

Abstract
Physiology and behaviour are critically dependent on circadian regulation via a core set of clock genes, dysregulation of which leads to metabolic and sleep disturbances. Metabolic and sleep perturbations occur in spinal muscular atrophy (SMA), a neuromuscular disorder caused by loss of the survival motor neuron (SMN) protein and characterized by motor neuron loss and muscle atrophy. We therefore investigated the expression of circadian rhythm genes in various metabolic tissues and spinal cord of the Taiwanese Smn−/−;SMN2 SMA animal model. We demonstrate a dysregulated expression of the core clock genes (clock, ARNTL/Bmal1, Cry1/2, Per1/2) and clock output genes (Nr1d1 and Dbp) in SMA tissues during disease progression. We also uncover an age- and tissue-dependent diurnal expression of the Smn gene. Importantly, we observe molecular and phenotypic corrections in SMA mice following direct light modulation. Our study identifies a key relationship between an SMA pathology and peripheral core clock gene dysregulation, highlights the influence of SMN on peripheral circadian regulation and metabolism and has significant implications for the development of peripheral therapeutic approaches and clinical care management of SMA patients.
Introduction
Circadian regulation is critical for many biological processes, disruption of which causes diverse metabolic disorders (1). The suprachiasmatic nucleus (SCN), whose major external cue is light, is the central pacemaker and synchronizes peripheral circadian oscillators (2), all of which are sustained in a cell-autonomous manner by core clock genes and their protein products (3). Circadian locomotor output cycles kaput (CLOCK) and aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL or BMAL1) activate transcription of Period (PER1, 2 and 3) and Cryptochrome (CRY1 and 2) genes, the protein products of which negatively regulate CLOCK-ARNTL/BMAL1 expression (4,5). Numerous clock gene mutants develop metabolic and muscle defects (6), highlighting the link between circadian regulation and metabolic homeostasis.
Spinal muscular atrophy (SMA) is a fatal autosomal recessive disorder in children characterized by spinal motor neuron degeneration and progressive muscle weakness (7,8). The disease-determining survival motor neuron 1 (SMN1) gene located on chromosome 5 is deleted or mutated on both alleles in SMA patients. While a highly homologous gene, SMN2, exists on the same chromosome, it undergoes alternative exon 7 splicing (9) to yield a truncated SMN2 protein (10) unable to compensate for loss of the full-length product (9). The SMN protein plays a role in small nuclear ribonucleoprotein (snRNP) assembly, pre-mRNA splicing and actin dynamics as well as in the regulation of axonal mRNA localization (11–13). Nonetheless, it is still not known why loss of the SMN protein leads to the specific pathophysiology of SMA.
Although motor neurons are amongst the most severely afflicted cells in SMA, tissues outside the central nervous system (CNS) including heart (14,15), pancreas (16), liver (17), skeletal muscle (18,19), spleen (20–22), thymus (22), the gastrointestinal tract (23) and lung (24) are also affected. Interestingly, many of these organs have metabolic functions and display intrinsic circadian gene expression (25). Various studies have reported significant metabolic abnormalities in SMA animal models and patients such as altered fatty acid metabolism, hyperlipidemia, hyperglycemia, hyperglucagonemia increased hepatic insulin sensitivity, glucose intolerance, development of diabetes mellitus, diabetic ketoacidosis as well as glucose and insulin aberrations (16,26–33). In addition to such metabolic perturbations, SMA patients display abnormal sleep (34), including altered sleep microstructure (35), nocturnal hypoxaemia and hypercapnia (36). These disruptions could point to a perturbed circadian phenotype. Indeed, we have recently demonstrated an aberrant diurnal regulation of the glucocorticoid-Krüppel-like factor 15-branched chain amino acid (GC-KLF15-BCAA) metabolic pathway in serum, skeletal muscle, spinal cord (SC), liver, heart, white adipose tissue (WAT) and brown adipose tissue (BAT) of the Taiwanese Smn−/−;SMN2 mice (37).
We thus evaluated the hypothesis of a generalized circadian dysregulation in the severe Taiwanese Smn−/−;SMN2 SMA mouse model (38) and uncover for the first time that the Smn gene displays a diurnal regulation in various metabolic tissues during early development. Further, we demonstrate disruption of the diurnal expression of core clock genes and clock output genes in metabolic tissues during SMA disease progression. Importantly, we find that controlled light (CL) exposure restores the expression of circadian rhythm genes and attenuates the severe SMA phenotype with beneficial effects on survival and weight. Combined, our results highlight a dysregulation of circadian rhythm genes in SMA metabolic tissues and suggest a functional relationship between the SMN gene, peripheral clock regulation and metabolic homeostasis.
Results
Altered diurnal expression of core clock genes in SMA metabolic tissues and SC during disease progression
To investigate the expression of the core clock genes in different SMA tissues, we used the severe Taiwanese Smn−/−;SMN2 SMA mouse model (38). Upon pairing, breeding pairs were continuously entrained to a 12-h light:12-h dark cycle (LD12:12). Metabolic tissues regulated by a peripheral clock (Tibialis anterior (TA), liver, heart, WAT, BAT) and SC were harvested from Smn−/−;SMN2 mice and Smn+/−;SMN2 healthy littermates every 4 hrs (Zeitgeber time, ZT) over a 24-h time course (ZT0 = 8 am, ZT1 = 9 am, ZT5 = 1 pm, ZT9 = 5 pm, ZT13 = 9 pm, ZT17 = 1 am, ZT21 = 5 am). The expression profiles of Per1/2, Cry1/2, Clock and Bmal1 were determined from pre-symptomatic postnatal day (P) 2 and post-symptomatic P7 mice (19).
In pre-symptomatic P2 Smn−/−;SMN2 animals, we detected a tissue-specific disruption of the diurnal expression of core clock genes compared to control littermates (Fig. 1). We observed changes in amplitude, whereby SMA and healthy littermates followed the same oscillation pattern but with differential expression at specific ZTs (SC: Bmal1 (ZT1); liver: Bmal1 (ZT9); heart: Per2 (ZT9, ZT17); WAT: Bmal1 (ZT13), Per2 (ZT5), Cry1 (ZT5)). We also identified changes in phase, whereby SMA and healthy littermates displayed distinct oscillation patterns, whether they cycle in either experimental groups or only in one (SC: Clock; liver: Clock, Bmal1, Per2; heart: Bmal1). As circadian patterns are still in development and not yet fully established at this early time point (39,40), many clock genes do not yet display a diurnal oscillatory pattern in P2 tissues from either SMA mice or healthy littermates. However, we still detect significant differences in clock gene expression levels in these instances (liver: Per1, Cry2; heart: Per1; BAT: Per1).
Figure 1.
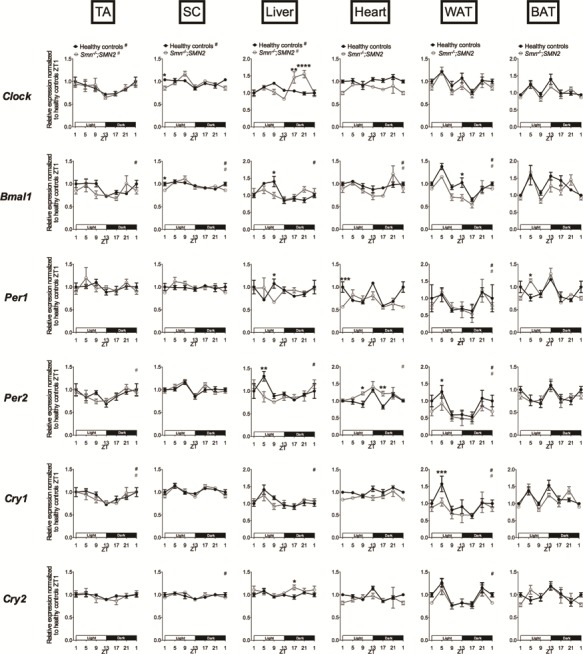
Dysregulation of diurnal expression of core clock genes in several tissues of pre-symptomatic SMA mice. Diurnal expression of core clock genes (Clock, Bmal1, Per1, Per2, Cry1 and Cry2) in TA, WAT, BAT, liver, heart and SC of postnatal day (P) 2 Smn−/−;SMN2 mice compared to healthy littermates. Data are mean ± SEM, n = 3–5 mice per ZT, *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ****P-value < 0.0001 (two-way ANOVA), # indicates circadian rhythmicity. ZT1 data are duplicated.
We next performed a similar analysis in symptomatic P7 Smn−/−;SMN2 mice and healthy littermates of the same age. Here, we found an even more prominent perturbation of clock gene expression in SMA tissues (Fig. 2), suggesting that diurnal expression defects in peripheral clocks increase with disease progression. Indeed, we observed several changes in amplitude, whereby SMA mice and healthy littermates followed the same oscillation patterns but with differential expression at specific ZTs (TA: Clock (ZT21); liver: Per1 (ZT17), Per2 (ZT1); heart: Bmal1 (ZT9), Per1 (ZT17), Per2 (ZT9, ZT13, ZT17), Cry2 (ZT17); WAT: Per1 (all ZTs), Per2 (ZT9, ZT13)). We also identified numerous changes in phase, whereby SMA mice and healthy littermates displayed distinct oscillation patterns, whether they cycle in either experimental groups or only in one (SC: Clock; liver: Clock; WAT: Cry2; BAT: Clock, Bmal1, Per2, Cry1). Similar to our P2 analysis, we find that many clock genes do not yet display a diurnal oscillatory pattern in P7 tissues from either SMA mice or healthy littermates. Nevertheless, differential expression between experimental groups is still observed, further highlighting dysregulated expression of core clock genes in SMA animals (TA: Per1; SC: Bmal1, Per1, Per2, Cry1; liver: Cry2; BAT: Per1, Cry2).
Figure 2.
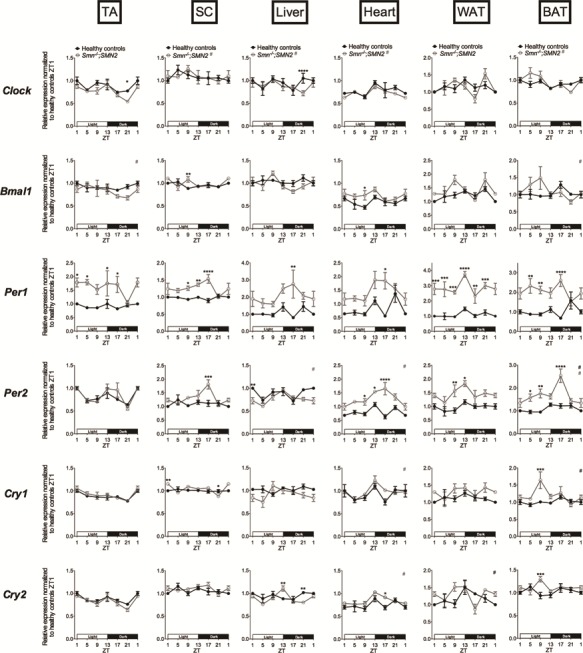
Dysregulation of diurnal expression of core clock genes in several tissues of symptomatic SMA mice. Diurnal expression of core clock genes (Clock, Bmal1, Per1, Per2, Cry1 and Cry2) in TA, WAT, BAT, liver, heart and SC of postnatal day (P) 7 Smn−/−;SMN2 mice compared to healthy controls. Data are mean ± SEM, n = 3–4 mice per ZT, *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ****P-value < 0.0001 (two-way ANOVA), # indicates circadian rhythmicity. ZT1 data are duplicated.
Our analysis of diurnal expression of core clock genes thus reveals a systemic dysregulation in pre- and post-symptomatic SMA tissues, which appears to worsen during disease progression.
Altered diurnal expression of clock output genes in SMA metabolic tissues and SC during disease progression
To determine if the perturbations observed with the core clock genes were reflected downstream in clock output genes, we evaluated the diurnal expression of Nr1d1 (also known as Rev-erb-α) and Dbp. Nr1d1 is a direct transcriptional target of the CLOCK/BMAL1 complex and modulates the interaction of circadian rhythms and metabolism (41). Dbp is also a transcriptional target of CLOCK/BMAL1, subsequently binding the Per1 promoter, thus promoting its cyclic behaviour (42).
In P2 pre-symptomatic animals, we only observed phase dist-inctions between Smn−/−;SMN2 animals and healthy littermates, whereby there was either a significant difference in expression level (Nr1d1: WAT, ZT13) or a diurnal oscillatory pattern in only one group (Nr1d1: liver, heart; Dbp: TA, SC, liver) (Fig. 3A).
Figure 3.
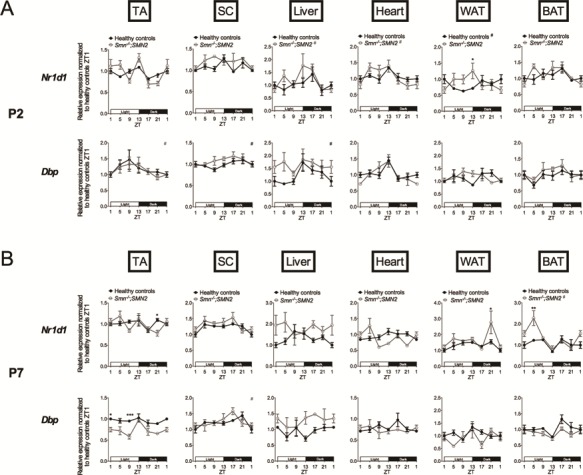
Dysregulation of diurnal expression of clock output genes in several tissues of pre-symptomatic and symptomatic SMA mice. Diurnal expression of clock output genes (Nr1d1, Dbp,) in TA, WAT, BAT, liver, heart and SC of postnatal day (P) 2 (A) and P7 (B) Smn−/−;SMN2 mice compared to healthy littermates. P2 data are mean ± SEM, n = 3–5 mice per ZT, P7 data are mean ± SEM, n = 3–4 per ZT, *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001 (two-way ANOVA), # indicates circadian rhythmicity. ZT1 data are duplicated.
In P7 symptomatic Smn−/−;SMN2 mice, we identified amplitude alterations when compared to healthy littermates (TA: Dbp (ZT1, ZT9); WAT: Nr1d1 (ZT21); BAT: Nr1d1 (ZT5)) (Fig. 3B). We also detect changes in phase, whether via significant differential expression levels (Nr1d1: TA, ZT21) or via a diurnal oscillatory pattern being detected in only one experimental group (Nr1d1: SC) (Fig. 3B).
Overall, our analysis of Nr1d1 and Dbp expression reveals that in addition to the core clock genes, the diurnal expression of canonical clock output effectors is also perturbed in SMA tissues during disease progression (Fig. 4).
Figure 4.
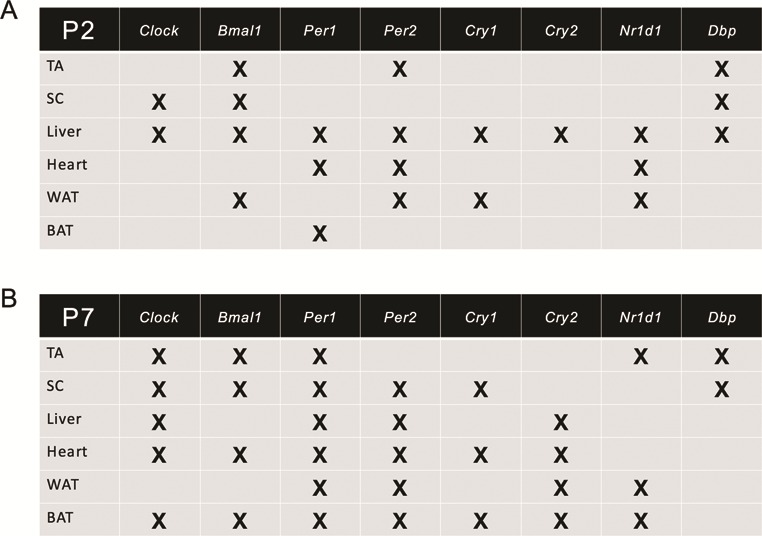
Summary of dysregulations of diurnal expression of clock and clock output genes in SMA tissues during disease progression. An X indicates either a change in phase, change in amplitude or differential expression of core clock and clock output genes in postnatal (P) 2 (A) and P7 (B) SMA mice compared to healthy littermates at one or more time points during a 24-h period.
Diurnal expression of myogenic regulatory factors is not impacted in muscle of SMA mice
In addition to the ubiquitously expressed core clock genes, muscle-specific myogenic regulatory factors such as MyoD and myogenin have also been reported as displaying a functional circadian expression profile in skeletal muscle of adult wild-type (WT) mice (43,44). As both MyoD and myogenin have been reported to be aberrantly expressed in Smn-depleted murine muscle tissue and cells (45,46), we assessed the diurnal expression of both genes in the TA of P2 and P7 Smn−/−;SMN2 mice and healthy littermates.
In P2 muscle, while MyoD did not cycle in either experimental group, we did detect a differential expression at ZT1, where MyoD levels were significantly greater in Smn−/−;SMN2 mice (Fig. 5A). Myogenin displayed a diurnal expression profile in healthy littermates only, accompanied by a significant decreased expression at ZT21 in Smn−/−;SMN2 mice (Fig. 5A).
Figure 5.
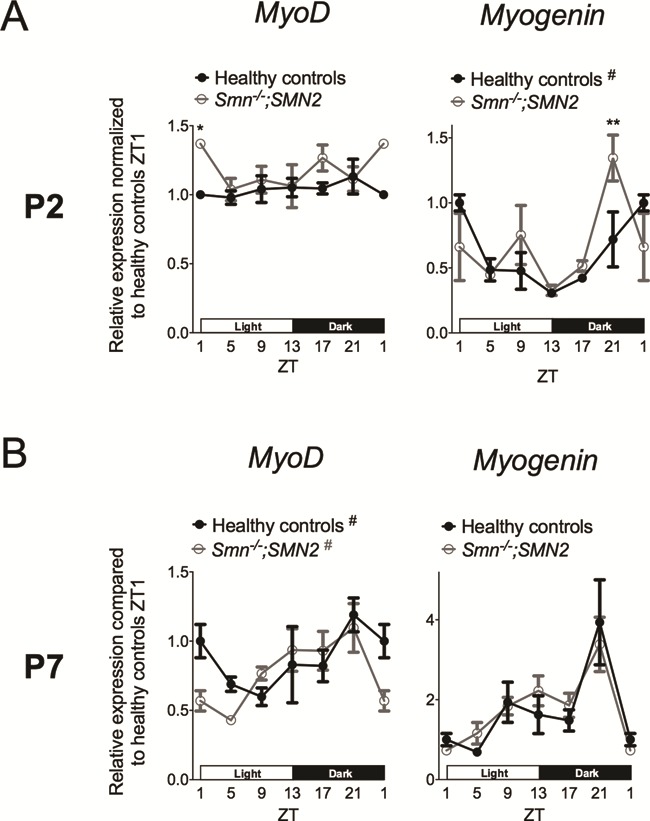
Diurnal expression of MyoD and myogenin is not significantly affected in skeletal muscle of SMA mice during disease progression. Diurnal expression of MyoD and myogenin in TA of postnatal day (P) 2 (A) and P7 (B) Smn−/−;SMN2 mice compared to healthy littermates. P2 data are mean ± SEM, n = 3–6 mice per ZT, P7 data are mean ± SEM, n = 3–5 mice per ZT, *P-value < 0.05, **P-value < 0.01 (two-way ANOVA), # indicates circadian rhythmicity. ZT1 data are duplicated.
At P7, a diurnal oscillatory pattern of MyoD was observed in both experimental groups while myogenin did not display a diurnal profile in either group (Fig. 5B). Furthermore, total expression levels between Smn−/−;SMN2 mice and healthy littermates were similar for both genes at all ZTs (Fig. 5B).
Thus, our analysis reveals that whilst small differences are observed in P2 muscle between Smn−/−;SMN2 mice and healthy littermates, SMA muscle pathology at P7 does not seem to impair the normal diurnal expression of MyoD and myogenin.
The Smn gene displays an age- and tissue-dependent diurnal expression profile
We next evaluated the diurnal expression of the disease-causing Smn gene in healthy littermates, which has never been assessed before. Surprisingly, we uncovered that Smn shows a 24-h differential expression pattern in P2 TA, liver, WAT and BAT (Fig. 6A). In P7 tissues, we found that while the expression patterns had somewhat changed between P2 and this later time point, the Smn gene presented a distinct 24-h differential expression pattern in TA, liver, heart, WAT and BAT (Fig. 6B).
Figure 6.
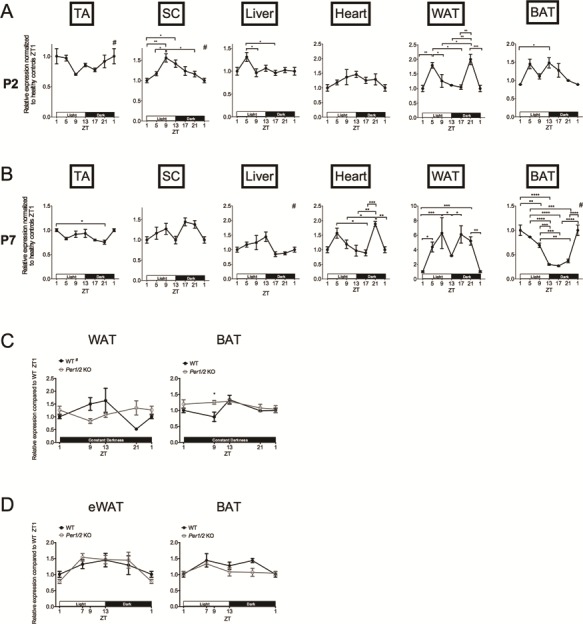
The Smn gene displays an age-, tissue- and light-dependent diurnal expression. Diurnal expression of Smn in TA, SC, liver, heart, WAT and BAT of postnatal day (P) 2 (A) and P7 (B) Smn+/−;SMN2 mice healthy control mice. P2 data are mean ± SEM, n = 3–4 mice per ZT, P7 data are mean ± SEM, n = 3–4 mice per ZT, *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001 (One-way ANOVA), # indicates circadian rhythmicity. ZT1 data is duplicated. (C) Diurnal expression of Smn in WAT and BAT from P7 WT and Per1/2 mutants (KO) exposed to constant darkness. Data are mean ± SEM, n = 3–7 mice per ZT, *P-value < 0.05 (two-way ANOVA), # indicates circadian rhythmicity. ZT1 data are duplicated. (D) Diurnal expression of Smn in epididymal eWAT and BAT from 3- to 4-month-old male WT and Per1/2 mutants (KO). Data are mean ± SEM, n = 3 mice per ZTs. ZT1 data ar duplicated.
As light is one of the most important external cues for the synchronization of internal peripheral clocks (47), we next determined if cycling of the Smn gene was dependent on light by investigating its expression in P7 tissues from WT animals that were exposed to constant darkness (D:D). As Per1 and Per2 were the clock genes whose expression levels were most significantly dysregulated in P7 SMA tissues (Fig. 2), we performed similar experiments with Per1/2 double mutants (48). WAT and BAT were chosen for analysis as they displayed the greatest variations in Smn, Per1 and Per2 expression over a 24-h period. Interestingly, we found that the diurnal expression of Smn is preserved in WAT of P7 WT D:D animals (Fig. 6C), albeit with a different pattern than observed in mice in LD12:12 (Fig. 6B). In BAT, however, the cyclic expression of Smn is completely lost in P7 WT D:D mice (Fig. 6C). Furthermore, the pattern of Smn expression is different in Per1/2 KO animals, reflected by a loss of cycling in WAT and a significantly greater expression in ZT9 BAT compared to WT animals (Fig. 6C). These results suggest that the diurnal expression of Smn in P7 adipose tissue may be partly regulated by light and intrinsic circadian regulators.
Finally, we investigated epididymal WAT (eWAT) and BAT from 3- to 4-month-old males to determine if cyclic expression of Smn is maintained in adulthood. We find that in adult mice, the Smn gene does not demonstrate a diurnal pattern (Fig. 6D). Furthermore, analysis of Smn expression in age- and gender-matched Per1/2 double mutants showed a similar expression pattern (Fig. 6D), further supporting that Smn levels are not influenced by circadian regulation in adult adipose tissue.
Our analysis of diurnal expression of core clock genes, clock output genes and Smn in P2 and P7 SMA and control mice suggests that SMN loss is associated with dysregulation of peripheral clock machinery and that this may be influenced by the fact that Smn itself demonstrates a diurnal pattern in highly metabolic tissues. Interestingly, the diurnal expression of Smn appears to be limited to early time points, pointing to a developmental interaction between Smn and circadian rhythmicity that could significantly impact the establishment of regulatory functions in these tissues.
A controlled-light environment improves phenotypic and molecular phenotypes of SMA mice
Given the impact of light on diurnal cycling of the Smn gene, we investigated the effect of light modulation on the phenotype of SMA mice. Breeding trio A (2 females and 1 male) and ensuing litters were first placed in our typical animal holding room, defined as the regular light (RL) environment. While these rooms were on an LD12:12, cages were not directly under the light source and light:dark disruptions could occur due to comings and goings of personnel. Breeding trio A was subsequently transferred and entrained to an LD12:12 in circadian isolation cages (defined as CL condition), where the light-emitting diode (LED) light source is directly above the cages and the LD12:12 phases are unperturbed. From birth, all pups were weighed daily and monitored for survival. Interestingly, when compared to SMA mice in the RL, SMA mice from the CL displayed a significantly enhanced lifespan and a significantly increased weight gain (Fig. 7A). Healthy littermates also showed increased weights in the CL environment, but to a lesser extent than SMA mice (Fig. 7B). Exposing the intermediate Smn2B/− SMA mouse model (49,50) to the same experimental paradigm also resulted in a significant increase in weight of Smn2B/− mice in CL (Supplementary Material, Fig. S1A), without a significant impact on survival (Supplementary Material, Fig. S1B). Intrinsic differences between both models have previously been reported (37,45,51) and the differential effect of CL on survival may be due to ranges in disease severity and overall metabolic influence on disease progression.
Figure 7.
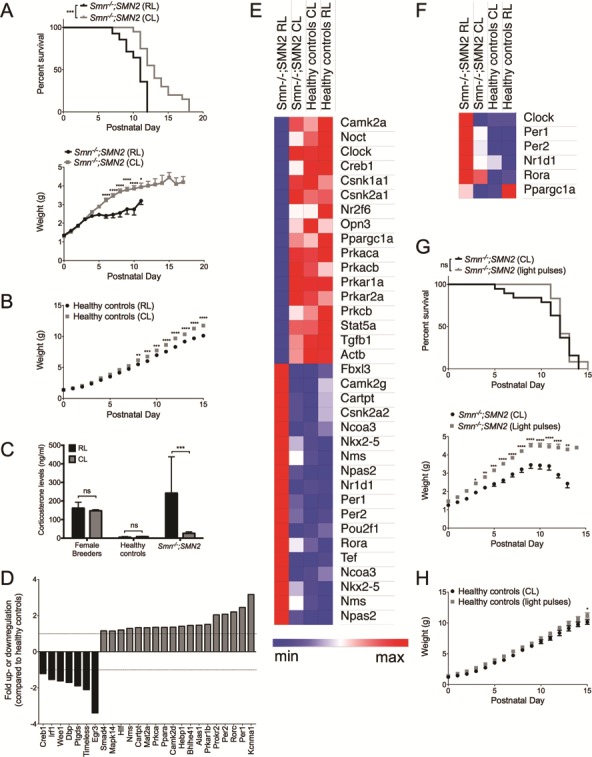
Light modulation impact molecular and phenotypic parameters in SMA mice. (A) Lifespan and weight development of Smn−/−;SMN2 mice in CL (n = 20) and RL (n = 14) conditions. P-value = 0.0003 for Kaplan–Meier log-rank (Mantel-Cox); Data are mean ± SEM, *P-value < 0.05, ****P-value < 0.0001 (two-way ANOVA). (B) Weight curves of healthy littermates in CL (n = 19) versus RL (n = 14). Data are mean ± SEM, **P-value < 0.01, ***P-value < 0.001, ****P-value < 0.0001 (two-way ANOVA). (C) Corticosterone levels in serum of female breeders (n = 2 for RL and CL), healthy littermates (n = 7 for RL and CL) and Smn−/−;SMN2 mice (n = 11 for RL and 9 for CL). Data are mean ± SEM, ***P-value < 0.001, ns = not significant (two-way ANOVA). (D) Circadian rhythm genes dysregulated in ZT9 BAT from P7 Smn−/−;SMN2 mice compared to healthy controls in CL conditions. (E) Heat map of circadian rhythm genes in ZT9 BAT of P7 Smn−/−;SMN2 mice and healthy littermates from regular (RL) and CL shows genes dysregulated in Smn−/−;SMN2 mice in RL and restored in Smn−/−;SMN2 mice in CL. n = 4 for all experimental groups. F. Heat map of circadian rhythm genes in ZT17 BAT of P7 Smn−/−;SMN2 mice and healthy littermates from RL and CL shows genes dysregulated in Smn−/−;SMN2 mice in RL and restored in Smn−/−;SMN2 mice in CL. n = 4 for all experimental groups. (G) Lifespan and weight development of Smn−/−;SMN2 mice in CL (n = 19) versus light pulses (n = 12). ns = not significant for Kaplan–Meier log-rank (Mantel-Cox); data are mean ± SEM, *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ****P-value < 0.0001 (two-way ANOVA). (H) Weight curves of healthy controls in CL (n = 16) and light pulses (n = 11). Data are mean ± SEM, *P-value < 0.05 (two-way ANOVA).
In addition to light, differences in parameters such as temperature and noise/stress between RL and CL conditions may have contributed to the improved phenotype of the SMA mice from the circadian isolation cages, thus confounding the intrinsic circadian influence. We firstly found that the temperatures in each environment were not significantly different from each other (RL: min 20.63°C ± 0.25, max 22.2°C ± 0.79; CL: min 20.4°C ± 0, max 23.1°C ± 0). To evaluate stress due to noise and human presence, we assessed serum corticosterone levels, as this glucocorticoid is a known stress indicator in rodents (52). Breeding trio B (2 females and 1 male) and ensuing litters were first placed in the CL, followed by a transition to the RL. Serum from both females and P7 pups was collected at the same time of day (ZT5) as GCs display a diurnal expression pattern (53). Corticosterone levels were not significantly different between female breeders and healthy littermates from both RL and CL (Fig. 7C). However, we did detect significantly reduced levels of corticosterone in serum of SMA mice from CL compared to RL (Fig. 7C), further supporting the beneficial impact of light modulation on SMA pathophysiology. Our results therefore suggest that differences in temperature and stress levels between RL and CL are not key mediators of the phenotypic changes observed in SMA mice from the circadian isolation cages.
We next wanted to determine if adapting the light environment had an effect at a molecular level. Firstly, we determined if additional circadian rhythm genes are dysregulated in SMA tissues, beyond the previously investigated clock and clock output genes (Figs. 1–3). For this purpose, we used a commercially available mouse circadian rhythm qPCR array (SABiosciences), which looks at a subset of 84 genes known to display and/or regulate circadian rhythmicity. For proof-of-concept, we used P7 ZT9 BAT, a time point and tissue where expression of many clock genes was significantly dysregulated in SMA mice compared to healthy littermates (Fig. 2). We observed a large number of dysregulated circadian rhythm genes in P7 BAT of SMA animals compared to healthy littermates (Fig. 7D and Supplementary Material, Table S1), further supporting a systemic dysregulation of circadian regulation in SMA.
We then compared the expression of 84 circadian rhythm genes in P7 ZT9 BAT from mice exposed to RL and CL (Supplementary Material, Table S1). Heat map comparison demonstrates a large number of genes specifically dysregulated in RL SMA mice that are restored to normalized levels in CL SMA mice (Fig. 7E). Using the publicly available PANTHER software (54), we interrogated the list of differentially expressed genes from Fig. 7E to determine specific protein classes, molecular functions, reactome pathways and GO biological processes (Table 1). Interestingly, in addition to the expected circadian rhythm-specific pathways, we identified functions such as glucocorticoid receptor signalling and regulation of transcription of RNA polymerase II, which we and others have demonstrated to be implicated in SMA pathogenesis (Fig. 7C) (37,55).
Table 1.
Function, protein class, Reactome pathway and GO biological process complete of dysregulated genes in P7 BAT from RL SMA mice generated with Panther software
| Genes dysregulated in SMA mice RL | Fold enrichment | P-value | |
|---|---|---|---|
| Molecular Function | Protein kinase activity | 8.16 | 1.40E-02 |
| Sequence-specific DNA binding transcription factor activity | 4.64 | 1.51E-02 | |
| Protein class | Non-receptor serine/threonine protein kinase | 11.78 | 1.24E-02 |
| Transcription factor | 5.41 | 5.06E-04 | |
| Reactome pathway | RORA activates gene expression | >100 | 3.14E-09 |
| Degradation of GLI2 by the proteasome | >100 | 2.35E-02 | |
| PPARA activates gene expression | >100 | 2.35E-02 | |
| Rora activates gene expression | >100 | 8.55E-09 | |
| Bmal1:Clock,Npas2 activates circadian gene expression | >100 | 9.35E-15 | |
| WNT mediated activation of DVL | >100 | 3.35E-04 | |
| PKA activation | >100 | 1.07E-05 | |
| PKA activation in glucagon signalling | >100 | 1.38E-05 | |
| DARPP-32 events | >100 | 2.21E-05 | |
| Trafficking of AMPA receptors | 69.76 | 1.73E-02 | |
| Vasopressin regulates renal water homeostasis via Aquaporins | 68.05 | 5.81E-04 | |
| Post NMDA receptor activation events | 63.41 | 2.29E-02 | |
| Factors involved in megakaryocyte development and platelet producti | 28.77 | 1.72E-02 | |
| MAPK family signaling cascades | 14.65 | 3.23E-02 | |
| Developmental Biology | 8.53 | 4.49E-03 | |
| GO biological process complete | Negative regulation of glucocorticoid receptor signaling pathway | >100 | 7.65E-04 |
| Development of secondary female sexual characteristics | >100 | 3.53E-03 | |
| Protein-chromophore linkage | >100 | 4.69E-03 | |
| Circadian regulation of gene expression | >100 | 9.62E-15 | |
| Entrainment of circadian clock by photoperiod | >100 | 2.40E-02 | |
| Circadian behavior | 95.12 | 3.71E-02 | |
| Peptidyl-serine phosphorylation | 25.37 | 1.00E-03 | |
| Positive regulation of sequence-specific DNA binding transcription fact | 19.84 | 4.18E-03 | |
| Cellular response to hormone stimulus | 15.5 | 2.72E-04 | |
| Morphogenesis of an epithelium | 10.09 | 3.76E-02 | |
| Embryonic morphogenesis | 9.47 | 1.12E-02 | |
| Cellular response to oxygen-containing compound | 8.52 | 2.46E-02 | |
| Regulation of secretion by cell | 8.29 | 3.00E-02 | |
| Positive regulation of transcription from RNA polymerase II promoter | 8.06 | 7.77E-05 | |
| Negative regulation of developmental process | 7.21 | 2.07E-02 | |
| Transcription, DNA-templated | 4.8 | 6.82E-03 | |
| Regulation of multicellular organismal process | 4.16 | 1.35E-03 | |
| Regulation of cellular protein metabolic process | 4.16 | 3.42E-02 | |
| Regulation of biological quality | 3.41 | 2.07E-02 | |
To evaluate if light modulation influences the diurnal expression of circadian rhythm genes, we compared the expression of a selected panel of genes in P7 ZT17 BAT from SMA mice and healthy littermates exposed to RL and CL. We chose to examine core clock genes (Clock, Per1 and Per2), clock output genes (Nr1d1 and Rora) and Ppargc1α, a transcriptional co-activator at the crossroads of circadian regulation and metabolism (56). Here, we found that CL did indeed restore the diurnal expression of Clock, Per1, Per2 and Nr1d1 genes in SMA mice (Fig. 7F). CL did not significantly impact Rora expression, while Ppargc1a levels seemed to be dependent on light environment irrespective of genotype (Fig. 7F). The considerable differences between RL and CL SMA BAT suggest that our reported differences in the diurnal expression profiles of clock and clock output genes between SMA mice and healthy littermates (Figs. 1–3) are most likely underestimated due to the experimental paradigm of synchronizing and regulating light exposure in the circadian isolation cages. Nevertheless, our results point to a beneficial influence of light modulation at a mechanistic and molecular level, particularly in restoring diurnal expression of circadian genes in BAT of symptomatic mice.
To further determine the role of light on the phenotypic changes in SMA mice, we transferred breeding trio C (2 females and 1 male) to LD12:12 in the CL and from birth, litters were exposed daily to light pulses during the dark cycle (1-h light pulses at ZTs 16 and 21). Breeding trio C and ensuing litters were subsequently maintained in the CL environment without light pulsing. From birth, all animals were weighed daily and monitored for survival. Disruption of the LD12:12 by light pulses had no effect on lifespan but led to enhanced weight gain in SMA animals compared to CL SMA mice (Fig. 7G). This effect was also observed in healthy littermates but to a much lesser extent (Fig. 7H). Several studies have reported an enhanced weight in adult rodents exposed to a disrupted LD cycle (57). As SMA mice display a more prominent weight gain in CL and light pulses conditions than healthy littermates, this suggests that SMN depletion increases sensitivity to changes in light modulation.
Finally, we set out to determine if light modulation influenced canonical pathologies in SMA SC and skeletal muscle, the two tissues that are the greatest contributors to SMA pathophysiology. The SC and TA of Smn−/−;SMN2 mice and healthy littermates in both RL and CL were harvested at ZT1. We first compared the expression of MuRF-1 and atrogin-1, well-characterized atrogenes (58), whose increase reflect the atrophy status of SMA muscle (37,51). While we observed that the CL environment did not influence the increased expression of MuRF-1 in the TA of SMA mice (Fig. 8A), atrogin-1 levels were significantly downregulated in muscle of CL SMA mice compared to RL SMA animals (Fig. 8B), indicating a reduced muscle atrophy. The expression of Fas and Pmaip1 genes have previously been demonstrated as being aberrantly regulated in SMA SC as well as markers of improved pathology (59,60). Here, we observed that Fas levels were not influenced by the light environment, regardless of the experimental group (Fig. 8C), whilst Pmaip1 levels displayed a non-significant trend towards a decreased expression in SC of CL SMA mice compared to RL SMA animals.
Figure 8.
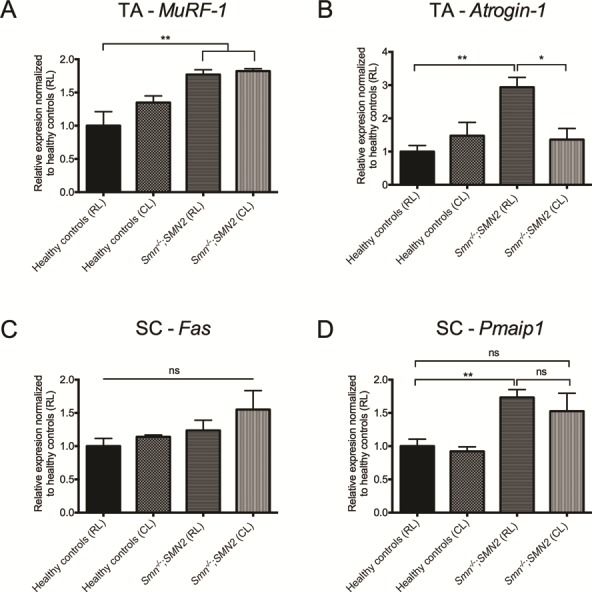
Light modulation impacts certain canonical pathological markers of SC and muscle pathology of SMA mice. TA and SC were harvested from postnatal (P) 7 Smn−/−;SMN2 mice (n = 7 for RL and 3 for CL) and healthy littermates (n = 5 for RL and 6 for CL) in CL and RL conditions at ZT1 (9 am). Expression of MuRF-1 (A) and atrogin-1 (B) in TA of P7 Smn−/−;SMN2 mice and healthy littermates from RL and CL. Expression of Fas (C) and Pmaip1 (D) in SC of P7 Smn−/−;SMN2 mice and healthy littermates from RL and CL. Data are mean ± SEM, *P-value < 0.05, **P-value < 0.01, ns = not significant (one-way ANOVA).
Combined, our results demonstrate that light modulation directly impacts several molecular and phenotypic parameters of SMA pathology and indicate that light as an external cue may have implications for overall metabolic health in SMA.
Discussion
Our work defines for the first time a perturbed regulation of circadian rhythm genes in SMA CNS and peripheral tissues, which may contribute to and/or be a consequence of the many metabolic and sleep dysfunctions reported in SMA mice and patients (27,28,30–32). We also demonstrate that Smn itself displays a diurnal expression pattern in a tissue- and age-dependent manner. Finally, we establish that SMA mice are sensitive to light modulation, resulting in significant molecular and phenotypic changes relevant to SMA pathology. Our recent report of dysregulated circadian regulation of the GC-KLF15-BCAA pathway in SMA mice (37) further supports a functional relationship between this neuromuscular disease and systemic disruption of peripheral core clock and circadian rhythm genes. As circadian defects have been reported in models of other neuromuscular disorders such as collagen VI myopathy (61) and amyotrophic lateral sclerosis (62), our results may also reflect a more general relationship between circadian homeostasis and neuromuscular health.
Very little is known on the establishment of peripheral clocks during early development. In the first weeks after birth, neonatal rodents are photo-sensitive but not yet entrained to a light:dark cycle and rather respond to external cues such as the mother’s presence/absence and feeding pattern (39,40). While rhythmic expression of peripheral clocks is detected in perinatal rodents, its development is tissue-specific and can take up to 3 weeks to fully be established (39,40). Furthermore, intrinsic peripheral clocks can also be influenced by extrinsic metabolic pathways (63), many of which are perturbed in SMA (64). In our study, diurnal expression is thus evidently still in development and highly malleable, reflected by the organ- and age-dependent expression patterns of the core clock, clock output, Smn, MyoD and myogenin genes. Nevertheless, we identify several amplitude and phase changes between SMA mice and healthy littermates in P2 and P7 tissues. Interestingly, early developmental defects have previously been reported in heart (15), liver (17), skeletal muscle (19) and SC (65) of SMA mice, which, in some cases, may be linked to metabolic and circadian disturbances. To date, besides reports of increased fat mass in SMA patients that may contribute to morbidity (66,67), not much is known about the role of adipose tissue in SMA pathogenesis. Our demonstration of circadian dysregulation combined with the diurnal expression profile of the Smn gene in adipose tissue highlights the need for further investigations on the relationship between this tissue, SMN function and SMA pathology. Of particular interest is BAT, which not only originates from the same progenitor cells as skeletal muscle (68,69) but also communicates with several tissues and cells that are pathologically affected in SMA (70).
Dysregulation of clock and clock output genes has recently been reported in a mouse model of muscle denervation (71), a phenomenon that typifies SMA pathogenesis (72). Indeed, surgically inducing denervation of skeletal muscle in adult mice revealed a decreased expression of Bmal1, Per1, Nr1d1 and Dbp and an increased expression of Per2 (71). While the relationship between circadian rhythm genes and denervation in neonatal muscle requires further investigation, this study may point to the potential contribution of intrinsic SMA muscle pathology to the perturbed expression of clock and clock output genes within this tissue.
Our analysis of core clock genes specifically highlights the diurnal expression of Per1 and Per2 as being significantly increased in most tissues of symptomatic mice. As this generalized transcriptional upregulation is observed for only Per1/2, there may be a clock machinery-independent process induced by the SMA phenotype. Indeed, the expression of Per1 and Per2 is influenced by glucose (73) and GCs (74), steroid hormones that regulate glucose homeostasis (75). Interestingly, we and others, have reported several glucose metabolism (16,32,33,76,77) and GC perturbations in SMA mice (Fig. 7C) (37). Future endeavours investigating the relationship between glycaemia, glucocorticoids and Per1/2 in SMA will therefore be of high interest.
Another key finding from our work is the observation that the Smn gene itself displays a diurnal expression pattern during early development in several tissues, which appears to be dependent on light and intrinsic circadian regulators. Previous studies have indicated the importance of SMN in early development (78), marked by high expression in fetal/perinatal tissues, followed thereafter by an age- and tissue-dependent decrease (79,80). This is reflected in our observed absence of diurnal Smn expression in eWAT and BAT of adult WT and Per1/2 KO mice. Early loss of diurnal Smn expression in the SC and key metabolic tissues may therefore contribute to the sleep, metabolism and peripheral perturbations reported in SMA. Interestingly, the diurnal oscillation observed in P2 SC is lost at P7 and may be related to the previously mentioned importance of SMN in very early stages of development (78–80), particularly in this key pathological tissue. However, diurnal oscillation of Smn, albeit with distinct patterns, is maintained throughout development in WAT and BAT, further highlighting the requirement for an in-depth investigation of the potential function that SMN may play in adipose tissue.
The use of PCR arrays to investigate light-dependent molecular changes in BAT of SMA mice has brought forward genes previously implicated in SMA pathology. Indeed, one of the genes downregulated in SMA mice in RL is Ppargc1a, which plays a critical role in energy metabolism (81) and is decreased in skeletal muscle from SMA patients (82). Importantly, we demonstrate that expression of Ppargc1a is normalized in CL SMA mice. Moreover, Prkacb and Prkaca, the catalytic subunits of protein kinase A (PKA), also display a reduced expression in RL SMA BAT, which is normalized in CL SMA mice. Interestingly, PKA is associated with an enhanced SMN stability (83) and its upregulation in BAT of CL mice may partly explain the beneficial effect of light modulation on SMA animals. Finally, Stat5a also displays a decreased expression in BAT from RL SMA mice and is upregulated in CL. Increased activation of Stat5 has previously been demonstrated to induce SMN expression (84) and pharmacological compounds that increase Stat5 improve disease phenotypes in SMA cellular and animal models (85,86). Thus, the beneficial influence of light modulation on SMA pathology may be due to the cumulative effect of not one, but several molecular changes. Our proof-of-concept comparison of BAT from SMA and healthy animals in CL and RL therefore uncovers several light-dependent molecular changes highly relevant to SMA pathogenesis. Moreover, our investigation of diurnal dysregulation in BAT highlights the importance for future in-depth assessments of circadian regulation in SMA tissues.
Our work thus uncovers for the first time a systemic dysregulation of circadian rhythm genes in several SMA tissues, which may have a direct impact on certain aspects of disease pathophysiology. Our study provides further evidence that SMA is a multi-system disease (87) and highlights circadian modulators (whether pharmacological, dietary or environmental) as potential novel therapeutic endeavours for the overall clinical management of patients.
Materials and Methods
Animals
Experiments were carried out in the Biomedical Sciences Unit, University of Oxford according to procedures authorized by the UK Home Office (Animal Scientific Procedures Act 1986). All experiments were performed in the severe Taiwanese Smn−/−;SMN2 SMA and the intermediate Smn2B/- mouse strains. Animals were housed either in the typical holding rooms of the animal facility (RL, LD12:12) or in circadian isolation cages (CL, LD12:12). For light pulse experiments, animals maintained in the circadian isolation cages were exposed to a 1-h light pulse during the dark phase at ZTs 16 and 21. For survival studies, animals were weighed daily and culled upon reaching their defined humane endpoint.
WT and Per1/2 double mutant mice (on a C57BL/6 J background) were bred in the animal facility of the University of Lübeck. All mice were individually housed under standard laboratory conditions under LD12:12 conditions, a room temperature of 22 ± 2°C and a relative humidity of 55 ± 5% with ad libitum access to food (breeding chow 3.457 kcal/g; Ssniff, Soest, Germany) and water. For constant darkness experiments, breeding pairs and pups were kept in constant darkness for 2 days prior to harvest. The experimental protocol was approved by the Committee on Animal Health and Care of the Government of Schleswig-Holstein, Germany.
qPCR
The liver, heart, WAT and BAT, SC and TA were harvested from P2 and P7 pups every 4 hrs over a 24-h period (ZT1 = 9 am, ZT5 = 1 pm, ZT9 = 5 pm, ZT13 = 9 pm, ZT17 = 1 am, ZT21 = 5 am). For single time point comparisons, tissues harvested at the same ZT were used. RNA was extracted with the RNeasy MiniKit (Qiagen) or the RNeasy Lipid Tissue MiniKit (Qiagen) for WAT and BAT. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCRs were performed using TaqMan Gene Expression Mastermix and Integrated DNA Technologies primers (see Supplementary Experimental Procedures). Housekeeping genes for each tissue was determined using the Mouse geNorm Kit and qbase+ software (Biogazelle) (see Supplementary Experimental Procedures).
Isolated WAT and BAT from WT and Per1/2 KO animals were harvested in 4- or 6-h intervals over a 24-h period (ZT1, 7, 13, 19) and stored in RNAlater solution (Life Technologies) at 4°C overnight. RNA was extracted using TRIzol reagent (Life Technologies) followed by cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). qPCRs were performed using TaqMan Gene Expression Mastermix and Integrated DNA Technologies primers (see Supplementary Experimental Procedures). Relative gene expression was quantified using the ΔΔ threshold cycle (Ct) method with adjustments for the amplification efficiencies of individual primer pairs. GAPDH was used as the reference gene.
PCR arrays
RNA from ZT9 BAT of P7 animals was extracted using the RNeasy Lipid Tissue MiniKit. cDNA was made using RT2 First Strand Kit. qPCRs were performed using Mouse Circadian Rhythm PCR arrays (PAMM-153Z, SABiosciences). Data was analysed with the RT Profiler PCR Array Data Analysis version 3.5 and mRNA expression was normalized to the two most stably expressed genes between all samples. Heat maps reflect log2 fold changes and were generated with MORPHEUS software. PANTHER software (54) was used to analyse differentially expressed genes.
Corticosterone ELISA
Analysis of corticosterone content in serum was performed with an ELISA kit (Abcam) following the manufacturer’s instructions. Serum samples were diluted 1:10 or 1:20.
Statistical analysis
All statistical analyses were done with Graphpad Prism software. When appropriate, a Student’s unpaired two-tail t-test, a one-way ANOVA followed by a Tukey’s multiple comparison test or a two-way ANOVA followed by a Sidak’s multiple comparison test was used. Outliers were identified via the Grubbs’ test. For the Kaplan–Meier survival analysis, the log-rank test was used and survival curves were considered significantly different at P-value < 0.05. CircWave v1.4 was used to determine circadian rhythmicity.
Supplementary Material
Acknowledgements
We are grateful to Dr Peter Oliver and the personnel at the Biomedical Sciences Unit at University of Oxford.
Conflict of Interest statement. None declared.
Funding
SMA Trust (to K.E.M. and M.B.); SMA Angels Charity (to M.B.); Association Française contre les Myopathies (Trampoline grant #20544 to M.B.); Muscular Dystrophy UK (to T.V.W. and K.E.M.); German Research Foundation (TR-SFB 134 to C.E.K.); Erasmus+ program scholarship (to L.M.W.).
References
- 1. Kim, T.W., Jeong, J.-H. and Hong, S.-C (2015) The impact of sleep and circadian disturbance on hormones and metabolism. Int. J. Endocrinol., 2015, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamazaki S., Numano R., Abe M., Hida A., Takahashi R., Ueda M., Block G.D., Sakaki Y., Menaker M. and Tei H. (2000) Resetting central and peripheral circadian oscillators in transgenic rats. Science, 288, 682–685. [DOI] [PubMed] [Google Scholar]
- 3. Dunlap J.C. (1999) Molecular bases for circadian clocks. Cell, 96, 271–290. [DOI] [PubMed] [Google Scholar]
- 4. Gekakis N., Staknis D., Nguyen H.B., Davis F.C., Wilsbacher L.D., King D.P., Takahashi J.S. and Weitz C.J. (1998) Role of the CLOCK protein in the mammalian circadian mechanism. Science, 280, 1564–1569. [DOI] [PubMed] [Google Scholar]
- 5. Kume K., Zylka M.J., Sriram S., Shearman L.P., Weaver D.R., Jin X., Maywood E.S., Hastings M.H. and Reppert S.M. (1999) mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell, 98, 193–205. [DOI] [PubMed] [Google Scholar]
- 6. Mayeuf-Louchart A., Staels B. and Duez H. (2015) Skeletal muscle functions around the clock. Diabetes Obes. Metab., 17, 39–46. [DOI] [PubMed] [Google Scholar]
- 7. Pearn J. (1978) Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet., 15, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crawford T.O. and Pardo C.A. (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis., 3, 97–110. [DOI] [PubMed] [Google Scholar]
- 9. Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 10. Lorson C.L. and Androphy E.J. (2000) An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet., 9, 259–265. [DOI] [PubMed] [Google Scholar]
- 11. Singh R.N., Howell M.D., Ottesen E.W. and Singh N.N. (2017) Diverse role of survival motor neuron protein. Biochim. Biophys. Acta, 1860, 299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hensel N. and Claus P. (2017) The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neuroscientist, 24, 54–72, 10.1177/1073858417705059. [DOI] [PubMed] [Google Scholar]
- 13. Donlin-Asp P.G., Bassell G.J. and Rossoll W. (2016) A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol., 39, 53–61. [DOI] [PubMed] [Google Scholar]
- 14. Rudnik-Schöneborn S., Heller R., Berg C., Betzler C., Grimm T., Eggermann T., Eggermann K., Wirth R., Wirth B. and Zerres K. (2008) Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet., 45, 635–638. [DOI] [PubMed] [Google Scholar]
- 15. Bevan A.K., Hutchinson K.R., Foust K.D., Braun L., McGovern V.L., Schmelzer L., Ward J.G., Petruska J.C., Lucchesi P.A., Burghes A.H.M. et al. (2010) Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet., 19, 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bowerman M., Swoboda K.J., Michalski J.-P., Wang G.-S., Reeks C., Beauvais A., Murphy K., Woulfe J., Screaton R.A., Scott F.W. et al. (2012) Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol, 72, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szunyogova E., Zhou H., Maxwell G.K., Powis R.A., Francesco M., Gillingwater T.H. and Parson S.H. (2016) Survival motor neuron (SMN) protein is required for normal mouse liver development. Sci. Rep., 6, 34635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walker M.P., Rajendra T.K., Saieva L., Fuentes J.L., Pellizzoni L. and Matera A.G. (2008) SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet., 17, 3399–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyer J.G., Murray L.M., Scott K., De Repentigny Y., Renaud J.-M. and Kothary R. (2013) Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomson A.K., Somers E., Powis R.A., Shorrock H.K., Murphy K., Swoboda K.J., Gillingwater T.H. and Parson S.H. (2017) Survival of motor neurone protein is required for normal postnatal development of the spleen. J. Anat., 230, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khairallah M.-T., Astroski J., Custer S.K., Androphy E.J., Franklin C.L. and Lorson C.L. (2017) SMN deficiency negatively impacts red pulp macrophages and spleen development in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 26, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deguise M.-O., De Repentigny Y., Mcfall E., Auclair N., Sad S. and Kothary R. (2017) Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet., 26, 801–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gombash S.E., Cowley C.J., Fitzgerald J.A., Iyer C.C., Fried D., Mcgovern V.L., Williams K.C., Burghes A.H.M., Christofi F.L., Gulbransen B.D. et al. (2015) SMN deficiency disrupts gastrointestinal and enteric nervous system function in mice. Hum. Mol. Genet., 24, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schreml J., Riessland M., Paterno M., Garbes L., Roßbach K., Ackermann B., Krämer J., Somers E., Parson S.H., Heller R. et al. (2013) Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. EJHG, 21, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoo S.-H., Yamazaki S., Lowrey P.L., Shimomura K., Ko C.H., Buhr E.D., Siepka S.M., Hong H.-K., Oh W.J., Yoo O.J. et al. (2004) PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U. S. A., 101, 5339–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quarfordt S.H., DeVivo D.C., Engel W.K., Levy R.I. and Fredrickson D.S. (1970) Familial adult-onset proximal spinal muscular atrophy. Report of a family with type II hyperlipoproteinemia. Arch. Neurol., 22, 541–549. [DOI] [PubMed] [Google Scholar]
- 27. Dahl D.S. and Peters H.A. (1975) Lipid disturbances associated with spiral muscular atrophy. Clinical, electromyographic, histochemical, and lipid studies. Arch. Neurol., 32, 195–203. [DOI] [PubMed] [Google Scholar]
- 28. Tein I., Sloane A.E., Donner E.J., Lehotay D.C., Millington D.S. and Kelley R.I. (1995) Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr. Neurol., 12, 21–30. [DOI] [PubMed] [Google Scholar]
- 29. Crawford T.O., Sladky J.T., Hurko O., Besner-Johnston A. and Kelley R.I. (1999) Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol., 45, 337–343. [DOI] [PubMed] [Google Scholar]
- 30. Lamarca N.H., Golden L., John R.M., Naini A., Vivo D.C. and Sproule D.M. (2013) Diabetic ketoacidosis in an adult patient with spinal muscular atrophy type II: further evidence of extraneural pathology due to survival motor neuron 1 mutation? J. Child Neurol., 28, 1517–1520. [DOI] [PubMed] [Google Scholar]
- 31. Borkowska A., Jankowska A., Szlagatys-Sidorkiewicz A., Sztangierska B., Liberek A., Plata-Nazar K. and Kamińska B. (2015) Coexistence of type 1 diabetes mellitus and spinal muscular atrophy in an 8-year-old girl: a case report. Acta Biochim. Pol., 62, 167–168. [DOI] [PubMed] [Google Scholar]
- 32. Davis R.H., Miller E.A., Zhang R.Z. and Swoboda K.J. (2015) Responses to fasting and glucose loading in a cohort of well children with spinal muscular atrophy type II. J. Pediatr., 167, 1362–1368.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bowerman M., Michalski J.-P., Beauvais A., Murray L.M., DeRepentigny Y. and Kothary R. (2014) Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet., 23, 3432–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pera M.C., Romeo D.M., Graziano A., Palermo C., Messina S., Baranello G., Coratti G., Massaro M., Sivo S., Arnoldi M.T. et al. (2017) Sleep disorders in spinal muscular atrophy. Sleep Med., 30, 160–163. [DOI] [PubMed] [Google Scholar]
- 35. Verrillo E., Bruni O., Pavone M., Ferri R., Caldarelli V., Novelli L., Testa M.B.C. and Cutrera R. (2014) Sleep architecture in infants with spinal muscular atrophy type 1. Sleep Med., 15, 1246–1250. [DOI] [PubMed] [Google Scholar]
- 36. Bersanini C., Khirani S., Ramirez A., Lofaso F., Aubertin G., Beydon N., Mayer M., Maincent K., Boulé M. and Fauroux B. (2012) Nocturnal hypoxaemia and hypercapnia in children with neuromuscular disorders. Eur Respir J, 39, 1206–1212. [DOI] [PubMed] [Google Scholar]
- 37. Walter L.M., Deguise M.-O., Meijboom K.E., Betts C.A., Ahlskog N., Westering T.L.E., Hazell G., McFall E., Kordala A., Hammond S.M. et al. (2018) Interventions targeting Glucocorticoid-Krüppel-like factor 15-branched-chain amino acid signaling improve disease phenotypes in spinal m uscular atrophy mice. EBioMedicine, 31, 226–242, 10.1016/j.ebiom.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H. and Li H. (2000) A mouse model for spinal muscular atrophy. Nat. Genet., 24, 66–70. [DOI] [PubMed] [Google Scholar]
- 39. Sumová A., Bendová Z., Sládek M., El-Hennamy R., Laurinová K., Jindráková Z. and Illnerová H. (2006) Setting the biological time in central and peripheral clocks during ontogenesis. FEBS Lett., 580, 2836–2842. [DOI] [PubMed] [Google Scholar]
- 40. Canaple L., Gréchez-Cassiau A., Delaunay F., Dkhissi-Benyahya O. and Samarut J. (2018) Maternal eating behavior is a major synchronizer of fetal and postnatal peripheral clocks in mice. Cell. Mol. Life Sci. CMLS, 10.1007/s00018-018-2845-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Duez H. and Staels B. (2009) Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J. Appl. Physiol. Bethesda Md 1985, 107, 1972–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamaguchi S., Mitsui S., Yan L., Yagita K., Miyake S. and Okamura H. (2000) Role of DBP in the circadian oscillatory mechanism. Mol. Cell. Biol., 20, 4773–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarthy J.J., Andrews J.L., McDearmon E.L., Campbell K.S., Barber B.K., Miller B.H., Walker J.R., Hogenesch J.B., Takahashi J.S. and Esser K.A. (2007) Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol. Genomics, 31, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shavlakadze T., Anwari T., Soffe Z., Cozens G., Mark P.J., Gondro C. and Grounds M.D. (2013) Impact of fasting on the rhythmic expression of myogenic and metabolic factors in skeletal muscle of adult mice. Am. J. Physiol.-Cell Physiol., 305, C26–C35. [DOI] [PubMed] [Google Scholar]
- 45. Boyer J.G., Deguise M.-O., Murray L.M., Yazdani A., De Repentigny Y., Boudreau-Larivière C. and Kothary R. (2014) Myogenic program dysregulation is contributory to disease pathogenesis in spinal muscular atrophy. Hum. Mol. Genet., 23, 4249–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bricceno K.V., Martinez T., Leikina E., Duguez S., Partridge T.A., Chernomordik L.V., Fischbeck K.H., Sumner C.J. and Burnett B.G. (2014) Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Hum. Mol. Genet., 23, 4745–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Freedman M.S., Lucas R.J., Soni B., Schantz M., Muñoz M., David-Gray Z. and Foster R. (1999) Regulation of mammalian circadian behavior by non-rod, non-cone, ocular photoreceptors. Science, 284. [DOI] [PubMed] [Google Scholar]
- 48. Zheng B., Albrecht U., Kaasik K., Sage M., Lu W., Vaishnav S., Li Q., Sun Z.S., Eichele G., Bradley A. et al. (2001) Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell, 105, 683–694. [DOI] [PubMed] [Google Scholar]
- 49. Bowerman M., Murray L.M., Beauvais A., Pinheiro B. and Kothary R. (2012) A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. NMD, 22, 263–276. [DOI] [PubMed] [Google Scholar]
- 50. Hammond S.M., Gogliotti R.G., Rao V., Beauvais A., Kothary R. and DiDonato C.J. (2010) Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PloS One, 5, e15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deguise M.-O., Boyer J.G., McFall E.R., Yazdani A., De Repentigny Y. and Kothary R. (2016) Differential induction of muscle atrophy pathways in two mouse models of spinal muscular atrophy. Sci. Rep., 6, 28846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Scheuer D.A. (2010) Regulation of the stress response in rats by central actions of glucocorticoids. Exp. Physiol., 95, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dickmeis T., Weger B.D. and Weger M. (2013) The circadian clock and glucocorticoids--interactions across many time scales. Mol. Cell. Endocrinol., 380, 2–15. [DOI] [PubMed] [Google Scholar]
- 54. Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D. and Thomas P.D. (2017) PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res., 45, D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pellizzoni L., Charroux B., Rappsilber J., Mann M. and Dreyfuss G. (2001) A functional interaction between the survival motor neuron complex and RNA polymerase II. J. Cell Biol., 152, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu C., Li S., Liu T., Borjigin J. and Lin J.D. (2007) Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature, 447, 477–481. [DOI] [PubMed] [Google Scholar]
- 57. Fonken L.K. and Nelson R.J. (2014) The effects of light at night on circadian clocks and metabolism. Endocr. Rev., 35, 648–670. [DOI] [PubMed] [Google Scholar]
- 58. Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K. et al. (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science, 294, 1704–1708. [DOI] [PubMed] [Google Scholar]
- 59. Hammond S.M., Hazell G., Shabanpoor F., Saleh A.F., Bowerman M., Sleigh J.N., Meijboom K.E., Zhou H., Muntoni F., Talbot K. et al. (2016) Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A., 113, 10962–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murray L.M., Beauvais A., Gibeault S., Courtney N.L. and Kothary R. (2015) Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/−) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun., 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scotton C., Bovolenta M., Schwartz E., Falzarano M.S., Martoni E., Passarelli C., Armaroli A., Osman H., Rodolico C., Messina S. et al. (2016) Deep RNA profiling identified CLOCK and molecular clock genes as pathophysiological signatures in collagen VI myopathy. J. Cell Sci., 129, 1671–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang Z., Liu Q., Peng Y., Dai J., Xie Y., Chen W., Long S., Pei Z., Su H. and Yao X. (2018) Circadian rhythm dysfunction accelerates disease progression in a mouse model with amyotrophic lateral sclerosis. Front. Neurol., 9, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kitazawa M. (2013) Circadian rhythms, metabolism, and insulin sensitivity: transcriptional networks in animal models. Curr. Diab. Rep., 13, 223–228. [DOI] [PubMed] [Google Scholar]
- 64. Wood M.J.A., Talbot K. and Bowerman M. (2017) Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet., 26, R151–R159. [DOI] [PubMed] [Google Scholar]
- 65. Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M. and Dreyfuss G. (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell, 133, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sproule D.M., Montes J., Montgomery M., Battista V., Koenigsberger D., Shen W., Punyanitya M., De Vivo D.C. and Kaufmann P. (2009) Increased fat mass and high incidence of overweight despite low body mass index in patients with spinal muscular atrophy. Neuromuscul. Disord. NMD, 19, 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leroy-Willig A., Willig T.N., Henry-Feugeas M.C., Frouin V., Marinier E., Boulier A., Barzic F., Schouman-Claeys E. and Syrota A. (1997) Body composition determined with MR in patients with Duchenne muscular dystrophy, spinal muscular atrophy, and normal subjects. Magn. Reson. Imaging, 15, 737–744. [DOI] [PubMed] [Google Scholar]
- 68. Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H.M., Erdjument-Bromage H. et al. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature, 454, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Farmer S.R. (2008) Brown fat and skeletal muscle: unlikely cousins? Cell, 134, 726–727. [DOI] [PubMed] [Google Scholar]
- 70. Harms M. and Seale P. (2013) Brown and beige fat: development, function and therapeutic potential. Nat. Med., 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- 71. Nakao R., Yamamoto S., Horikawa K., Yasumoto Y., Nikawa T., Mukai C. and Oishi K. (2015) Atypical expression of circadian clock genes in denervated mouse skeletal muscle. Chronobiol. Int., 32, 486–496. [DOI] [PubMed] [Google Scholar]
- 72. Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K. and Gillingwater T.H. (2008) Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 17, 949–962. [DOI] [PubMed] [Google Scholar]
- 73. Hirota T., Okano T., Kokame K., Shirotani-Ikejima H., Miyata T. and Fukada Y. (2002) Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. JBC, 277, 44244–44251. [DOI] [PubMed] [Google Scholar]
- 74. Cheon S., Park N., Cho S. and Kim K. (2013) Glucocorticoid-mediated Period2 induction delays the phase of circadian rhythm. Nucleic Acids Res., 41, 6161–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tanaka H., Shimizu N. and Yoshikawa N. (2017) Role of skeletal muscle glucocorticoid receptor in systemic energy homeostasis. Exp. Cell Res., 360, 24–26, 10.1016/j.yexcr.2017.03.049. [DOI] [PubMed] [Google Scholar]
- 76. Bruce A.K., Jacobsen E., Dossing H. and Kondrup J. (1995) Hypoglycaemia in spinal muscular atrophy. Lancet Lond. Engl., 346, 609–610. [DOI] [PubMed] [Google Scholar]
- 77. Butchbach M.E.R., Rose F.F., Rhoades S., Marston J., McCrone J.T., Sinnott R. and Lorson C.L. (2010) Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun., 391, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Le T.T., Mcgovern V.L., Alwine I.E., Wang X., Massoni-Laporte A., Rich M.M. and Burghes A.H.M. (2011) Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet., 20, 3578–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bella V., Cisterni C., Salaün D. and Pettmann B. (1998) Survival motor neuron (SMN) protein in rat is expressed as different molecular forms and is developmentally regulated. Eur. J. Neurosci., 10, 2913–2923. [DOI] [PubMed] [Google Scholar]
- 80. Burlet P., Huber C., Bertrandy S., Ludosky M.A., Zwaenepoel I., Clermont O., Roume J., Delezoide A.L., Cartaud J., Munnich A. et al. (1998) The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum. Mol. Genet., 7, 1927–1933. [DOI] [PubMed] [Google Scholar]
- 81. Liang H. and Ward W.F. (2006) PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ, 30, 145–151. [DOI] [PubMed] [Google Scholar]
- 82. Ripolone M., Ronchi D., Violano R., Vallejo D., Fagiolari G., Barca E., Lucchini V., Colombo I., Villa L., Berardinelli A. et al. (2015) Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol2, 72, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Burnett B.G., Muñoz E., Tandon A., Kwon D.Y., Sumner C.J. and Fischbeck K.H. (2009) Regulation of SMN protein stability. Mol. Cell. Biol., 29, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. MacKenzie D., Shamim F., Mongeon K., Trivedi A., MacKenzie A. and Farooq F. (2014) Human growth hormone increases SMN expression and survival in severe spinal muscular atrophy mouse model. J. Neuromuscul. Dis., 1, 65–74. [PubMed] [Google Scholar]
- 85. Farooq F., Molina F.A., Hadwen J., MacKenzie D., Witherspoon L., Osmond M., Holcik M. and MacKenzie A. (2011) Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J. Clin. Invest., 121, 3042–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ting C.-H., Lin C.-W., Wen S.-L., Hsieh-Li H.-M. and Li H. (2007) Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum. Mol. Genet., 16, 499–514. [DOI] [PubMed] [Google Scholar]
- 87. Hamilton G. and Gillingwater T.H. (2013) Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med., 19, 40–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.