

Abstract
In humans, poor nutrition, malabsorption and variation in cobalamin (vitamin B12) metabolic genes are associated with hematological, neurological and developmental pathologies. Cobalamin is transported from blood into tissues via the transcobalamin (TC) receptor encoded by the CD320 gene. We created mice carrying a targeted deletion of the mouse ortholog, Cd320. Knockout (KO) mice lacking this TC receptor have elevated levels of plasma methylmalonic acid and homocysteine but are otherwise healthy, viable, fertile and not anemic. To challenge the Cd320 KO mice we maintained them on a vitamin B12-deficient diet. After 5 weeks on this diet, reproductive failure develops in Cd320 KO females but not males. In vitro, homozygous Cd320 KO embryos from cobalamin-deficient Cd320 KO dams develop normally to embryonic day (E) 3.5, while in vivo, few uterine decidual implantation sites are observed at E7.5, suggesting that embryos perish around the time of implantation. Dietary restriction of vitamin B12 induces a severe macrocytic anemia in Cd320 KO mice after 10–12 months while control mice on this diet are anemia-free up to 2 years. Despite the severe anemia, cobalamin-deficient KO mice do not exhibit obvious neurological symptoms. Our results with Cd320 KO mice suggest that an alternative mechanism exists for mice to transport cobalamin independent of the Cd320 encoded receptor. Our findings with deficient diet are consistent with historical and epidemiological data suggesting that low vitamin B12 levels in humans are associated with infertility and developmental abnormalities. Our Cd320 KO mouse model is an ideal model system for studying vitamin B12 deficiency.
Introduction
In animals, two critical enzymatic processes—the one-carbon metabolism pathway leading to nucleic acid synthesis and cellular methylation reactions (1) and propionic acid oxidation, which converts odd-chain fatty acids and branched-chain amino acids into energy (2,3)—rely on the presence of an adequate and continued supply of cobalamin (vitamin B12), a nutrient that is acquired through dietary intake (4). Methylcobalamin is required by 5-methyltetrahydrofolate-homocysteine methyltransferase (MTR) in the formation of tetrahydrofolate and methionine (5). Decreased activity of MTR results in increased levels of plasma homocysteine (Hcy) (6,7). Adenosylcobalamin is required by methylmalonyl-CoA mutase (MUT), a mitochondrial enzyme that converts methylmalonyl CoA to succinyl-CoA for entry into the Krebs cycle (3). Reduced MUT activity in humans leads to increased concentrations of plasma methylmalonic acid (MMA) that can cause fatal ketoacidosis if not treated in newborns (8). While loss-of-function mutations in MTR or MUT are lethal, a range of nonlethal phenotypes results from deficiency or absence of cobalamin or the loss of other genes involved in the transport and processing of cobalamin (7). Dietary uptake of cobalamin requires the sequential binding of haptocorrin (HC) followed by intrinsic factor (IF) before transport into the distal ileal epithelium, degradation of IF and release of cobalamin into the blood stream where it is bound by transcobalamin (TC) (4,9). The cobalamin-TC complex (holotranscobalamin or holoTC) forms the pool of cobalamin in the peripheral blood that is actively available for tissue uptake via the transcobalamin receptor protein CD320 (also known as TcblR) encoded by the CD320 gene (10).
In humans, acquired cobalamin deficiency occurs when dietary cobalamin is not available (11) or, more commonly in the elderly, owing to decreased absorption efficiency (9). The inability to extract cobalamin from food leads to megaloblastic anemia resulting in general feelings of weakness, malaise and ill health (12). These symptoms may present before frank anemia develops. Prolonged cobalamin deficiency may result in neurological abnormalities such as peripheral neuropathy, cognitive decline, depression and memory loss (9,13). In pregnant women, low levels of maternal cobalamin are associated with an increased risk of giving birth to a child with a neural tube defect (NTD) (14). If left untreated, cobalamin deficiency in humans is fatal (15,16).
The study of cobalamin deficiency has been hampered by the lack of an adequate animal model. Although monkeys, pigs, fruit bats, rats and mice have all been used, creating a graded and manipulatable deficiency in these animals remains challenging (17–25). For example, a mouse model creating a null mutation at the Mtr locus causes embryonic lethality between embryonic day (E) 4.5 and E8.5 (26) while targeted mutation of the Mut gene results in the perinatal death of homozygous KO mice (27,28). Human genetic studies found that a three-base pair (bp) deletion in the CD320 receptor protein, p.Glu88del (CD320 c.262_264delGAG), is associated with risk of NTDs in an Irish population (29) and correlates with elevated plasma acylcarnitine at birth but otherwise may be asymptomatic (30). A mouse containing a gene-trapped KO allele of Cd320 results in a nonlethal phenotype with moderately increased concentrations of plasma Hcy and MMA, reduced concentrations of serum cobalamin and greatly reduced cobalamin levels in the central nervous system (31,32). A recent report further demonstrates that these mice suffer from anxiety, behavioral changes and memory deficits (33) but do not develop anemia or developmental defects. To further investigate the functional importance of the TC receptor, CD320, and identify associated pathologies, we hypothesized that removing the receptor would mimic cobalamin deficiency, either alone or in combination with dietary restriction. We therefore engineered a mouse model that carries a conditional Cd320 null allele and have evaluated the metabolic, reproductive, developmental and hematopoietic phenotypes in mice lacking the CD320 protein. We modeled the gene-environmental interactions by examining the impact of dietary vitamin B12 restriction on isogenic mice with and without this receptor.
Results
Creating a targeted deletion in the Cd320 gene
Genetic variants in the CD320 gene, encoding the receptor for TC-bound vitamin B12, may play a role in human health and disease. To further explore the function of the TC receptor, we created a mouse model lacking the Cd320 gene. Given that previously published mouse models lacking either of the two cobalamin-dependent enzymes result in embryonic or perinatal lethality (26,28), we created a conditionally targeted deletion in C57BL/6J mice of the first two exons out of the total 5 exons in the mouse Cd320 gene (Supplementary Material, Fig. S1). Mice carrying the conditionally targeted Cd320 allele (mouse strain nomenclature C57BL/6J Cd320tm1Lcb) were crossed to B6.FVB-Tg(EIIa-Cre)C5379Lmgd/J mice engineered to express Cre recombinase in the female germ cell and early embryo. Surprisingly, global homozygous germline C57BL/6J Cd320tm1Lcb.Lcb KO mice (henceforth referred to as Cd320 KO) were viable, born in the normal Mendelian ratio and appeared normal compared to the wild type (WT) or control mice (Supplementary Material, Fig. S2). Furthermore, pairs of homozygous KO mice fed with standard high-fat breeder chow containing 51 μg vitamin B12/kg of chow were fertile and produced normal-sized litters through the first 16 generations (mean [SD] control litter size = 5.62 [2.28], n = 158 and mean Cd320 KO litter size = 5.64 [2.18], n = 187, p = 0.93) (Supplementary Material, Table S1).
To confirm that the targeted deletion results in a null Cd320 allele, reverse transcription-polymerase chain reaction (RT-PCR) was performed on RNA prepared from mouse kidney and liver (Fig. 1). No product was observed from Cd320 exons 1 and 2, the two exons targeted for deletion from the homozygous KO mice. WT, homozygous floxed (control) and heterozygous KO mice yielded the expected 205 bp exon1–2 product. In liver tissue, a mispriming product of approximately 125 bp was also amplified containing a LINE1 repeat. An RT-PCR product spanning portions of exons 4 and 5 yielded a 468 bp product in all the mouse genotypes tested, although this band was reduced in intensity in the KO mice. An examination of the Cd320 coding sequence reveals an in-frame ATG (c.627-629) in exon 4 with a partial Kozak consensus sequence that might function as a second translation start site. This hypothetical transcript would not include the membrane targeting signal sequence, either of the LDL-A repeat domains or most of the extracellular portion of the transmembrane receptor, suggesting that this transcript would be nonfunctional.
Figure 1.
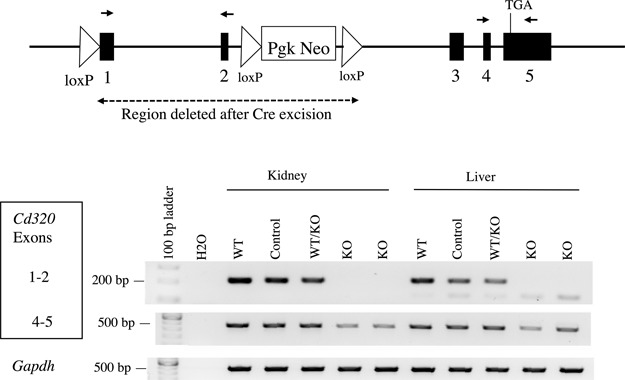
RT-PCR confirms the deletion and lack of expression of exons 1 and 2 in Cd320 KO mice but reveals a persistent but less abundant expression product from exons 4 and 5. The genomic Cd320 region in our KO mice is represented by a map with exons (filled rectangles), introns (black line), loxP sites (triangles), PgK-Neo (selection cassette), TGA (stop codon) and region deleted in the KO mouse (dashed line). The locations of two PCR primer sets (small black arrows) that amplify portions of exons 1 and 2 or exons 4 and 5 are shown. The exon 4–5 product is reduced in intensity in the KO tissues, indicating that residual RNA message is less abundant but present in the KO mice. Controls include the Gapdh housekeeping gene positive control, the no template RT-PCR (H2O) lane negative control and lanes with (+RT). The lanes without reverse transcriptase (−RT) did not amplify any bands.
Biochemical characterization of Cd320 KO mice
Blood and tissues were harvested from animals using approved protocols. Tissues were not perfused (to remove blood) prior to freezing. Cobalamin concentrations in plasma and tissues were measured in 27-week-old male Cd320 KO mice and control mice fed standard mouse chow containing 50 μg vitamin B12/kg of chow (Fig. 2A). The cobalamin concentrations were normal in the plasma of KO mice. In the same animals, cobalamin concentrations in the kidney were normal, reflecting that the kidney expresses high levels of megalin, the multi-ligand receptor important in the retention of cobalamin and other biomolecules. Cobalamin concentrations were significantly decreased in all other tissues from Cd320 KO mice. Notably, cobalamin concentrations were reduced by nearly 60% in brain tissue from KO mice compared to controls.
Figure 2.
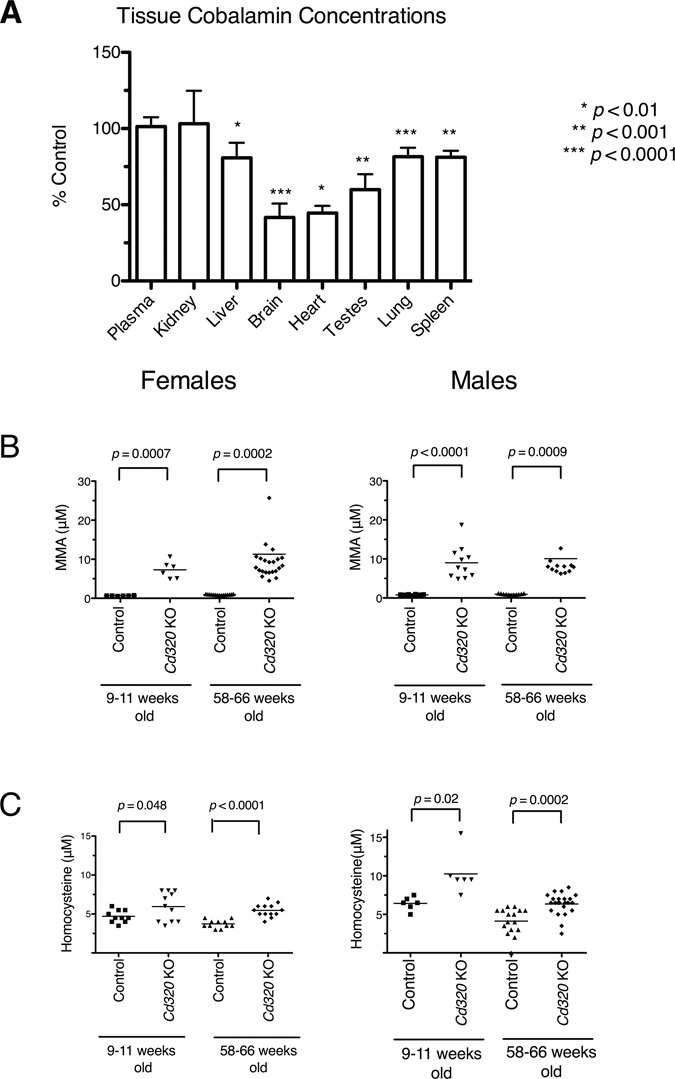
Tissue cobalamin concentrations are reduced and plasma MMA and plasma Hcy concentrations are increased in Cd320 KO mice fed with standard mouse chow. (A) Cobalamin concentrations in plasma and tissues from male 27-week-old Cd320 KO mice (n = 7) were normalized to the values from age-matched control male mice (n = 7, brain tissue n = 6) and plotted to display mean and standard deviation (SD). Cobalamin concentrations in blood plasma and kidney did not differ from control, but all other tissues tested had significantly reduced levels of cobalamin. (Unpaired Student’s t-test, error bars represent SD). One extreme outlier value was excluded from the control brain cobalamin analysis. (B) Plasma MMA and (C) plasma Hcy concentrations were significantly elevated in both sexes of Cd320 KO mice as compared to age-matched controls in young (9–11 weeks old) and aged mice (58–66 weeks old). (Solid line denotes mean value, unpaired Student’s t-test with Welch’s correction).
Plasma MMA concentrations were significantly elevated in both sexes of 9- to 11-week-old Cd320 KO mice fed with standard chow as compared to control mice and remained elevated beyond 1 year in age (Fig. 2B). Likewise, plasma Hcy concentrations were also elevated in both sexes of the KO mice fed with the standard chow at both 9–11 weeks and 58–66 weeks of age (Fig. 2C).
Despite the low tissue cobalamin and increased MMA and Hcy concentrations, the Cd320 KO mice exhibited no overt signs of ill health or pathology. These KO mice were maintained on a standard laboratory mouse chow containing ∼50 μg vitamin B12/kg of chow. To further reduce intracellular cobalamin availability by dietary restriction, both control and Cd320 KO weanling-aged (21 days old) mice were placed on a soy protein-based diet containing no vitamin B12 (vitamin B12-deficient diet). As a control diet, an identical soy formulation was produced and supplemented with 50 μg vitamin B12/kg of diet (vitamin B12-supplemented diet). Regardless of genotype, the vitamin B12-deficient diet was associated with changes in plasma concentrations of cobalamin, MMA and Hcy. After 12 weeks, mice of both genotypes maintained on the deficient diet had greatly reduced plasma cobalamin (Fig. 3A) and increased plasma MMA and plasma Hcy concentrations (Supplementary Material, Fig. S3) as compared to mice fed with the vitamin B12-supplemented control diet. The size of the effect based on genotype was less striking. Comparing between genotypes in mice fed with the vitamin B12-deficient diet for 12 weeks, Cd320 KO mice had significantly higher plasma cobalamin concentration than the controls (Fig. 3A,P-value < 0.0001, Mann–Whitney test). This suggests that the reduced amount of cobalamin available remained in the blood compartment due to the engineered defect in CD320-mediated transport. Assayed after 24 weeks on the deficient diet, plasma MMA and Hcy concentrations continued to be elevated in KO and control mice (Fig. 3B and C).
Figure 3.
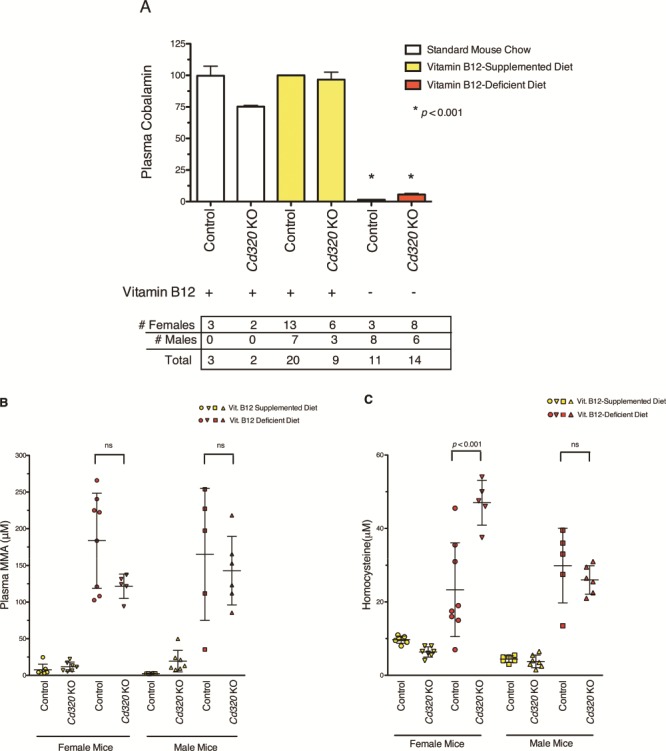
Mice maintained on vitamin B12-deficient diet have reduced plasma cobalamin concentrations and increased plasma MMA and plasma Hcy concentrations. (A) The plasma cobalamin concentration is greatly reduced in all mice fed with the vitamin B12-deficient diet (red bars) for 12 weeks (P-value < 0.001). Plasma cobalamin concentrations were normalized to control mice fed with the vitamin B12-supplemented diet (control-yellow bar) and plotted to show mean and SD. Mice fed with the vitamin B12-supplemented diet (yellow bars) had equivalent levels of plasma cobalamin as control mice fed with the standard mouse chow (white bar). Cd320 KO mice fed with standard mouse chow appear to have lower plasma cobalamin concentration (P-value < 0.01), although only a small number of mice were tested (n = 2). (One-way ANOVA with Tukey’s multiple comparison test.) Comparing between the two mouse genotypes fed with the vitamin B12-deficient diet (red bars), plasma cobalamin concentrations are significantly increased in Cd320 KO mice compared to controls (P-value < 0.0001 Mann–Whitney test). (B) After 24 weeks, all mice maintained on the vitamin B12-deficient diet (red symbols) had highly elevated plasma MMA concentrations compared to mice fed with the vitamin B12-supplemented diet (yellow symbols). However, no significant difference in plasma MMA was found between genotypes fed with the same diet for 24 weeks (one-way ANOVA with Tukey’s multiple comparisons test, mean with SD). (C) All mice maintained on the vitamin B12-deficient diet (red symbols) for 24 weeks had elevated concentrations of plasma Hcy compared to mice fed with the vitamin B12-supplemented diet (yellow symbols) (one-way ANOVA with Tukey’s multiple comparison test, mean with SD).
Reproductive deficits in Cd320 KO female mice maintained on a vitamin B12-deficient diet
All mice fed with the vitamin B12-supplemented diet were fertile and produced normal-sized litters (Table 1, rows 1 and 3). Mating pairs of control mice fed with the vitamin B12-deficient diet were fertile and produced normal-sized liters (Table 1, row 2). However, homozygous Cd320 KO breeding pairs failed to produce any litters when fed with the vitamin B12-deficient diet (Table 1, row 4). Breeding experiments demonstrated that male Cd320 KO mice fed with the vitamin B12-deficient diet impregnated WT females, and gave rise to viable offspring (Table 1, row 5). In contrast, 8-week-old homozygous KO females (n = 21) fed with the vitamin B12-deficient diet beginning at 21 days old (i.e. starting at weaning and fed continuously) never gave birth to offspring regardless of the genotype of the male in the mating pair (Table 1, rows 4, 6 and 7). Histological sections of ovaries from Cd320 KO females fed with the vitamin B12-deficient diet showed no gross abnormalities (data not shown). To determine if infertility was due to embryonic loss, homozygous KO females and control females fed with the vitamin B12-deficient diet for 8 weeks or more were time-mated to KO males, euthanized at E7.5 to E9.5 and dissected for embryo collection. Control females (n = 4) had on average 8 decidua. Only a single homozygous KO dam, mated at 11-weeks of age, had uterine decidual swellings, evidence of embryonic implantations in the uterine lumen, but no identifiable embryonic material at E7.5. All other KO females (n = 5) in this mating experiment, between 16–25 weeks old, lacked decidua. Homozygous Cd320 KO females continuously maintained on the vitamin B12-deficient diet for a minimum of 8 weeks failed to reproduce, with no live embryos observed beyond E7.5.
Table 1.
Litters produced from mice maintained on vitamin B12-supplemented or vitamin B12-deficient diet
| Breeding Cross Female X Male | Dietary Vit. B12 | # dams Mated | # litters | Mean pups/litter | S.D. | P-value* | ||
|---|---|---|---|---|---|---|---|---|
| 1 | Control | Control | + | 4 | 22 | 5.27 | 2.59 | |
| 2 | Control | Control | - | 6 | 18 | 4.39 | 2.06 | 0.25 |
| 3 | KO | KO | + | 7 | 27 | 3.96 | 2.18 | 0.066 |
| 4 | KO | KO | - | 14 | 0 | n/a | n/a | |
| 5 | Control | KO | - | 2 | 9 | 4.33 | 2.87 | |
| 6 | KO | Control | - | 4 | 0 | n/a | n/a | |
| 7 | KO | Control/KO | - | 3 | 0 | n/a | n/a | |
*Two tailed t-test compared to the control mice on vitamin B12-replete diet in line 1
The apparent lack of fertility in the Cd320 KO females fed with the vitamin B12-deficient diet may be attributable to changes in the maternal uterine environment or in the embryo proper. To isolate the cause of the infertility, we crossed mice of various genotypes. Crosses between homozygous KO females and control males, both on vitamin B12-deficient diet, were expected to produce heterozygous offspring but were unsuccessful, suggesting that the maternal uterine environment did not sustain development (n = 4, Table 1, row 6). Crossing heterozygous Cd320 KO females to homozygous KO males, both fed with the vitamin B12-deficient diet from weaning age, resulted in the birth of neonatal homozygous Cd320 KO embryos but at reduced Mendelian frequencies (Chi-squared test, P-value = 0.0006) (Table 2). Regardless of genotype, none of the pups born to these vitamin B12-deficient heterozygous mothers survived beyond two days after birth. No milk spots were observed in the stomachs of these pups and no gross birth defects or malformations were observed.
Table 2.
Mating results from 10 heterozygous females on vitamin B12-deficient diet
| Cd320 Genotype Female X Male | Observeda Control/KO KO | Expected Control/KO KO | Total | P-value* | |||
|---|---|---|---|---|---|---|---|
| Control/KO | KO | 29 | 8 | 18.5 | 18.5 | 37 | 0.0006 |
aall pups perished within the first 24–48 h after birth
*Chi-Squared Test
Table 3.
Mating results of Cd320 KO females on vitamin B12-deficient diet given weekly intraperitoneal injections of vitamin B12
| Chow | Female X | Male | # dams mated | # litters | Mean Pups/litter | SD | P-value |
|---|---|---|---|---|---|---|---|
| Deficient | KO + B12 IP injection | KO | 3 | 5a | 3.0 | 2.0 | 0.38b |
aall pups perished within the first 24–48 h after birth
bcompared to KO on replete chow
To test whether these neonatal deaths were due to poor maternal care we transferred pups from WT parents and cross-fostered them onto heterozygous Cd320 KO dams fed with the deficient diet. These fostered WT pups were able to nurse and develop normally, indicating that heterozygous dams on deficient diet lactate normally and that abnormal maternal behavior was not the cause of neonatal death. Two surviving newborn offspring, both heterozygous KOs by genotyping, from mating heterozygous Cd320 control/KO dams to Cd320 KO males, were cross-fostered onto control mice fed with standard mouse chow but died within 1day of birth, suggesting that a defect may lie in the offspring.
The ability, albeit compromised, of homozygous Cd320 KO embryos to develop to full term in vitamin B12-deficient heterozygous females but not in vitamin B12-deficient KO dams (Table 2) suggests that maternal effects may also be important. To compare the ovulation capacity and embryonic viability from mice on vitamin B12-deficient diet, control and KO females were mated to KO males and the embryos were flushed from the fallopian tubes, collected, pooled and cultured until the blastocyst stage. We observed that 34.6% of the homozygous Cd320 KO one-cell embryo (25 of the 72 ova pooled from six KO females) reached the blastocyst stage in culture. In comparison, 34.7% of the heterozygous embryos from controls dams (34 of the 105 embryos pooled from seven control females) reached the blastocyst stage. No difference in embryo viability up to the blastocyst stage was found in vitro between KO and control dams both fed vitamin B12-deficient diet.
To assess the ability of embryos from homozygous Cd320 KO mice fed with the vitamin B12-deficient diet to develop in a vitamin B12-replete WT uterine environment, two-cell embryos were transplanted into pseudo-pregnant WT mice maintained on standard chow (Supplementary Material, Table S2, row 1). In one of these embryo transfer experiments, four of the five recipient mice appeared pregnant at mid-gestation. Three of these recipients resorbed their litters and had no decidual evidence of pregnancy on their expected delivery date. A cesarean section was performed on one female that still appeared pregnant at day E20 which yielded two pups that were both alive and respiring normally but died after approximately 30 minutes. In another experiment, one recipient female naturally delivered two homozygous KO pups, one of which died and the other survived (Supplementary Material, Table S2 row 1). Additional embryo transfer experiments demonstrated that homozygous KO pups could be produced from embryos flushed from homozygous KO dams fed with the vitamin B12-deficient diet (and crossed to heterozygous KO males) but at rates significantly below the expected Mendelian ratio (Supplementary Material, Table S2 row 2). Importantly, embryo transfer experiments with embryos from heterozygous KO dams (crossed to KO males) resulted in the expected ratio (Supplementary Material, Table S2 row 3). Taken together, these results suggest that embryos derived from Cd320 KO dams and exposed to cobalamin deficiency can develop in a normal uterine environment but survive to term at a reduced rate.
In an attempt to reverse the infertility phenotype associated with the homozygous Cd320 KO females fed with the vitamin B12-deficient diet, three female KO mice were maintained for more than 11 weeks on vitamin B12-deficient diet and then given weekly intraperitoneal injections of vitamin B12. These females were mated to Cd320 KO males on deficient diet. By the fifth week after the first vitamin B12 injection, two of three homozygous KO females were visibly pregnant. These two females both delivered multiple litters in succession ranging from one to six pups per litter, however all the pups perished within the first 24–48 h after birth. The third female was obviously pregnant at least twice, but no pups were ever observed (Table 3).
Macrocytic anemia develops in Cd320 KO mice fed a B12-deficient diet
We also examined the impact of Cd320 loss in older mice. Macrocytic anemia is a hallmark of humans suffering from vitamin B12 deficiency. To examine the hematological parameters in our mice, we performed complete blood count (CBC), hematocrit and mean corpuscular volume assays on 12-week-old male mice fed with the standard mouse chow. These assays failed to distinguish the Cd320 KO mice from WT or control mice, indicating that the global KO mice are not anemic at this age (Supplementary Material, Fig. S4).
These assays were repeated with mice maintained on the vitamin B12-deficient diet. Control and Cd320 KO mice continued to have normal hematocrit values when continuously maintained on the vitamin B12-supplemented diet for extended lengths of time (Fig. 4A). Despite dramatically reduced cobalamin concentrations after 12 weeks of deficient diet (Fig. 3A), Cd320 KO and control mice exclusively fed with the vitamin B12-deficient diet for 27–43 weeks had normal CBC, normal hematocrit values and were not anemic (data not shown). However, after 40–44 weeks on deficient diet, plasma hematocrit values began to decline in the Cd320 KO mice, indicating the onset of anemia (Fig. 4B). While the age at onset is variable, all of the KO mice eventually became anemic, lost body weight and reached a moribund state. The onset of anemia in female Cd320 KO mice preceded the onset in KO males by 2–4 weeks (data not shown). Anemic mice were lethargic, had rapid respiration rates and pale toes and foot pads. Control mice on the vitamin B12-deficient diet continued to have normal robust hematocrit values and were not anemic even after 90 weeks on deficient diet (n = 10). CBC assays (Fig. 5) performed on 56- to 72-week-old Cd320 KO mice fed with the vitamin B12-deficient diet since weaning had low hematocrit values (Fig. 5A) (P-value = 0.016), increased mean corpuscular volumes (MCV) (Fig. 5B) (P-value = 0 .0036), decreased red blood cell (RBC) counts (Fig. 5C) (P-value = 0.0059) and decreased hemoglobin concentrations (Fig. 5D) (P-value = 0.0083) compared to controls (Student’s t-test with Welch’s correction).
Figure 4.
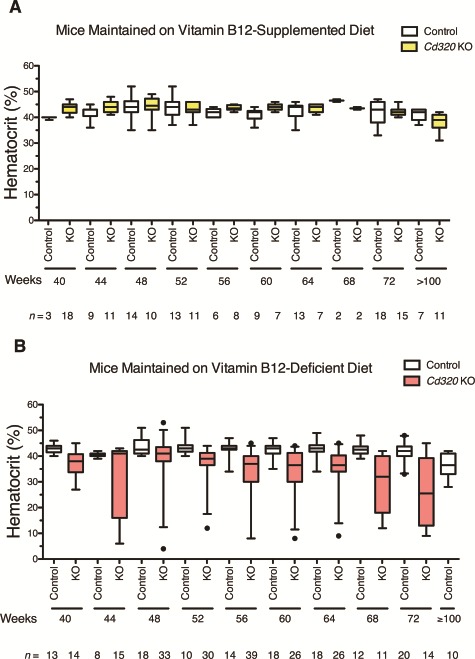
Hematocrit values in control and Cd320 KO mice maintained for 40–100 weeks on vitamin B12-supplemented or vitamin B12-deficient diet. (A) Hematocrit measurements from peripheral blood in mice maintained continuously on the vitamin B12-supplemented diet for up to 100 weeks were normal for both genotypes, control and Cd320 KO. (B) After 40–44 weeks of the vitamin B12-deficient diet, Cd320 KO but not control mice became anemic and eventually succumbed to severe anemia. Hematocrit values from both male and female mice are presented as box and whiskers plots depicting the 25th to 75th percentiles as the box, median value as the middle line, whiskers as the 5th–95th percentiles and closed circles as outlier values.
Figure 5.
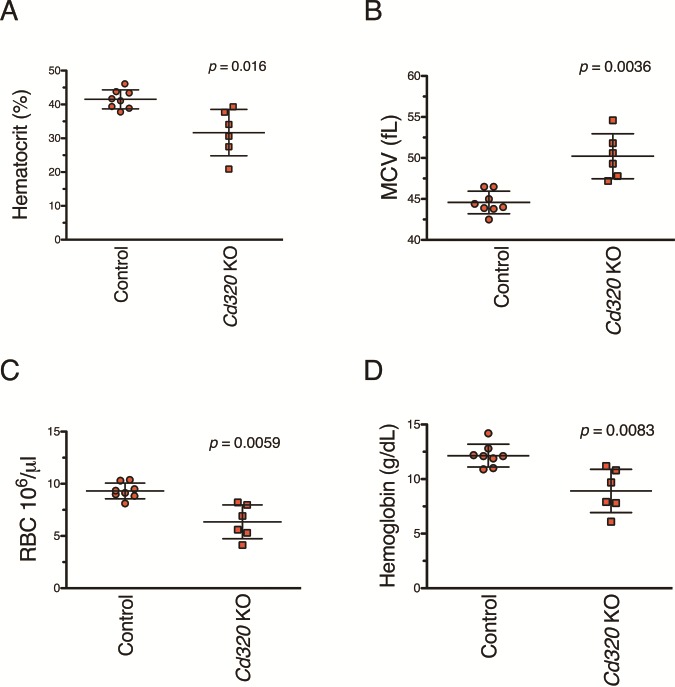
Results from CBC assays performed on mice maintained on the vitamin B12-deficient diet for 56–72 weeks revealed a macrocytic anemia, a hallmark of vitamin B12 deficiency, in the Cd320 KO mice. (A) Aged Cd320 KO mice maintained on the vitamin B12-deficient diet have decreased hematocrit values (P-value = 0.016), (B) increased MCV (P-value = 0.0036), (C) decreased RBC counts (P-value = 0.0059) and (D) decreased hemoglobin concentrations (P-value = 0.0083) compared to diet-matched aged controls. Results are from both male and female mice assayed with a mean time maintained on vitamin B12-deficient diet of 60 weeks for KO mice and 66 weeks for control mice. (Student’s t-test with Welch’s correction, mean with SD).
To confirm that the illness associated with low hematocrit values was due to a cobalamin deficiency, a single intraperitoneal injection containing 1.25 μg of cyanocobalamin was administered to five anemic Cd320 KO mice with hematocrit values ranging from 12–32%. Within approximately 10 days (mean [SD], 10 [3] days), the vitamin B12 injection restored the low hematocrit values to near normal range (mean [SD], pre-injection hematocrit = 22 [10], post-injection hematocrit = 35 [5], n = 5, P-value = 0.03 two-tailed t-test). Vitamin B12-treated mice gained body weight, regained color in the foot pads and returned to normal cage activity. This rescue effect from the vitamin B12 injections persisted for several months before body weight and hematocrit values began to decline again. However, severely anemic Cd320 KO mice with extremely low hematocrit values rapidly became moribund and were unable to be rescued by vitamin B12 injections.
Discussion
Multiple targeted mouse models lacking vitamin B12-related genes result in embryonic lethality (23). In particular, the KO models of the two known cobalamin-dependent enzymes, methylmalonyl-Coenzyme A mutase and methionine synthase, result in perinatal and embryonic death, respectively (26,28). Given this precedent, and to better understand the role of the TC receptor in vitamin B12 metabolism, we created a KO mouse model of Cd320. We expected that Cd320 KO mice would present with an early lethal phenotype regardless of vitamin B12 availability. Surprisingly, when maintained on a vitamin B12-replete diet, Cd320 KO mice survive, reproduce and live up to 2 years without becoming anemic. The viability of Cd320 KO mice is consistent with reports by Quadros and colleagues of a gene trap engineered Cd320 locus that also resulted in viable mice (31,32).
The lack of a severe phenotype in our mice prompted characterization of the metabolic hallmarks observed in human vitamin B12 deficiency. Our Cd320 KO mice present with biochemical abnormalities, i.e. increased concentrations of plasma MMA and plasma Hcy, consistent with vitamin B12 deficiency. Plasma cobalamin concentrations are normal in our Cd320 KO mice. Others have reported normal to increased mouse serum cobalamin levels when using cobalamin analogues that bind TC but block CD320-mediated transport (34). Interestingly, a mouse KO model of gastric intrinsic factor (Gif) is not fully depleted of circulating vitamin B12 in the first generation and these mice are able to reproduce (35). Mice do not have HC (36), suggesting that the Gif KO mice and the Cd320 KO mice are capable of utilizing dietary cobalamin through some other mechanism or other receptor protein.
CD320, at 260 amino acids, is one of the smaller proteins in the low-density lipoprotein (LDL) receptor family of transmembrane proteins. There are at least 13 members of the LDL receptor protein family with some of the larger proteins, such as megalin and LRP1, known to bind and transport scores of different ligands (37,38). One of these other LDL family members may retain an unrecognized ability to function in vitamin B12 transport. In the kidney the product of the Lrp2 gene, megalin, is known to bind and transport holoTC (39,40). Arora et al. (41) suggest that the presence and function of megalin in the yolk sac may compensate for the loss of CD320 in the placenta and developing embryo.
Several reports suggest that nonspecific or low-affinity uptake of free- or protein-bound vitamin B12 may be due to fluid phase transfer during pinocytosis, simple diffusion or other mechanisms. Zhao et al. (42) suggest that pinocytosis may account for small but rapid amounts of cobalamin uptake in their cell culture models of cobalamin transport using heat-denatured serum. Berliner and Rosenberg (43) studied a human fibroblast cell line from a patient with congenital TC deficiency and found evidence of free vitamin B12 uptake. Finally, the clinical treatment of congenital TC-deficient patients with massive doses of vitamin B12 suggests that another mechanism exists besides CD320 transport of holo-TC transport (44).
In a recent report, WT C57BL/6 mice fed a diet with reduced amounts of vitamin B12 were found to have smaller than average litter sizes, smaller pups and an increased rate perinatal death (45). In our study, the combination of vitamin B12 dietary restriction with the loss of the CD320 receptor produced female infertility. The vitamin B12 deficiency appears to cause embryo mortality around the time of implantation to placentation in our Cd320 KO females, phenocopying the methionine synthase KO mouse (26). Cyanocobalamin injections restore the ability of these Cd320 KO females fed with vitamin B12-deficient diet to carry pregnancies and deliver live-born pups, although all of these pups died within the day of birth. Strikingly, these dams were rapidly impregnated within the first few weeks after receiving the first vitamin B12 injection. This partial rescue of the reproductive defect is proof that vitamin B12 deficiency is an underlying cause of infertility in our mice. Our results suggest that adequate cellular levels of cobalamin are critical for implantation. Pharmacologic intervention in the context of long-term deficiency does eliminate the early fetal loss and produces full-term offspring. The cause of the perinatal lethality remains unexplained. These results are consistent with independent embryonic and maternal deficits due to the combination of the dietary cobalamin restriction and the lack of the CD320 receptor most likely manifesting from the loss of one-carbon metabolism affecting methylation reactions and nucleotide synthesis.
Embryonic death is a common phenotype in other mouse models of vitamin B12 deficiency. Cubn, Amn, Lmbrd1 and Mmachc KO mice all die between E3.5 and gastrulation stage (46–50). However, mice deficient for Lrp2, the multiligand receptor megalin that also binds the TC-cobalamin complex, die perinatally of respiratory insufficiency (51). Additional studies examining the precise timing of developmental arrest in the Cd320 KO embryos in dams fed with the vitamin B12-deficient diet would be necessary to better understand the nature of the embryonic defect.
Our findings are largely consistent with observations in humans and other species. Numerous reports suggest that infertility in women suffering from pernicious anemia and vitamin B12 deficiency can be rapidly reversed with vitamin B12 supplementation (52–55). Furthermore, women with higher serum vitamin B12 and folate levels undergoing assisted reproductive treatments had increased rates of successful live births (56). In Caenorhabditis elegans, vitamin B12 deficiency may also lead to the loss of fertility (57). Although male Cd320 KO mice, even when stressed with the vitamin B12-deficient diet, were fertile and productive breeders, male infertility in humans has been associated with low serum folate and CD320 variations (58). Female control mice maintained on the vitamin B12-deficient diet retained their reproductive success despite extremely low plasma cobalamin concentrations. Perhaps, further cobalamin restriction in these control mice might affect reproductive performance. Experiments to address reproductive capacity could include placing control mice on deficient diets at earlier ages, continuously maintaining several successive generations of control mice on deficient diet and housing control mice on screened flooring to limit fecal sources of cobalamin.
When the environmental availability of cobalamin is limited through dietary restrictions, a fatal macrocytic anemia develops in Cd320 KO mice as they approach one year in age. In contrast, control mice fed with a diet lacking vitamin B12 for over 2 years never became anemic. A single vitamin B12 injection swiftly reverses the Cd320 KO anemia and restores the hematocrit values to the normal range for several months. This reversal by vitamin B12 injection demonstrates that cobalamin deficiency is the primary cause of the macrocytic anemia in these KO mice on vitamin B12-deficient diet.
There were several limitations to our mouse model of cobalamin deficiency. Although our mice were treated with antibiotics to reduce the likelihood that gut flora could serve as a source of vitamin B12, mice were not housed in cages that prevent coprophagy. The average age of onset of anemia in KO mice maintained on vitamin B12-deficient diet has a large variance. The inability to accurately predict the phenotype of an individual mouse in our environmentally controlled and genetically identical vitamin B12-deficient Cd320 KO mice suggests that stochastic elements may play a role in the delicate metabolic balance that tips an animal into an anemic state. This may be an example of variable expressivity and is consistent with observations in humans where a minority of patients with vitamin B12 insufficiency present with pernicious anemia (9,59,60).
Although we did not observe obvious signs of neuropathology, we did not directly test for peripheral neuropathy nor did we perform behavioral assays to evaluate subtle neurological defects. Arora et al. (33) reported subtle anxiety and learning and memory abnormalities in 5-month-old Cd320 gene-trapped KO mice on standard mouse chow. It is possible that our mice perish from severe anemia prior to the onset of overt neuropathology. Folic acid supplementation in human vitamin B12-deficient patients reverses anemia but fails to inhibit neurological disease progression (61). Folic acid supplementation might reverse the fatal anemia in our aged KO mice fed with the vitamin B12-deficient diet and allow neuropathies to develop. It is also possible that extremely low amounts of vitamin B12 are sufficient to prevent neuropathology, that mice are resistant to the vitamin B12 associated neuropathies, or that the time required for these to develop is longer than the lifespan of a mouse.
In conclusion, we have created a mouse model of vitamin B12 deficiency that will be useful for the study pernicious anemia during all stages of life. This model will also permit the exploration of the biology of CD320-independent vitamin B12 transport. Importantly, we now have a system in which we can study the impact of a specific gene in a controlled environment. In the context of pregnancy, this system allows us to study maternal–fetal interactions in a controlled way. As with nearly all traits, the ultimate phenotype is the product of the interplay of gene activity and its interaction with environmental conditions. The use of inbred mouse lines and the controlled laboratory environment allows us to precisely manipulate allelic variation and the exposure of a critical micronutrient. Systems such as this could be developed for many combinations of genes and micronutrients. Such studies should yield insights into the impact of a gene-environment interactions that are not tractable when using less-controlled systems such as humans and outbred animals.
Materials and Methods
Mouse husbandry
All animal protocols were reviewed and approved by the National Human Genome Research Institute (NHGRI) Animal Care and Use Committee prior to animal experiments. Mice were housed in shoe box cages and fed ProLab RMH 1800 diet (PMI Nutrition International) containing 50 μg vitamin B12/kg of diet and 3.3 mg folic acid/kg of diet. Breeding mice were fed Picolab Mouse Diet 20, containing 51 μg vitamin B12/kg diet and 2.9 mg folic acid/kg of diet.
Vitamin B12-free diet
Mice require a minimum of approximately 5–10 μg of vitamin B12 per kg diet for reproduction and growth (62). Given that C57BL/6J male mice consume an average of 3.6 g of diet per day (63), this translates to a daily intake of approximately 18–36 ng of vitamin B12. As vitamin B12 is not found in plant proteins, a soy-based diet lacking vitamin B12 was custom formulated by Teklad Diets (Envigo). A replete version of this soy diet was supplemented with 50 μg of vitamin B12 per kg of diet. Both formulations contained 3 mg folic acid/kg of diet and included 1% by weight of the antibiotic succinylsulfathiazole. Different colors of food dyes were added to each formulation to avoid feeding mistakes. See Supplementary Material, Table S3 for custom diet formulation.
Designing the Cd320 gene KO mice
The murine Cd320 gene was targeted in mouse embryonic stem cells using the Cre-loxP system (see Supplementary Material, Fig. S1). The Cd320tm1Lcb-targeted mice were engineered to carry a conditional deletion of the first two of the five exons in the Cd320 gene by flanking exons 1 and 2 with loxP sites. This region targeted for deletion includes the transcription start codon in exon 1 and is predicted to delete the first 83 amino acids of the protein including the sequences for the transmembrane targeting signal and the first LDL-A repeat domain (10). Southern hybridization using a DNA probe located in the genomic region just outside of the targeted region identified 11 out of 240 C57BL/6J ES cell clones that were homologously recombined (see Supplementary Material, Fig. S5). PCR genotyping strategies were used to identify WT, targeted and deleted alleles (see Supplementary Material, Fig. S6).
Synthesis of gene targeting construct
Long-range PCR utilizing the ABI rTth polymerase was used to clone the genomic regions of the mouse C57BL/6J RPCI-23 BAC 213I16 (64) for the left arm, floxed region and right arm of the Cd320 targeting construct.
Right Arm cloning: Ex3-5 F1 primer (5′-AAGGCCCTGTGATGAAAGCGAGTC-3′) and Ex3-5 R1 primer (5′-AAAGATGAGGCAGGAAGATTACCC-3′) were used to clone a 6.8 kb genomic region of BAC 213I16 spanning exons 3, 4 and 5 of the Cd320 gene. The resulting amplicon was TA cloned into pCR2.1, excised with restriction enzymes XbaI and BamHI and cloned into the XbaI/BamHI sites of the pPNT vector (65) modified to have loxP sites flanking the Neo gene.
Two synthetic loxP oligos (loxP F 5′- GGCCGCATAACTTCGTATAATGTATGCTATACGAAGTTAT -3′, loxP R 5′-GGCCATAACTT-CGTATAGCATACATTATACGAAGTTATGC -3′) were annealed to create a double-stranded loxP fragment. A microcentrifuge tube containing 50 ng of each oligo in a reaction volume of 100 μl with 1X NEB (New England Biolabs) restriction buffer #2 was boiled in a beaker of 300 ml of water on a hot plate for 5 min. The hot plate was turned off and the beaker of water with the primers was allowed to slowly cool to room temperature. The resulting double-stranded loxP oligo was designed to destroy the downstream NotI site but retain the NotI site on the upstream side of the vector.
Floxed region: The 3.8 kb genomic region of BAC 213I16 was cloned using Ex1-2 cloning F primer containing an EagI site underlined (5′-CCCCGGCCGCATTGGGACCATTGGGCAGTGATA-3′) and Ex1-2 cloning R primer containing an XhoI site as underlined (5′- CCCCTCGAGAGCAATCCCAACAGTCCAGAC -3′). The amplicon was digested with XhoI and EagI restriction enzymes and ligated into the XhoI/EagI sites in pBSII SK(+) (Stratagene). The resulting plasmid, named pBSEx1-2, was digested with NotI and the annealed loxP oligos were ligated into the vector producing the plasmid pBS loxP Ex1-2. The ligation destroyed the NotI site downstream of the loxP site but retained the upstream NotI site which was screened by PCR, restriction digestion and sequencing to confirm loxP insertion and orientation.
Left arm: The Left arm cloning F primer, containing the underlined NotI site (5′-CCCCCCGCGGCCGCGGAGGTGAAAGGGCAAGCGAGATTC-3′), and Left arm R2 primer, containing the underlined EagI site (5′-CCCCGGCCGATGGCAGCCACACCCTGTGAACACT-3′), amplified a 3.2 kb region upstream of the Cd320 gene and was TA cloned into pCR2.1 Topo vector (Life Technologies). The orientation of the insert was verified by NotI digestion. EagI digestion of pCR2.1 Left Arm was used to excise the 3.2 kb EagI fragment for cloning into the NotI site of the pBS loxP Ex1-2 plasmid. The new plasmid, pBS Left loxP Ex1-2, was screened for proper left arm orientation by double digest with NotI plus XhoI and sequence verified. Excision of the 7 kb Left loxP Ex1-2 fragment was accomplished with NotI and XhoI double digestion and cloned into the NotI/XhoI cut pPNT dloxP Ex3-5 to create the final targeting vector. Digestion with NotI/XhoI verified the correct insertion. The ends of the targeting vector, exons and intron boundaries, loxP sites and selection cassettes were confirmed by sequencing prior to transfecting the construct into ES cells.
Gene targeting in ES cells
The NotI-linearized and phenol/chloroform purified targeting vector was electroporated into the C57BL/6J ES cell line HGTC-8 (66) using a positive/negative double selection strategy (G418+FIAU) that produced a 1:6 enrichment of doubly selected clones compared to the G418 single selection. 240 ES cell colonies were cloned and 11 homologously targeted clones were identified by Southern blotting. Two clones, A67 and A79, were expanded and injected into Balb/C blastocysts. Both clones produced founder chimeras, passed the targeted allele through the germline and gave rise to mice with identical phenotypes, thus line A67 from founder ‘D’was used for further experiments. These C57BL/6J Cd320tm1Lcb mice on a pure C57BL/6J background were bred to homozygosity, having both alleles flanked by loxP sites (floxed), and are used as controls.
Cre mediated deletion of floxed region
Mice were mated to the B6.FVB-Tg(EIIa-Cre)C5379Lmgd/J (Jackson Labs stock number 003724) which express the Cre transgene in the ova and early embryo under the control of the adenovirus EIIA promoter (67). By passing the deletion through the germline, intercrossing the resulting offspring and selecting offspring lacking the Cre transgene, we generated Cd320tm1Lcb.1Lcb as the homozygous deleted KO mouse strain.
Southern blot
10 μg of ES cell DNA was digested overnight with XmnI restriction enzyme (NEB) plus 1mm spermidine in 100 μl total reaction volume. Following ethanol precipitation and resuspension in 25 μl TE, the genomic DNA was separated on a 0.6% agarose (Seakem ME agarose) gel overnight at 40 V in 1X TAE buffer. The gel was soaked in 0.25N HCl for 7–8 min, twice in 1.0 M NaCl/0.5M NaOH for 15 min and twice in 1.5 M NaCl/0.5M Tris pH = 7.4 for 15 min. The DNA was blotted onto Optitran nitrocellulose (Whatman) by overnight capillary transfer in 20X SSC followed by UV crosslinking. 50 ng of probe was labeled with α-32P dCTP (Perkin Elmer) using Ready-To-Go-DNA Labeling Beads (Amersham Pharmacia). Labeled probes were separated from unincorporated radioactivity using NICK column purification (GE Healthcare). Southern hybridizations were performed overnight at 42°C in 50% formamide, 5X SSPE, 1X Denhart’s, 1% SDS and 10% dextran sulfate and blocked with sheared herring sperm DNA. The hybridized membranes were twice washed in 2X SSC/0.1% SDS at room temperature followed by two washes at 65°C and imaged on a PhosphoImager (Fuji).
Southern probes
The 696 bp 3′ outside probe was amplified from C57Bl/6N RPCI-23 BAC 213I16 (64) using the primers 3′Probe F (5′-GTTGTGATCTCCTGTCTACTT-3′) and 3′Probe R (5′-AACTAACCCTTCTTCTTCAACTTC-3′). The 3′probe PCR product was TA cloned into pCR2.1 Topo (Life Technologies), sequence verified and excised from the plasmid with EcoRI restriction digestion. Genomic mouse DNA digested with XmnI and probed with the 3′probe yields a WT band of 17.5kb while the Cd320 targeted allele gives a smaller band at 11.3 kb due to the insertion of a novel XmnI site in the targeting construct.
The 470 bp 5′ outside probe was amplified from C57BL/6N BAC 213I16 using the primers 5′Probe F1 (5′-CTGGGGAGAGTGTGACC-3′) and 5′Probe R1 (5′-AAAACCAATTCCCAAAGT-3′). The 5′ probe PCR product was TA cloned into pCR2.1 Topo plasmid vector (Life Technologies), sequence verified and excised by restriction digest with BstXI. Genomic mouse DNA digested with EcoRV yields a WT band of 32 kb while the Cd320-targeted band is 25 kb due to the insertion of a novel EcoRV site in the targeting construct.
Blood chemistry
Mouse peripheral blood was collected from anesthetized mice via the retro-orbital sinus. Blood was collected into EDTA tubes (EDTA capillary collectors, Sarstedt catalog # 20-1288-100) and analyzed using a CBC Hemavet blood counter at the Pathology and Histotechnology Laboratory of the Laboratory of Animal Sciences Program at the NCI-Frederick.
Hematocrit measurements
Mouse peripheral blood was collected from anesthetized mice via the retro-orbital sinus into heparinized microhematocrit tubes (Drummond Scientific Company) and sealed with critoseal StatSpin sealant (Iris Sample Processing). Tubes were centrifuged for 5 min in a micro-capillary centrifuge (International Equipment Company Model MB) and packed red cell volume was measured manually by comparison to a hematocrit reading chart (Thompson Scientific). Normal mouse hematocrits values are in the range of 45–35%. Values below 35% were considered to be indicative of anemia. Mice with extremely low hematocrit values reaching a moribund state were euthanized.
Vitamin B12 assay
Tissues were collected from non-perfused mice, drop-frozen in liquid nitrogen and stored at −80°C. Extraction of vitamin B12 from mouse tissues was carried out by homogenization of tissues in 20 volumes of sodium acetate/sodium cyanide buffer (consisting of 8.3 mmol/L NaOH, 20.7 mmol/L acetic acid and 0.45 mmol/L NaCN, pH 4.5) and autoclaving at 121°C for 10 min. After cooling, the precipitate was loosened from the walls of the tubes by vortexing and the tubes were centrifuged at 2500 RPM for 15 min. After centrifugation, supernatants were assayed for vitamin B12 using a microtiter plate adaptation of the colistin resistant microorganism Lactobacillus leichmannii (68).
Plasma MMA and Hcy measurements
Retro-orbital blood samples were collected from anesthetized mice into heparinized microcapillary tubes. The blood was expelled into microcentrifuge tubes, placed on ice and centrifuged for 10 min at 6 800x g at 4°C. The upper plasma layer was transferred to a clean tube, diluted 1:5 in deionized water and frozen at −80°C. Diluted plasma samples were sent to Metabolite Laboratories, Inc. in Denver, CO, USA for quantitation of MMA and total Hcy using stable isotope dilution capillary gas chromatography/mass spectrometry (69,70).
PCR genotyping
PCR was performed on mouse tail DNA according to standard protocols. Primers flanking the first loxP site (1st loxP F2 5′ -TAGGGGGCAATCAAGACAATAAAG-3′ and 1st loxP R2 5′-AGGACACCGGGGGCTACAAAGAGA-3′, Ta=55°C) were used to detect both the WT (247 bp) and floxed alleles (290 bp) by separation on 2% agarose (Seakem ME) gel. Detection of the deleted allele, generated by the Cre excision of the floxed region, produced a 281 bp amplicon from primers that flank the entire floxed region (Ex1+2 F 5′-TTCCCTCACTATTTTCGCCATTCA -3′ and Ex2SeqR 5′ -TACGCCTGCCTACCTATGTCTCTA -3′, Ta=55°C, 30 s extension).
RT-PCR
RNA was isolated from mouse tissues homogenized in Trizol (Invitrogen), DNase treated with amplification grade DNaseI (Life Technologies) and amplified using the SuperScript First-Strand cDNA synthesis kit (Invitrogen) and an oligo dT primer. A 205 bp fragment spanning Cd320 Exons 1 and 2 was amplified using 119F primer (5′-CCTGGTGCTGCGGCTCCTCTT-3′) and 323R primer (5′- GTCTTCCTCGTCACTGCCATCA-3′). A 468 bp fragment spanning portions of exons 4 and 5 was amplified using Northern ProbeF (5′-GAATGCCACAACTACAAGGAT-3′) and Northern ProbeR (5′-AGCCAAGCTCACCACATACA-3′) primers. As a positive control, amplification of a 452 bp fragment of the GAPDH cDNA was performed with GAPDH-F (5′-ACCACAGTCCATGCCATCAC-3′) and GAPDH-R (5′-TCCACCACCCTGTTGCTGTA-3′) primers.
Embryo harvest
Female mice were superovulated by injection with five units of pregnant mare serum and approximately 50 h later injected with five units of human chorionic gonadotropin. The females were mated to single housed Cd320 KO or control males and monitored for copulation plugs. Mated females were euthanized at 0.5–1.5 days post coitum and the dissected fallopian tubes were flushed with M2 media.
Embryo culture
The eggs from multiple females (seven control females fed vitamin B12-deficient diet for an average of 16.9 +/−0.5 weeks and six Cd320 KO females fed vitamin B12-deficient diet for an average 16.0 +/−2.3 weeks) were collected, pooled by genotype, cultured in serum-free KSOM and incubated in 5% CO2 at 37°C. Embryos were counted and staged every day until the late blastocyst stage.
Embryo transfer
Approximately 12- to 15 two-cell embryos were surgically transferred into the oviducts of pseudo-pregnant female mice according to published methods (71).
Vitamin B12 injections
Sterile injectable cyanocobalamin (Vet One) was diluted in sterile 0.9% sodium chloride (B. Braun Medical Inc.) such that 200 μl containing 1.25 μg of vitamin B12 were injected once a week for 13 weeks. As a control experiment, KO mice were injected with 200 μl of 0.9% sodium chloride once a week for 13 weeks.
Supplementary Material
Acknowledgements
The authors wish to thank the NHGRI transgenic and embryonic stem cell core for their invaluable assistance, Karen Hazzard for expertise with embryonic transplant experiments, Irene Ginty for valuable assistance bleeding and caring for our mice, Clare Horan for performing the vitamin B12 assays and Barry Shane for a critical reading of the manuscript.
Conflict of Interest statement. None declared.
Funding
Division of Intramural Research of the National Human Genome Research Institute, National Institutes of Health.
References
- 1. Ducker G.S. and Rabinowitz J.D. (2017) One-carbon metabolism in health and disease - ScienceDirect. Cell Metab., 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lehninger A.L., Nelson D.L. and Cox M.M. (2005) Lehninger Principles of Biochemistry, 4th edn. W.H. Freeman and Company, New York, NY. [Google Scholar]
- 3. Fenton W.A., Gravel R.A. and Rosenblatt D.S. (2014) Disorders of propionate and methylmalonate metabolism. In Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M., Mitchell, G. (eds) In The Online Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, New York, NY. [Google Scholar]
- 4. Nielsen M.J., Rasmussen M.R., Andersen C.B.F., Nexo E. and Moestrup S.K. (2012) Vitamin B12 transport from food to the body's cells–a sophisticated, multistep pathway. Nat. Rev. Gastroenterol. Hepatol., 9, 345–354. [DOI] [PubMed] [Google Scholar]
- 5. Allen R.H., Stabler S.P., Savage D.G. and Lindenbaum J. (1993) Metabolic abnormalities in cobalamin (vitamin B12) and folate deficiency. FASEB J., 7, 1344–1353. [DOI] [PubMed] [Google Scholar]
- 6. Li Y.N., Gulati S., Baker P.J., Brody L.C., Banerjee R. and Kruger W.D. (1996) Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum. Mol. Genet., 5, 1851–1858. [DOI] [PubMed] [Google Scholar]
- 7. Watkins D. and Rosenblatt D.S.. Regardless of Genotype and Cobalamin Transport and Metabolism, Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M., Mitchell, G. (eds), New York, NY. [Google Scholar]
- 8. Baumgartner M.R., Hörster F., Dionisi-Vici C., Haliloglu G., Karall D., Chapman K.A., Huemer M., Hochuli M., Assoun M., Ballhausen D. et al. (2014) Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis., 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Green R., Allen L.H., Bjørke-Monsen A.-L., Brito A., Guéant J.-L., Miller J.W., Molloy A.M., Nexo E., Stabler S., Toh B.-H. et al. (2017) Vitamin B12 deficiency. Nat. Rev. Dis. Primers, 3, 17040. [DOI] [PubMed] [Google Scholar]
- 10. Quadros E.V., Nakayama Y. and Sequeira J.M. (2009) The protein and the gene encoding the receptor for the cellular uptake of transcobalamin-bound cobalamin. Blood, 113, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen L.H. (2009) How common is vitamin B-12 deficiency? Am. J. Clin. Nutr., 89, 693S–696S. [DOI] [PubMed] [Google Scholar]
- 12. Green R. (2017) Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood, 129, 2603–2611. [DOI] [PubMed] [Google Scholar]
- 13. Reynolds E. (2006) Vitamin B12, folic acid, and the nervous system. Lancet Neurol., 5, 949–960. [DOI] [PubMed] [Google Scholar]
- 14. Molloy A.M., Kirke P.N., Troendle J.F., Burke H., Sutton M., Brody L.C., Scott J.M. and Mills J.L. (2009) Maternal vitamin B12 status and risk of neural tube defects in a population with high neural tube defect prevalence and no folic acid fortification. Pediatrics, 123, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott J.M. and Molloy A.M. (2012) The discovery of vitamin B12. Ann. Nutr. Metab., 61, 239–245. [DOI] [PubMed] [Google Scholar]
- 16. Minot G.R. (1926) Treatement of pernicious anemia by a special diet. JAMA, 87, 470. [PMC free article] [PubMed] [Google Scholar]
- 17. Agamanolis D.P., Chester E.M., Victor M., Kark J.A., Hines J.D. and Harris J.W. (1976) Neuropathology of experi-mental vitamin B12 deficiency in monkeys. Neurology, 26, 905–914. [DOI] [PubMed] [Google Scholar]
- 18. Weir D.G., Keating S., Molloy A., McPartlin J., Kennedy S., Blanchflower J., Kennedy D.G., Rice D. and Scott J.M. (1988) Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem., 51, 1949–1952. [DOI] [PubMed] [Google Scholar]
- 19. Van Tonder S.V., Metz J. and Green R. (1975) Vitamin B12 metabolism in the fruit bat (Rousettus aegyptiacus). The induction of vitamin B12 deficiency and its effect on folate levels. Br. J. Nutr., 34, 397–410. [DOI] [PubMed] [Google Scholar]
- 20. Green R., Van Tonder S.V., Oettle G.J., Cole G. and Metz J. (1975) Neurological changes in fruit bats deficient in vitamin B12. Nature, 254, 148–150. [DOI] [PubMed] [Google Scholar]
- 21. Westhuyzen J., Cantrill R.C., Fernandes-Costa F. and Metz J. (1983) Effect of a vitamin B-12-deficient diet on lipid and fatty acid composition of spinal cord myelin in the fruit bat. J. Nutr., 113, 531–537. [DOI] [PubMed] [Google Scholar]
- 22. Duffield M.S., Phillips J.I., Vieira-Makings E., Westhuyzen J. and Metz J. (1990) Demyelinisation in the spinal cord of vitamin B12 deficient fruit bats. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol., 96, 291–297. [DOI] [PubMed] [Google Scholar]
- 23. Peng L., Dreumont N., Coelho D., Guéant J.-L. and Arnold C. (2016) Genetic animal models to decipher the pathogenic effects of vitamin B12 and folate deficiency. Biochimie, 126, 43–51. [DOI] [PubMed] [Google Scholar]
- 24. Choi S.-W., Friso S., Ghandour H., Bagley P.J., Selhub J. and Mason J.B. (2004) Vitamin B-12 deficiency induces anomalies of base substitution and methylation in the DNA of rat colonic epithelium. J. Nutr., 134, 750–755. [DOI] [PubMed] [Google Scholar]
- 25. Buccellato F.R., Miloso M., Braga M., Nicolini G., Morabito A., Pravettoni G., Tredici G. and Scalabrino G. (1999) Myelinolytic lesions in spinal cord of cobalamin-deficient rats are TNF-alpha-mediated. FASEB J., 13, 297–304. [DOI] [PubMed] [Google Scholar]
- 26. Swanson D.A., Liu M.L., Baker P.J., Garrett L., Stitzel M., Wu J., Harris M., Banerjee R., Shane B. and Brody L.C. (2001) Targeted disruption of the methionine synthase gene in mice. Mol. Cell. Biol., 21, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chandler R.J., Sloan J., Fu H., Tsai M., Stabler S., Allen R., Kaestner K.H., Kazazian H.H. and Venditti C.P. (2007) Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle. BMC Med. Genet., 8, 64–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peters H., Nefedov M., Sarsero J., Pitt J., Fowler K.J., Gazeas S., Kahler S.G. and Ioannou P.A. (2003) A knock-out mouse model for methylmalonic aciduria resulting in neonatal lethality. J. Biol. Chem., 278, 52909–52913. [DOI] [PubMed] [Google Scholar]
- 29. Pangilinan F., Mitchell A., VanderMeer J., Molloy A.M., Troendle J., Conley M., Kirke P.N., Sutton M., Sequeira J.M., Quadros E.V. et al. (2010) Transcobalamin II receptor polymorphisms are associated with increased risk for neural tube defects. J. Med. Genet., 47, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quadros E.V., Lai S.-C., Nakayama Y., Sequeira J.M., Hannibal L., Wang S., Jacobsen D.W., Fedosov S., Wright E., Gallagher R.C. et al. (2010) Positive newborn screen for methylmalonic aciduria identifies the first mutation in TCblR/CD320, the gene for cellular uptake of transcobalamin-bound vitamin B(12). Hum. Mutat., 31, 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fernàndez-Roig S., Lai S.-C., Murphy M.M., Fernandez-Ballart J. and Quadros E.V. (2012) Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr. Metab. (Lond), 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lai S.-C., Nakayama Y., Sequeira J.M., Wlodarczyk B.J., Cabrera R.M., Finnell R.H., Bottiglieri T. and Quadros E.V. (2013) The transcobalamin receptor knockout mouse: a model for vitamin B12 deficiency in the central nervous system. FASEB J., 27, 2468–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arora K., Sequeira J.M., Hernández A.I., Alarcon J.M. and Quadros E.V. (2017) Behavioral alterations are associated with vitamin B12 deficiency in the transcobalamin receptor/CD320 KO mouse. PLoS ONE, 12, e0177156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mutti E., Ruetz M., Birn H., Kräutler B. and Nexo E. (2013) 4-ethylphenyl-cobalamin impairs tissue uptake of vitamin B12 and causes vitamin B12 deficiency in mice. PLoS ONE, 8, e75312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roman-Garcia P., Quiros-Gonzalez I., Mottram L., Lieben L., Sharan K., Wangwiwatsin A., Tubio J., Lewis K., Wilkinson D., Santhanam B. et al. (2014) Vitamin B12-dependent taurine synthesis regulates growth and bone mass. J. Clin. Invest., 124, 2988–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fràter-Schröder M., Haller O., Gmür R., Kierat L. and Anastasi S. (1982) Allelic forms of mouse transcobalamin 2. Biochem. Genet., 20, 1001–1014. [DOI] [PubMed] [Google Scholar]
- 37. May P., Woldt E., Matz R.L. and Boucher P. (2007) The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann. Med., 39, 219–228. [DOI] [PubMed] [Google Scholar]
- 38. Nykjaer A. and Willnow T.E. (2002) The low-density lipoprotein receptor gene family: a cellular Swiss army knife? Trends Cell Biol., 12, 273–280. [DOI] [PubMed] [Google Scholar]
- 39. Moestrup S.K., Birn H., Fischer P.B., Petersen C.M., Verroust P.J., Sim R.B., Christensen E.I. and Nexø E. (1996) Megalin-mediated endocytosis of transcobalamin-vitamin-B12 complexes suggests a role of the receptor in vitamin-B12 homeostasis. Proc. Natl. Acad. Sci. U.S.A., 93, 8612–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birn H., Willnow T.E., Nielsen R., Norden A.G.W., Bönsch C., Moestrup S.K., Nexo E. and Christensen E.I. (2002) Megalin is essential for renal proximal tubule reabsorption and accumulation of transcobalamin-B(12). Am. J. Physiol. Renal Physiol., 282, F408–F416. [DOI] [PubMed] [Google Scholar]
- 41. Arora K., Sequeira J.M. and Quadros E.V. (2017) Maternofetal transport of vitamin B12: role of TCblR/CD320 and megalin. FASEB J., 31, 3098–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao H., Ruberu K., Li H. and Garner B. (2016) Cell type-specific modulation of cobalamin uptake by bovine serum. PLoS ONE, 11, e0167044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berliner N. and Rosenberg L.E. (1981) Uptake and metabolism of free cyanocobalamin by cultured human fibroblasts from controls and a patient with transcobalamin II deficiency. Metab. Clin. Exp., 30, 230–236. [DOI] [PubMed] [Google Scholar]
- 44. Hakami N., Neiman P.E., Canellos G.P. and Lazerson J. (1971) Neonatal megaloblastic anemia due to inherited transcobalamin II deficiency in two siblings. N. Engl. J. Med., 285, 1163–1170. [DOI] [PubMed] [Google Scholar]
- 45. Ghosh S., Sinha J.K., Putcha U.K. and Raghunath M. (2016) Severe but not moderate vitamin B12 deficiency impairs lipid profile, induces adiposity, and leads to adverse gestational outcome in female C57BL/6 mice. Front. Nutr., 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fyfe J.C., Madsen M., Hojrup P., Christensen E.I., Tanner S.M., de la Chapelle A., He Q. and Moestrup S.K. (2004) The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood, 103, 1573–1579. [DOI] [PubMed] [Google Scholar]
- 47. Smith B.T., Mussell J.C., Fleming P.A., Barth J.L., Spyropoulos D.D., Cooley M.A., Drake C.J. and Argraves W.S. (2006) Targeted disruption of cubilin reveals essential developmental roles in the structure and function of endoderm and in somite formation. BMC Dev. Biol., 6, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Bornslaeger E.A., Haub O., Tomihara-Newberger C., Lonberg N., Dinulos M.B., Disteche C.M., Copeland N., Gilbert D.J., Jenkins N.A. et al. (1996) A candidate gene for the amnionless gastrulation stage mouse mutation encodes a TRAF-related protein. Dev. Biol., 177, 274–290. [DOI] [PubMed] [Google Scholar]
- 49. Buers I., Pennekamp P., Nitschke Y., Lowe C., Skryabin B.V. and Rutsch F. (2016) Lmbrd1 expression is essential for the initiation of gastrulation. J. Cell. Mol. Med., 20, 1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moreno-Garcia M.A., Pupavac M., Rosenblatt D.S., Tremblay M.L. and Jerome-Majewska L.A. (2014) The Mmachc gene is required for pre-implantation embryogenesis in the mouse. Mol. Genet. Metab., 112, 198–204. [DOI] [PubMed] [Google Scholar]
- 51. Willnow T.E., Hilpert J., Armstrong S.A., Rohlmann A., Hammer R.E., Burns D.K. and Herz J. (1996) Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. U.S.A., 93, 8460–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jackson I.M., Doig W.B. and McDonald G. (1967) Pernicious anaemia as a cause of infertility. Lancet, 2, 1159–1160. [DOI] [PubMed] [Google Scholar]
- 53. Hall M. and Davidson R.J. (1968) Prophylactic folic acid in women with pernicious anaemia pregnant after periods of infertility. J. Clin. Pathol., 21, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Girdwood R.H., Eastwood M.A., Finlayson N.D. and Graham G.S. (1971) Pernicious anaemia as a cause of infertility in twins. Lancet, 1, 528–530. [DOI] [PubMed] [Google Scholar]
- 55. Gulden K.D. (1990) Pernicious anemia, vitiligo, and infertility. J. Am. Board Fam. Pract., 3, 217–220. [PubMed] [Google Scholar]
- 56. Gaskins A.J., Chiu Y.-H., Williams P.L., Ford J.B., Toth T.L., Hauser R., Chavarro J.E., and EARTH Study Team (2015) Association between serum folate and vitamin B-12 and outcomes of assisted reproductive technologies. Am. J. Clin. Nutr., 102, 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bito T., Matsunaga Y., Yabuta Y., Kawano T. and Watanabe F. (2013) Vitamin B12 deficiency in Caenorhabditis elegans results in loss of fertility, extended life cycle, and reduced lifespan. FEBS Open Bio, 3, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murphy L.E., Mills J.L., Molloy A.M., Qian C., Carter T.C., Strevens H., Wide-Swensson D., Giwercman A. and Levine R.J. (2011) Folate and vitamin B12 in idiopathic male infertility. Asian J. Androl., 13, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carmel R. (2012) Subclinical cobalamin deficiency. Curr. Opin. Gastroenterol., 28, 151–158. [DOI] [PubMed] [Google Scholar]
- 60. Sun A., Chang J.Y.-F., Wang Y.-P., Cheng S.-J., Chen H.-M. and Chiang C.-P. (2016) Do all the patients with vitamin B12 deficiency have pernicious anemia? J. Oral Pathol. Med., 45, 23–27. [DOI] [PubMed] [Google Scholar]
- 61. Rothenberg S.P. (1999) Increasing the dietary intake of folate: pros and cons. Semin. Hematol., 36, 65–74. [PubMed] [Google Scholar]
- 62. National Research Council (US) Subcommittee on Laboratory Animal Nutrition (1995) Nutrient Requirements of Laboratory Animals: Fourth Revised Edition, 1995. Washington (DC): National Academies Press (US) https://www.ncbi.nlm.nih.gov/books/NBK231927/ doi:10.17226/4758. Accessed June 21, 2018.10.17226/4758. [PubMed] [Google Scholar]
- 63. Champy M.-F., Selloum M., Zeitler V., Caradec C., Jung B., Rousseau S., Pouilly L., Sorg T. and Auwerx J. (2008) Genetic background determines metabolic phenotypes in the mouse. Mamm. Genome, 19, 318–331. [DOI] [PubMed] [Google Scholar]
- 64. Osoegawa K., Tateno M., Woon P.Y., Frengen E., Mammoser A.G., Catanese J.J., Hayashizaki Y. and Jong P.J. (2000) Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res., 10, 116–128. [PMC free article] [PubMed] [Google Scholar]
- 65. Tybulewicz V.L., Crawford C.E., Jackson P.K., Bronson R.T. and Mulligan R.C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell, 65, 1153–1163. [DOI] [PubMed] [Google Scholar]
- 66. Cheng J., Dutra A., Takesono A., Garrett-Beal L. and Schwartzberg P.L. (2004) Improved generation of C57BL/6J mouse embryonic stem cells in a defined serum-free media. Genesis, 39, 100–104. [DOI] [PubMed] [Google Scholar]
- 67. Lakso M., Pichel J.G., Gorman J.R., Sauer B., Okamoto Y., Lee E., Alt F.W. and Westphal H. (1996) Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U.S.A., 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kelleher B.P. and Broin S.D. (1991) Microbiological assay for vitamin B12 performed in 96-well microtitre plates. J. Clin. Pathol., 44, 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stabler S.P., Lindenbaum J., Savage D.G. and Allen R.H. (1993) Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood, 81, 3404–3413. [PubMed] [Google Scholar]
- 70. Allen R.H., Stabler S.P., Savage D.G. and Lindenbaum J. (1993) Elevation of 2-methylcitric acid I and II levels in serum, urine, and cerebrospinal fluid of patients with cobalamin deficiency. Metab. Clin. Exp., 42, 978–988. [DOI] [PubMed] [Google Scholar]
- 71. Hogan B., Costantini F. and Lacy E. (1986) Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.