protein p62 expression in infected Huh-7.5 cells by flow cytometry. Infected cells were collected by trypsin-EDTA treatment and washed with phosphate-buffered saline (PBS). Infected cells were fixed for 10 minutes with 2% paraformaldehyde and permeabilized for 15 minutes using ice-cold methanol. For p62 detection, cells were incubated with a rabbit monoclonal antibody tagged with Alexa Fluor 488 (1:100) for 60 minutes. For detection of HCV core, cells were incubated with mouse monoclonal antibody (1:200) for 60 minutes. After three washes, cells were incubated with Alexa Fluor 488–conjugated secondary antibody (1:500) for 60 minutes. After these steps, cells were washed three times with PBS and analyzed separately by flow analysis. A: Percentage of core-positive and p62-positive cells were compared at each time points. B: Histogram of p62-positive cells from 0 to 30 days. P62 positivity decreases until day 12; after that p62 expression increases. C: Histogram showing the expression levels of HCV core. The number of core-positive cells gradually increases from 0 to 12 days. D: Expression levels of p62 and HCV NS3 in infected Huh-7.5 cultures by Western blot analysis. d, days; FITC-A, fluorescein isothiocyanate A.
Figure 6.

Beclin1 silencing and overexpression modulates autophagy flux in infected Huh-7.5 cells. A: Uninfected and persistently infected Huh-7.5 cells at day 21 were treated with Bafilomycin A for 24 hours. Cell lysates were examined for the expression of p62 and light chain 3B II (LC3-II) levels by Western blot analysis to determine the effect of persistent hepatitis C virus (HCV) replication on autophagy flux. B: Uninfected Huh-7.5 cells were treated with different concentration of beclin1 siRNA for 48 hours. Cell lysate were examined for p62 and LC3 by Western blot analysis. C: HCV-infected Huh-7.5 cells at day 21 were transfected with beclin1 cDNA (pcDNA Beclin1) for 72 hours. Cell lysate were prepared and examined for expression of p62 and LC3B-I/II by Western blotting.
Beclin1 Loss Inhibits Autophagy Initiation
Beclin1 is involved in the initiation of the canonical form of autophagy and autophagosome-endosome fusion. It was examined whether beclin1 loss through CMA inhibited autophagy at the level of initiation (isolation of autophagosome membrane) or conjugation (autophagosome formation). To study the initiation stage of autophagy, a green fluorescent protein (GFP)–ATG5 (K130R) mutant plasmid that is incapable of conjugating to ATG12 was used.52, 53 Uninfected and infected Huh-7.5 cells were transfected with the GFP-ATG5 (K130R) plasmid clone for 24 hours, and cells were examined using fluorescence microscopy. Numerous strong isolation membrane GFP signals punctate fluorescence signals in uninfected Huh-7.5 cells, and diffuse cytoplasmic GFP signals in infected cells were observed 6 days after infection (Figure 7A). By contrast, localized crescent-shaped structures with GFP-ATG5 puncta were observed in late-infected HCV cultures (day 21). These results are consistent with previous reports suggesting that the GFP-ATG5 (K130R) mutant is associated with the membrane without ATG12 conjugation.52, 53 The number of membrane-associated puncta is significantly lower in late-infected culture than in uninfected culture, suggesting that beclin1 loss also reduced autophagy initiation (Figure 7B). The expression levels of ATG16L1 and ATG7 were comparable in uninfected and HCV infected cultures, suggesting that beclin1 loss did not affect the elongation step of autophagy machinery or autophagosome formation (Figure 7C).
Figure 7.

Hepatitis C virus (HCV)–infected Huh-7.5 cells at day 6 and day 21 were transfected with green fluorescent protein (GFP)–autophagy-related 5 (ATG5) (K130R) mutant plasmid DNA for 24 hours. As a control, uninfected Huh-7.5 cells treated with 400 nmol/L Torin 1 or 5 mmol/L 3-methyladenine (3-MA) for 6 hours. Nuclei were stained with Hoechst 33342. Fluorescence images were generated at 60×. A: Accumulation of ATG-GFP puncta formation in uninfected Huh-7.5 cells, in early- and late-infected culture, and under different treatment conditions. B: Quantification of GFP puncta using three high-power fields were measured using ImageJ version 1.52c. C: Expression levels of autophagy-related 16-like 1 (ATG16L1) and ATG7 by Western blot analysis. ∗∗∗∗P < 0.0001. Scale bars = 5 μm. Original magnification, ×60.
Beclin1 Degradation by CMA Inhibits Autophagosome-Lysosome Fusion
Autophagy proceeds through several steps, beginning with the formation of a phagophore complex, which then elongates to form a complete double-membrane vesicle called an autophagosome. The fusion of an autophagosome with a lysosome forms an autolysosome, where contents are degraded (Figure 8A). The presence of HCV-induced autophagic vacuoles and the ultrastructure of HCV-infected Huh-7.5 cells were determined using transmission electron microscopy. Cytoplasmic areas of 10 different cells were imaged under the grid, and the number of autophagosomes were counted in uninfected, early-infected, and late-infected Huh-7.5 cells. The number of autophagic vacuoles was increased in early-infected Huh-7.5 cells compared with uninfected Huh-7.5 cells (Figure 8B). Double-membrane autophagosomes were quantified by counting 10 early-infected (day 6) and 10 late-infected (day 21) Huh-7.5 cells using electron microscopy. Autophagic vacuoles per field per cell were compared between uninfected, early-infected, and late-infected Huh-7.5 cells. The number of autophagic vacuoles was decreased in late-infected cells (Figure 8C). Lysosomes are small and autophagosome-lysosome fusion occurs rapidly, so these events are rarely seen under electron microscopy. Autophagosomes can be distinguished from autolysosome vesicles because most autolysosomes show a small break in their double-membrane wall. Electron microscopy was used to compare autophagy induction in early-infected and late-infected culture. Large numbers of autolysosome-like structures with partial dissolution of the double membrane were present exclusively in early-infected culture (Figure 8D). By contrast, most autophagosomes present in the late-infected culture exhibited unbroken complete double-membrane structures (Figure 8E). Next, the effect of stress-induced beclin1 loss was examined on autophagosome-lysosome fusion in early-infected and late-infected Huh-7.5 cells using a monomeric red fluorescent protein (RFP) –GFP tandem fluorescent-tagged LC3.54 LC3 is known to specifically associate with autophagosome membranes and is the standard marker used to monitor autophagy in liver cells. This construct contains a GFP moiety that is sensitive to lysosomal degradation and is quenched at low pH and an RFP moiety that is stable under lysosomal conditions. Therefore, autophagosomes will appear as yellow puncta when examined using fluorescence microscopy, whereas autolysosomes will appear as red puncta (Figure 9A). Red puncta were exclusively seen in early-infected culture, whereas most late-infected cells showed yellow puncta, suggesting that autophagosome-lysosome fusion was inhibited in late infection (Figure 9B). The numbers of green and red puncta were calculated under 10 different high-powered fields, and the number of red puncta was significantly higher in early-infected Huh-7.5 cells, comparable with cells treated with Torin 1 (Figure 9C). Impaired fusion of autophagosomes and lysosomes was also seen in HCC cells. We posit that the HCV-induced impaired autophagic flux was not due to lysosomal dysfunction because equal levels of DQ-BSA degradation was found between early (day 6) and late (day 21) infected culture (Figure 9D). Finally, a flow-based assay was used to compare autophagy induction between early- and late-infected Huh-7.5 cultures using monodansylcadaverine (MDC). This compound accurately detects autophagy induction in living cells based on the incorporation of MDC after amino acid starvation.55 MDC-labeled vesicles are induced after starvation, and autophagy inhibitors and phosphatidylinositol 3- kinase inhibitors effectively inhibit MDC incorporation. Infected cells were treated with MDC at 0.05 mmol/L MDC for 10 minutes at 37°C and then washed four times with PBS. Intracellular fluorescence and MDC uptake were quantified by flow cytometry. The mean positivity of MDC fluorescence increased from 3% to 44.7% at day 6 of infection (early), indicating autolysosome formation (Figure 9E). By contrast, the late culture shows only 0.2% fluorescence compared with the untreated culture. The mean fluorescence intensity of MDC was significantly higher in early-infected culture (Figure 9F). These results suggest that autophagy is induced in early-infected cultures and the autophagosome-lysosome fusion was inhibited in late-infected cultures.
Figure 8.

The effect of beclin1 loss on autophagosome-lysosome formation by transmission electron microscopy (TEM). A: Illustration showing steps involved in autophagosome-lysosome fusion that lead to formation of autolysosome during macroautophagy. B: Representative image of TEM between uninfected, early hepatitis C virus (HCV) infection, and late HCV infection. C: During TEM studies, cytoplasmic areas of 10 different cells were captured under the grid, and the number of autophagosomes were counted in uninfected, early-infected, and late-infected Huh-7.5 cells. D: Autophagic vacuoles with a partial degradation of double membrane (boxed areas in enlarged image) are seen in early infected culture. E: In contrast, autophagic vacuoles with complete double membrane are seen in late infected culture. ∗∗∗P < 0.001. Scale bars: 2 μm (B); 200 nm (D and E).
Figure 9.

Persistent hepatitis C virus (HCV) infection impairs autophagosome-lysosome fusion. A: Plasmid construct used for transfection. B: Early-infected and late-infected cells were transfected with monomeric red fluorescent protein (RFP)–green fluorescent protein (GFP) tandem fluorescent-tagged light chain 3 (LC3) plasmid and after 24 hours examined under fluorescent microscopy. GFP and RFP puncta were counted under a fluorescence microscope. C: DQ-BSA degradation by lysosome protease remains the same between early- and late-infected culture. D: Torin 1–treated and early-infected culture only shows red fluorescence because the GFP signal was quenched due to low pH associated with efficient autophagosome-lysosome fusion. Uninfected Huh-7.5 cells and late-infected Huh-7.5 cells show both green and red, meaning impaired autophagosome-lysosome fusion. Impaired autophagy flux is not due to lysosomal dysfunction. E: HCV-infected Huh-7.5 cells at day 6 and day 21 were incubated with 0.05 mmol/L monodansylcadaverine (MDC) for 10 minutes at 37°C. After this treatment, cells were washed three times with phosphate-buffered saline (PBS) (pH 7.4) and then suspended in 0.5 mL of PBS and subjected to flow analysis. F: Graph presenting mean fluorescence intensity (MFI) of MDC. ∗∗∗P < 0.001 versus uninfected. Scale bars = 10 μm (C and D).
Beclin1 Loss by CMA Inhibits Autophagosome-Endosome Fusion and Endocytosis
Beclin1is a member of the phosphatidylinositol 3-kinase complex, which is involved in vesicle trafficking during autophagy and other cellular processes. Whether beclin1 loss blocks the fusion of autophagosomes with endosomes and lysosomes was examined. Interactions among beclin1, ATG14L, and UV radiation resistance-associated gene protein are needed for autophagosome-endosome fusion, whereas Rab7, various tethering proteins, and soluble N-ethylmaleimide-sensitive factor activating protein receptor proteins are required for autophagosome-lysosome fusion (Figure 10A). The colocalization of Rab7 and LC3-GFP was examined in uninfected, early-infected, and late- infected Huh-7.5 cells by confocal microscopy (Figure 10B). Confocal microscopy was used to examine the colocalization of beclin1 and LC3 in uninfected, early-infected, and late-infected culture (Figure 10C). Confocal images were quantified using ImageJ. LC3 puncta were increased in early- and late-infected Huh-7.5 cells (Figure 10D). Colocalization of LC3/Rab7 and LC3/beclin1 was observed in uninfected culture and early-infected culture but decreased in late HCV infection (Figure 10, E and F). In the late-infected culture, co-localization of LC3 and beclin1 was almost completely abolished because only green puncta were observed. The colocalization of Rab7 with LC3-GFP was also completely lost in late-infected culture. The expression of Rab5 and Rab7 was also found to be decreased during persistent HCV infection without altering the expression of VPS34 (Figure 10G). Immunostaining showed increased accumulation of the EGFR on the surface of infected Huh-7.5 cells 21 days after infection compared with uninfected Huh-7.5 cells (Figure 11A). Receptor-mediated endocytosis and lysosomal degradation of EGFR are thought to be necessary for attenuation of ligand-induced EGFR signaling. It was then examined whether the surface expression of EGFR in HCV-infected cells could activate EGFR signaling by inhibiting its degradation. Western blot analysis shows that EGF ligand treatment induced EGFR degradation without HCV infection but inhibited EGFR degradation in HCV-infected Huh-7.5 cells (Figure 11, B and C). EGF stimulation induced EGFR degradation in uninfected cells as seen through fluorescence puncta, and this pathway was inhibited in HCV-infected culture (Figure 11D). Taken together, our data suggest that beclin1 degradation by CMA inhibits endocytosis by specifically blocking autophagosomes and endocytic vesicle fusion.
Figure 10.

Beclin1 and Rab7 degradation inhibits autolysosome formation. A: Diagram showing involvement of beclin1 and Rab7 in autophagosome-endosome-lysosome fusion during the formation of autolysosome. B: Hepatitis C virus (HCV)–infected Huh-7.5 cells were harvested at day 6 and day 21 and transfected with light chain 3 (LC3) green fluorescent protein (GFP). Cells were stained with DAPI (blue), antibodies against Rab7 (red), and antibodies against beclin1 (red). Co-localization of beclin1 with LC3 was examined under fluorescence microscope. C: Co-localization of Rab7 with LC3 was examined under confocal microscopy. D: Quantification of LC3 puncta in uninfected and HCV-infected Huh-7.5 cells. E: Co-localization of LC3 with Rab7 quantified by ImageJ version 1.52c. F: Co-localization of beclin1 and LC3 in uninfected, early-infected, and late-infected Huh-7.5 cells by ImageJ. G: Western blot showing the expression of Rab7, Rab5, and VPS34 in HCV-infected Huh-7.5 cells. Arrows represent LC3 GFP puncta in autophagosomes. ∗P < 0.05. Scale bars = 10 μm (B and C). Original magnification, ×60.
Figure 11.

Persistent hepatitis C virus (HCV) infection blocks endocytosis and epidermal growth factor receptor (EGFR) degradation. A: Uninfected and HCV-infected Huh-7.5 cells (day 21) were trypsinized and immobilized onto a glass slide, and the membrane expression of EGFR was examined by immunostaining. Expression of HCV core was used to monitor infection efficiency. B and C: The ligand-induced EGFR degradation between uninfected and HCV-infected culture was examined at day 21 infected culture by Western blot. D: Fluorescence microscopy images of double staining (red indicates HCV; green, EGFR; and blue, DAPI) shows ligand-induced EGFR degradation in uninfected cells; HCV infection prevents EGFR degradation. Scale bars = 200 μm (A and D). ATG14L, autophagy-related 14-like; LC3, light chain 3.
Discussion
The UPR improves ER function through activation of autophagy. Adaptive ER stress promotes cell survival, whereas nonadaptive ER stress can promote cell death. All three branches of the UPR were induced in 15 to 21 days, suggesting that persistent HCV replication induces the UPR to improve cell survival under chronic ER stress. The PERK axis of the UPR was activated more strongly than the IRE1α and ATF6α branches in persistent HCV infection.9 This finding is consistent with previous findings, such as the activation of PERK and phosphorylation of eukaryotic translation initiation factor-2α seen in chronically infected liver biopsy specimens and explanted cirrhotic livers.22 UPR activation, particularly in the PERK axis, resulted in Nrf2 phosphorylation and nuclear translocation in persistently infected Huh-7.5 cells. The levels of Nrf2 activation correlated with PERK activation, and silencing PERK inhibited Nrf2 activation. These results are consistent with one of our previous studies, which found that treatment with a PERK inhibitor or taurodeoxycholic acid, an ER-stress inhibitor, prevented Nrf2 nuclear translocation, suggesting that activation of the PERK pathway is responsible for the sustained nuclear translocation of Nrf2.25 Nrf2 induced expression of LAMP2A and Hsc70 at the mRNA and protein levels in HCV-infected culture. ER stress–induced CMA activation leads to the degradation of both mutant and wild-type p53 as a mechanism of cell survival during chronic HCV infection.24 Using siRNA and co-immunoprecipitation assays, CMA was found to promote LAMP2A-associated beclin1 degradation in the lysosome. Although no difference in the levels of autophagy proteins (ATG7, ATG16L1) between early and late infection were observed, the expression levels of Rab5 and Rab7 decreased in late-infected culture. This finding is consistent with a previous report.52 It is possible that increased stress or the CMA response also degrades other proteins involved in autophagosome-endosome-lysosome fusion to impair autophagy. Because CMA is responsible for the degradation of 30% of cytosolic proteins under stress conditions, future investigations will address whether CMA is also involved in degradation of other autophagy proteins that contribute to the autophagy inhibition.
Beclin1 is a component of the phosphatidylinositol 3-kinase complex, which mediates vesicle and endolysosomal trafficking as well as the fusion of autophagosomes with endosomes and lysosomes.56 Beclin1 has been implicated in several disease-associated processes, such as neurodegeneration and tumorigenesis, and plays a role in apoptosis.56 A previous report claims that monoallelic deletion of beclin1 results in increased incidence of spontaneous malignancies, including lung cancer, lymphoma, and HCC.57 Moreover, loss of beclin1 has been correlated with poor prognosis in various cancers, including HCC, in clinical studies.58, 59 Similarly, many earlier studies reported spontaneous HCC development in mice as a result of a mosaic deletion of ATG5 or hepatic disruption of ATG7, indicating that impairment in autophagy promotes HCC development.60, 61 p62 accumulates in most HCC cases with viral and nonviral origins, suggesting that there may be a mechanism common to viral and nonviral insults promoting HCC.62 CMA is induced in HCC as a compensatory mechanism for impaired autophagy flux.23
To study the effect of beclin1 loss on autophagy during persistent infection, autophagy flux protein p62 was quantitatively measured using flow cytometry. HCV induced autophagy and decreased p62 expression up to day 12. Expression of p62 increased in HCV culture after day 12, corresponding to high levels of HCV replication and loss of beclin1. After day 17, the level of HCV decreased slightly because of impaired autophagy. These results suggest that HCV can replicate in the presence of impaired autophagy flux up to day 30. Beclin1 loss appears to inhibit autophagy at the initiation step, as evidenced by the decrease in ATG5 puncta, and at the autolysosome formation step. Impaired autolysosome formation was confirmed using electron microscopy, flow cytometry with MDC, and transfection with an LC3-GFP-RFP plasmid. The DQ-BSA assay results support the conclusion that the impaired autophagy flux seen in late infection was not due to decreased lysosomal activity. The significance of Nrf2 activation and LAMP2A induction in high ER stress was examined by measuring cell viability after Nrf2 and LAMP2A silencing. Silencing Nrf2 and LAMP2A decreased cell survival, suggesting that high ER stress inhibits autophagy to enhance cell survival during persistent infection. The effect of HCV infection on the cellular autophagy response has been reported by numerous publications over the years.63, 64, 65 We speculate that HCC development during chronic HCV infection occurs during a long period as a cellular adaptive response to a highly stressed ER, which occurs during the stage of liver cirrhosis. Our results provide an explanation for how different autophagy processes compensate for each other to support cell survival under high ER stress. The autophagy compensatory cell survival mechanism is relevant for HCC development in the highly stressed cirrhotic liver microenvironment because Nrf2 translocation and degradation of beclin1 and accumulation of autophagy flux p62 occur during HCC development in liver cirrhosis.
Acknowledgments
We thank the Xuejun Jiang Laboratory at the Memorial Sloan Kettering Cancer Center (New York, NY) for supplying the ATG-GFP K130R mutant clone, and Marco Corazzari, Ph.D., (Cell Biology Laboratory, INMI-IRCCS) and Lazzaro Spallanzani and Via Portuense (Rome, Italy) for the RFP-GFP-LC3 plasmid construct.
Y.A., C.M.S., S.C., D.D.W., R.P., Z.H., K.W, A.B., and W.W. performed experiments; K.R. prepared the confocal images used in this paper; Y.A. prepared figures for publication; S.D. analyzed the results and wrote the paper.
Footnotes
Supported by NIH grants CA089121 (S.D.), AI103106 (S.D.), and 1P20 GM121288-01 (Y.A.) and Louisiana Clinical and Translational Science Center grant U54 GM104940.
Disclosures: None declared.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2018.06.022.
Supplemental Data
Supplemental Figure S1.

Hepatitis C virus (HCV) replication in nonproliferative primary human hepatocytes and proliferative Huh-7.5 cell cultures. Primary human hepatocytes (PHHs) were infected with cell culture–grown JFH-ΔV3-Rluc virus (HCV genotype 2a) at a multiplicity of infection of 0.1 by overnight incubation. The next day cells were incubated with fresh media with 10% human serum. A: HCV replication in the PHH model was confirmed by the measurement of core protein levels by Western blot. B: Western blot analysis shows time-dependent increase in the expression of NS3 protein in Huh-7.5 cells infected with JFH-ΔV3-Rluc virus (HCV genotype 2a). C: Renilla luciferase activity in infected Huh-7.5 cells for 21 days. D: Immunostaining for infected Huh-7.5 cells using HCV core monoclonal antibody. E: Quantification of core protein positive cells by ImageJ version 1.52c. Scale bar = 200 μm. Original magnification, ×100.
Supplemental Figure S2.

A: Western blot analysis shows basal expression levels of protein kinase RNA-activated–like endoplasmic reticulum kinase (PERK), activating transcription factor-6α (ATF6α), inositol-requiring enzyme-1α (IRE1α), and β-actin in uninfected primary human hepatocytes (PHHs) collected at different time points as shown in Figure 1. B: Expression levels of PERK, IRE1α, ATF6α, and β-actin in uninfected Huh-7.5 cells at similar time points described in Figure 1. HCV, hepatitis C virus.
Supplemental Figure S3.

Silencing of protein kinase RNA-activated–like endoplasmic reticulum kinase (PERK) inhibits nuclear factor erythroid 2–related factor (Nrf2) signaling in hepatitis C virus–infected Huh-7.5 cells. Infected Huh-7.5 cells (day 21) were treated with different concentrations of siRNAs targeted to three branches of unfolded protein response using lipofectamine for 72 hours. Twenty micrograms of cell lysate proteins was separated by SDS-PAGE. The Nrf2 level was examined by Western blotting. Knockdown efficiency of PERK (A), inositol-requiring enzyme-1α (IRE1α) (B), and activating transcription factor-6α (ATF6α) (C) and their effect on the expression levels of Nrf2, heat shock cognate 70 kDa (Hsc70), and lysosome-associated membrane protein 2A (LAMP2A).
Supplemental Figure S4.
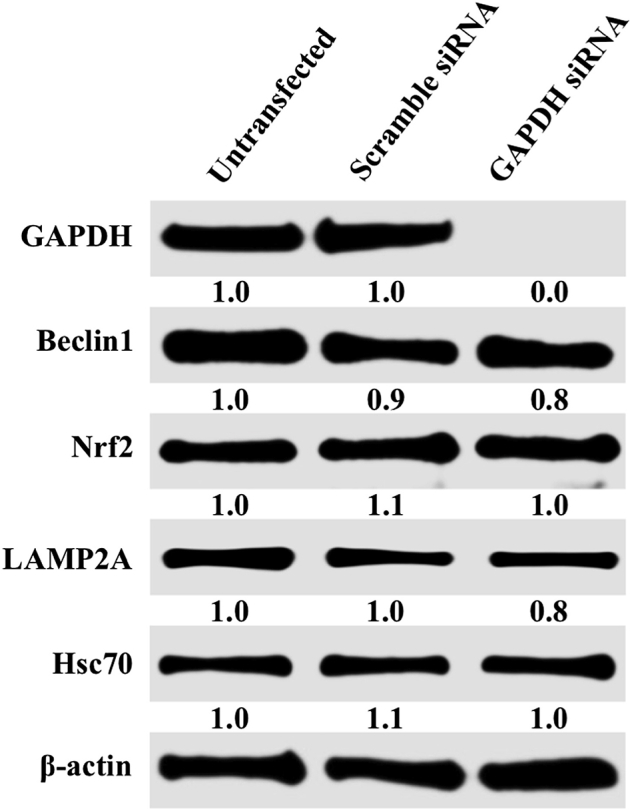
Western blotting shows silencing-unrelated siRNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) siRNA transfection does not alter beclin1, nuclear factor erythroid 2–related factor (Nrf2), lysosome-associated membrane protein 2A (LAMP2A), and heat shock cognate 70 kDa (Hsc70). β-Actin was used as an internal control.
References
- 1.Petruzziello A., Marigliano S., Loquercio G., Cozzolino A., Cacciapuoti C. Global epidemiology of hepatitis C virus infection: an up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol. 2016;22:7824–7840. doi: 10.3748/wjg.v22.i34.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.AASLD/ADSA HCV Guidance Panel Hepatitis C guidance: AASLF-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology. 2015;62:932–954. doi: 10.1002/hep.27950. [DOI] [PubMed] [Google Scholar]
- 3.Rusyn I., Lemon S.M. Mechanisms of HCV induced liver cancer: what did we learn from in vitro and animal studies. Cancer Lett. 2014;345:210–215. doi: 10.1016/j.canlet.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheel T.K., Rice C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartenschlager R., Penin F., Lohmann V., Andre P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 7.Baiceanu A., Mesdom P., Lagouge M., Foufelle F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol. 2016;12:710–722. doi: 10.1038/nrendo.2016.124. [DOI] [PubMed] [Google Scholar]
- 8.Tardif K.D., Mori K., Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J Virol. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tardif K.D., Mori K., Kaufman R.J., Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem. 2004;279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 10.Tardif K.D., Waris G., Siddiqui A. Hepatitis C virus, ER-stress, and oxidative stress. Trends Microbiol. 2005;13:159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Waris G., Turhson J., Hassanein T., Siddiqui A. Hepatitis C virus (HCV) constitutively activates Stat-3 via oxidative stress: role of Stat-3 in HCV replication. J Virol. 2005;79:1569–1580. doi: 10.1128/JVI.79.3.1569-1580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Gong G., Waris G., Tanveer R., Siddiqui A. Human hepatitic C virus NS5A protein alters intracellular calcium levels, induces oxidative stress and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci U S A. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joyce M.A., Walters K.A., Lamb S.E., Yeh M.M., Zhu L.F., Knetman N., Doyle J.S., Katze M.G., Tyrrell D.L. HCV induces oxidative and ER-stress and sensitize infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009;5:e1000291. doi: 10.1371/journal.ppat.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sir D., Chen W.L., Choi J., Wakita T., Yen T.S., Ou J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ke P.Y., Chen S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shinohara Y., Imajo K., Yoneda M., Tomeno W., Ogawa Y., Kirikoshi H., Funakoshi K., Ikeda M., Kato N., Nakajima A., Saito S. Unfolded protein response pathways regulate hepatitis C virus replication via modulation of autophagy. Biochem Biophys Res Commun. 2013;432:326–332. doi: 10.1016/j.bbrc.2013.01.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merquiol E., Uzi D., Mueller T., Goldenberg D., Nahmias Y., Xavier R.J., Tirosh B., Shibolet O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One. 2011;6:e24660. doi: 10.1371/journal.pone.0024660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandra P.K., Bao L., Song K., Aboulnasr F.M., Baker D.P., Shores N., Wimley W.C., Liu S., Hagedorn C.H., Fuchs S.Y., Wu T., Balart L.A., Dash S. HCV infection selectively impairs type I but not the type III IFN signaling. Am J Pathol. 2014;184:214–229. doi: 10.1016/j.ajpath.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurt R., Chandra P.K., Aboulnasr F., Panigrahi R., Ferraris P., Aydin Y., Reiss K., Wu T., Balart L.A., Dash S. Chaperone-mediated autophagy targets IFNAR1 for lysosomal degradation in free fatty acid treated HCV cell culture. PLoS One. 2015;10:e0125962. doi: 10.1371/journal.pone.0125962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shuda M., Kondoh N., Imazeki N., Tanaka K., Okada T., Mori K., Hada A., Arai M., Wakstsuki T., Matsubara O., Yamamoto N., Yamamoto M. Activation of the ATF6, XBP1 and GRP78 genes in human hepatocellular carcinoma: a possible involvement of the ER-stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–614. doi: 10.1016/s0168-8278(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 21.Dash S., Chava S., Aydin Y., Chandra P., Ferraris P., Chen W., Balart L.A., Wu T., Garry R.F. Review: Hepatitis C virus infection induces autophagy as a pro-survival mechanism to alleviate hepatic ER-stress response. Viruses. 2016;23:8. doi: 10.3390/v8050150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandra P.K., Gunduz F., Hazari S., Ramazan K., Panigrahi R., Poat B., Bruce D., Cohen A.J., Behorquez H.E., Carmody I., Loss G., Balart L.A., Wu T., Dash S. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS One. 2014;9:e108616. doi: 10.1371/journal.pone.0108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chava S., Lee C., Aydin Y., Chandra P.K., Dash A., Chedid M., Thung S.N., Nayak N.C., Wu T., Dash S. Chaperone-mediated autophagy compensate for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget. 2017;8:40019–40036. doi: 10.18632/oncotarget.16685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aydin Y., Chatterjee A., Chandra P.K., Chava S., Chen W., tendon A., Dash A., Chedid M., Moehlen M.W., Regenstein F., Balart L.A., Cohen A., Lu H., Wu T., Dash S. Interferon-alpha induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol Commun. 2017;1:256–269. doi: 10.1002/hep4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aydin Y., Chedid M., Chava S., Williams D.D., Liu S., Hagedorn C.H., Sumitran-Holgersson S., Reiss K., Balart L.A., Lu H., Dash S. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci Rep. 2017;23:9223. doi: 10.1038/s41598-017-10087-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S., Massey D.C., Menzies F.M., Moreau K., Narayan U., Renna M., Siddiqi F.H., Underwood B.R., Winslow A.R., Rubinsztein D.C. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 27.Dreux M., Chisari F.V. Viruses and the autophagy machinery. Cell Cycle. 2010;9:1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 28.Galluzzi L., Baehrecke E.H., Ballabio A., Boya P., Bravo-San Pedro J.M., Cecconi F. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control center for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahu R., Kaushik S., Clement C.C., Cannizzo E.S., Scharf B., Follenzi A., Potolicchio I., Nleves E., Cuervo A.M., Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu P., Li X., Ruan L., Xu H., Liu Q. Effect of siRNA PERK on fluoride-induced osteoblastic differentiation in OS732 cells. Biol Trace Elem Res. 2014;159:434–439. doi: 10.1007/s12011-014-9988-5. [DOI] [PubMed] [Google Scholar]
- 32.Rohde M., Daugaard M., Jensen M.H., Helin K., Nylandsted J., Jaattela M. Members of heat-shock protein 70 family promote cancer growth by distinct mechanisms. Genes Dev. 2005;19:570–582. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji G.-R., Yu N.-C., Xue X., Li Z.-G. PERK-mediated autophagy in osteosarcoma cells resists ER stress-induced cell apoptosis. Int J Biol Sci. 2015;11:803–812. doi: 10.7150/ijbs.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiffin R., Christian C., Knecht E., Cuervo A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rah B., Rasool R., Nayak D., Yousuf S.K., Mukherjee D., Kumar L.D., Goswami A. PAWR-mediated suppression of BCL2 promotes switching of 3-azido withaferin A (3-AWA)-induced autophagy to apoptosis in prostate cancer cells. Autophagy. 2015;11:314–331. doi: 10.1080/15548627.2015.1017182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd-Lewis B., Krueger C.C., Sargeant T.J., D'Angelo M.E., Deery M.J., Feret R., Howard J.A., Lilley K.S., Watson C.J. Stat3-mediated alterations in lysosomal membrane protein composition. J Biol Chem. 2018;293:4244–4261. doi: 10.1074/jbc.RA118.001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macri C., Wang F., Tasset I., Schall N., Page N., Briand J.-P., Cuervo A.M., Muller S. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy. 2015;113:472–486. doi: 10.1080/15548627.2015.1017179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang J., Gao N., Kaufman R.J., Ron D., Harding H.P., Lehman M.A. Translation attenuation by PERK balances ER-glycoprotein synthesis with lipid-linked oligosaccharide flux. J Cell Biol. 2007;176:605–616. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugiura K., Moro Y., Futamura K., Matsumoto K., Hashimoto N., Nishizawa Y., Nagasaka T., Saito H., Tomita Y., Usukuma J. The unfolded protein response is activated in differentiating epidermal keratinocytes. J Invest Dermatol. 2009;129:2126–2135. doi: 10.1038/jid.2009.51. [DOI] [PubMed] [Google Scholar]
- 40.Saeed M., Suzuki R., Watanabe N., Masaki T., Tomonaga M., Muhammad A., Kato T., Matsuura Y., Watanabe H., Wakita T., Suzuki T. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem. 2011;286:37264–37273. doi: 10.1074/jbc.M111.259085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Florczyk U., Czauderna S., Stachurska A., Tertil M., Nowak W., Kozakowska M., Poellinger L., Jozkowicz A., Loboda A., Dulak J. Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic Biol Med. 2011;51:1882–1892. doi: 10.1016/j.freeradbiomed.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Son Y.O., Pratheeshkumar P., Roy R.V., Hitron J.A., Wang L., Zhang Z., Shi X. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J Biol Chem. 2014;289:18660–28675. doi: 10.1074/jbc.M114.595496. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Li D.-D., Wang L.-L., Deng R., Tang J., Shen Y., Guo J.-F., Wang Y., Xia L.-P., Feng G.-K., Liu Q.Q., Huang W.-L., Zeng Y.-X., Zhu X.-F. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–898. doi: 10.1038/onc.2008.441. [DOI] [PubMed] [Google Scholar]
- 44.McCloy R.A., Rogers S., Caldon C.E., Lorca T., Castro A., Burgess A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13:1400–1412. doi: 10.4161/cc.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen E.J. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec (Hoboken) 2013;296:378–381. doi: 10.1002/ar.22641. [DOI] [PubMed] [Google Scholar]
- 46.Cullinan S.B., Zhang D., Hannink M., Arvisais E., Kaufman R.J., Diehl A.J. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menegon S., Columbano A., Giordano S. The dual roles of NRf2 in cancer. Trends Mol Med. 2016;22:578–593. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Karin M., Dhar D. Liver carcinogenesis: from naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis. 2016;37:541–546. doi: 10.1093/carcin/bgw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L., Tan J., Miao Y., Lei P., Zhang Q. ROS and autophagy: interactions and molecular mechanisms. Cell Mol Neurobiol. 2015;35:615–621. doi: 10.1007/s10571-015-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitsuishi Y., Motohashi H., Yamamoto M. The Keap-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:1–13. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaushik S., Cuervo A.M. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganley I.G., Wong P.M., Gammoh N., Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–743. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 55.Munato D.B., Colombo M.I. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 56.Levine B., Liu R., Dong X., Zhong Q. Beclin orthologys: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015;25:9. doi: 10.1016/j.tcb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qu X., Yu J., Bhagat G., Furuya N., Hibshoosh H., Troxel A., Rosen J., Eskelinen E.L., Mizushima N., Ohsumi Y., Cattoretti G., Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi Y.H., Ding Z.B., Zhou J., Qiu S.J., Fan J. Prognostic significance of beclin1 dependent apoptotic activity in hepatocellular carcinoma. Autophagy. 2009;5:380–382. doi: 10.4161/auto.5.3.7658. [DOI] [PubMed] [Google Scholar]
- 59.Qiu D.M., Wang G.L., Chen L., Xu Y.Y., He S., Cao X.L., Qin J., Zhou J.M., Zhang Y.X., Q E. The expression of beclin-1, an autophagy gene in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer. 2014;14:327. doi: 10.1186/1471-2407-14-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takamura A., Komatsu M., Hara T., Sakamoto A., Kishi C., Waguri S., Eishi Y., Hino O., Tanaka K., Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Umemura A., he F., Taniguchi K., Nakagawa H., Yamachika S., Font-Burgada J., Zhong Z., Subramaniam S., Raghunadan S., Duran A., Linares J.F., Reina-Campos M., Umemura S., Valasek M.A., Seki E., Yamaguchi K., Koike K., Itoh Y., Diaz-Meco M.T., Moscat J., Karin M. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival if stresses HCC-initiating cells. Cancer Cell. 2016;29:935–948. doi: 10.1016/j.ccell.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bao L., Chandra P.K., Moroz K., Zhang X., Thung S.N., Wu T., Dash S. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014;96:149–154. doi: 10.1016/j.yexmp.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang L., Tian Y., Ou J.H. HCV induces the expression of Rubicon and UVRAG to temporarily regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015;11:e1004764. doi: 10.1371/journal.ppat.1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sir D., Kuo C.F., Tian Y., Liu H.M., Huang E.J., Jung J.U., Machida K., Ou J.H. Replication of hepatitis C virus on autophagosomal membranes. J Biol Chem. 2012;287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sir D., Liang C., Chen W.L., Jung J.U., Ou J.H. Perturbation of autophagy pathway by hepatitis C virus. Autophagy. 2008;4:8330–8831. doi: 10.4161/auto.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.