

MyoD upstream noncoding RNA (MUNC) initiates in the distal regulatory region (DRR) enhancer of MYOD and is formally classified as an enhancer RNA (DRReRNA). MUNC is required for optimal myogenic differentiation, induces specific myogenic transcripts in trans (MYOD, MYOGENIN, and MYH3), and has a functional human homolog.
KEYWORDS: MUNC, MyoD, eRNA, enhancer, lncRNA, myogenesis, skeletal muscles
ABSTRACT
MyoD upstream noncoding RNA (MUNC) initiates in the distal regulatory region (DRR) enhancer of MYOD and is formally classified as an enhancer RNA (DRReRNA). MUNC is required for optimal myogenic differentiation, induces specific myogenic transcripts in trans (MYOD, MYOGENIN, and MYH3), and has a functional human homolog. The vast majority of eRNAs are believed to act in cis primarily on their neighboring genes (1, 2), making it likely that MUNC action is dependent on the induction of MYOD RNA. Surprisingly, MUNC overexpression in MYOD−/− C2C12 cells induces many myogenic transcripts in the complete absence of MyoD protein. Genomewide analysis showed that, while many genes are regulated by MUNC in a MyoD-dependent manner, there is a set of genes that are regulated by MUNC, both upward and downward, independently of MyoD. MUNC and MyoD even appear to act antagonistically on certain transcripts. Deletion mutagenesis showed that there are at least two independent functional sites on the MUNC long noncoding RNA (lncRNA), with exon 1 more active than exon 2 and with very little activity from the intron. Thus, although MUNC is an eRNA of MYOD, it is also a trans-acting lncRNA whose sequence, structure, and cooperating factors, which include but are not limited to MyoD, determine the regulation of many myogenic genes.
INTRODUCTION
Myogenesis is a process of skeletal muscle differentiation occurring during vertebrate embryo development and during regeneration of muscle fibers after injury in the adult. During embryonic development, muscles derive from the mesoderm, where myoblasts, embryonic progenitor cells, give rise to muscle fibers (3). Myogenesis requires a network of muscle-specific transcription factors composed of four muscle regulatory factors (MRFs) from the basic helix-loop-helix (bHLH) family of transcription factors (myogenic factor 5 [Myf5], myoblast determination protein [MyoD], myogenin, and muscle-specific regulatory factor 4 [MRF4]). When myogenesis is activated, MyoD-MyoE protein heterodimers bind to E-box sequences in promoters of genes, driving their transcription and setting off a transcriptional cascade (4). This activation leads to the expression of several muscle-specific target genes, such as MYOGENIN, M-CADHERIN, myosin heavy and light chains (such as MYH3), and the muscle creatine kinase gene (5).
Three DNA sequence elements regulate MYOD expression in mice: a proximal regulatory region (PRR) that is adjacent to the transcription start site (TSS) of MYOD, a 720-bp-long distal regulatory region (DRR) located ∼5 kb upstream from the MYOD TSS, and a core enhancer region (CER) located ∼23 kb upstream from the MYOD TSS (6–8). The DRR sequence is functionally conserved between mouse and human, sharing blocks of sequence identity over a 445-bp region between the two species. DRR deletion reduces MYOD RNA and the protein level in adult muscle (9, 10). The DRR contains consensus binding sites for MyoD, MEF-2, and SRF (10, 11), explaining how it positively regulates MYOD expression like a classic enhancer. The DRR is essential as an enhancer for skeletal muscle differentiation, but it also serves as the initiation site of a myogenic enhancer RNA (eRNA), MyoD upstream noncoding RNA (MUNC), or DRReRNA, which plays a positive regulatory role during muscle development (12, 13).
Long noncoding RNAs (lncRNAs) form a diverse family of RNA transcripts longer than 200 nucleotides (nt) that do not encode proteins but have different functions in the cell as RNA molecules (reviewed in reference 14). High-throughput RNA sequencing (RNA-Seq) analysis in mice suggests that lncRNAs are a major component of the transcriptome (15). Mainly transcribed by RNA polymerase II (RNA Pol II), lncRNA can be intergenic, multiexonic, antisense to known genes, or from regulatory elements located distal to a known TSS. High-throughput RNA sequencing identified many novel lncRNAs specifically expressed during skeletal muscle differentiation (16). Their mechanisms of action are heterogeneous, and they are localized differently in cells (reviewed in references 14 and 17). Nuclear lncRNAs can mediate epigenetic changes by recruiting chromatin-remodeling complexes to specific genomic loci. Muscle-specific steroid receptor RNA activator (SRA) RNA promotes muscle differentiation through its interactions with RNA helicase coregulators p68, p72, and MyoD (18). Another example of a promyogenic lncRNA functioning in cis is Dum (developmental pluripotency-associated 2 [Dppa2] upstream binding muscle RNA), which silences its neighboring gene, DPPA2, by recruiting Dnmts to its locus (19). DBE-T, a lncRNA produced selectively in patients with facioscapulohumeral muscular dystrophy (FSHD), binds to the chromatin and recruits transcriptional activator Ash1L to derepress the FSHD locus (20).
An important group of nuclear lncRNAs work as eRNAs, stimulating transcription of adjacent genes (1). A recent study of 12 mouse lncRNAs identified 5 of them that act as eRNAs stimulating the transcription of the adjoining gene in cis by a process that involves the transcription and splicing of the eRNA but is not dependent on the sequence of the actual RNA transcript (2). Myogenic eRNAs include DRReRNA, or MUNC, and CEReRNA, which, consistent with current models of eRNA function, stimulate expression of the adjoining MYOD gene in cis by increasing chromatin accessibility for transcriptional factors. DRReRNA, or MUNC, is already a little atypical as an eRNA because it can induce expression not only of the MYOD gene located in cis but also of MYOGENIN and MYH3, which are located on different chromosomes (12, 13).
In this study, we show that MUNC has a function independent of its action as an eRNA stimulating expression of MYOD. Specifically, MUNC has a MyoD-independent promyogenic function during skeletal muscle differentiation, has multiple separate functional regions, and can act in trans on multiple genes on different chromosomes. These findings raise the possibility that, although many eRNAs act as classic enhancer RNAs that stimulate transcription of adjoining genes merely by the acts of transcription and splicing, some of them have additional roles as trans-acting lncRNAs, where the sequence of the RNA matters for its function.
RESULTS
MUNC as a lncRNA has multiple domains important for its function.
In the previous study, we showed that stable overexpression of MUNC from a heterologous site in C2C12 cells increases the levels of three myogenic RNAs, MYOD, MYOGENIN, and MYH3 (13). This in itself is at odds with the prevailing model, in which the acts of transcription and splicing at the endogenous eRNA locus are important for the action of the eRNA. We therefore decided to investigate the second tenet of the eRNA hypothesis: is the specific sequence of the MUNC transcript irrelevant for stimulating the myogenic transcripts? Fragments of MUNC containing different parts of the RNA were stably overexpressed in C2C12 cells (Fig. 1A). The overexpression was confirmed both in proliferating myoblasts (Fig. 1C to E) and in differentiating myotubes (Fig. 1F to H). In addition, we used C2C12 cells stably transfected with the spliced isoform of MUNC and with the genomic sequence of MUNC (overexpressing both spliced and unspliced isoforms). We compared the expression levels of MYOD, MYOGENIN, and MYH3 RNAs in cells overexpressing MUNC or fragments of MUNC relative to control cells transfected with the empty vector (EV). We performed the analysis under two conditions: in proliferating myoblasts (growth medium [GM]) to see whether MUNC is able to induce myogenic factors when cells proliferate, and after 3 days of differentiation (DM3) in differentiation medium (DM) to see whether overexpression of MUNC is still able to change myogenic RNA levels when other myogenic factors have already been induced (Fig. 1B). Several interesting points emerge from consideration of the results.
FIG 1.

MUNC has at least two domains important for its function. (A) Schematic illustrating MUNC structure. The red lines indicate three potential micropeptides coded by MUNC spliced sequence: two of 20 amino acids and one of 60 amino acids. The micropeptides were defined using a translation tool (http://web.expasy.org/translate/). (B) Heat maps showing summaries of qRT-PCR analyses of C2C12 mutant cells stably overexpressing different truncated MUNC sequences. Levels of myogenic factor transcripts were measured in three biological runs and normalized to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) level and to control cells under each condition, and mean values were calculated. The colors used in the heat maps correspond to fold changes according to the legend. N.S., not significant. Analysis of proliferating cells and differentiating cells. (C to H) qRT-PCR analysis of mutant cells overexpressing truncated MUNC sequences showing levels of different parts of the transcript (exon 1, intron, and exon 2) in GM (C to E) and in DM3 (F to H). The data were normalized to GAPDH and to control cells transfected with an EV. The values represent three biological replicates and are presented as means and standard errors of the mean (SEM). (I) Predicted structures of different mutants of MUNC generated using the Forna RNA prediction tool.
First, in differentiating cells, MUNC induced MYOGENIN and MYH3 to much higher levels than in proliferating cells, suggesting that differentiating cells may express additional factors that facilitate MUNC's action. Second, MYOD induction by exon 1, intron plus exon 2, or unspliced or spliced MUNC was much lower in DM3 (10 to 61 times than in cells without MUNC overexpression) than in GM (26 to 214 times), yet the reverse was true for MYOGENIN and MYH3 (12 to 600 times in DM3 versus 1 to 8 times in GM). This suggests that there is not a linear correlation between the fold induction of MYOD and that of MYOGENIN and MYH3, as would have been expected if MUNC worked solely by inducing MYOD to induce MYOGENIN or MYH3. This lack of correlation is consistent with our earlier observation that MUNC overexpression induced MYOGENIN and MYH3 mRNAs without inducing MyoD protein (despite the induction of MYOD mRNA) (13).
Third, spliced MUNC was always better than genomic MUNC at inducing MYOD. We know from RNA-Seq that genomic MUNC expresses mostly unspliced MUNC in these cells, so the difference is probably attributable either to the presence of inhibitory sequences in the intron or to different folding of the exonic sequences in unspliced and spliced MUNC. Differences in folding of the two isoforms were predicted by the Forna tool (21) (Fig. 1I). Among truncated mutants of MUNC, exon 1 was the most potent at inducing MYOD. Although the intron and exon 2 by themselves were mostly ineffective, addition of the intron to exon 2 made it more effective at inducing MYOD than either of them alone. As Fig. 1I shows, exon 2, intron, and exon 2 plus intron fragments of MUNC have different predicted RNA-folding structures.
In summary, these studies suggest that the simple act of transcription of MUNC (as suggested for eRNAs) cannot be enough for the stimulation of MYOD, MYOGENIN, or MYH3. Instead, as isolated fragments, exon 1 has the most significant stimulatory activity, although a second domain with activity became evident in the intron plus exon 2 fragment. Finally, the high degree of activity of the intron plus exon 2 fragment compared to either part alone (intron or exon 2) or of spliced MUNC (exon 1 plus exon 2) compared to unspliced MUNC (exon 1 plus intron plus exon 2) suggests that the folding of the RNA is important for this activity.
There have been a few reports of lncRNAs encoding micropeptides with biological functions (22). Spliced MUNC transcripts could code for three such micropeptides unrelated to each other in sequence (underlined in red in Fig. 1A). The structure-function analysis mentioned above rules out the possibility that the induction of the three genes is due to any of these micropeptides.
MYOD knockout (KO) diminishes muscle differentiation in vitro.
A crucial role of MyoD during skeletal muscle differentiation was established both in vitro and in vivo. Skeletal muscles of MYOD−/− mice displayed reduced capacity for regeneration following injury (23), and in vitro knockdown of MYOD in differentiating C2C12 cells decreased the efficiency of differentiation (13, 24). It is also known that knockdown of MUNC decreases expression of MYOD and negatively affects other downstream effectors of muscle differentiation (13). To investigate whether the role of MUNC during muscle differentiation is through the induction of MyoD or whether MUNC has activities independent of MyoD, we engineered MYOD−/− C2C12 cells. Using clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 technology (25), both alleles of MYOD were knocked out by deletion of 149 bp of MYOD exon 1 (corresponding to amino acids P7 to L57 of the MyoD protein and throwing the rest of the protein out of frame) (Fig. 2A). The deletion was confirmed by PCR of the genomic DNA (Fig. 2B) and by Sanger sequencing of the PCR products (Fig. 2A). RNA-Seq data provided additional corroboration of the deletion by showing the complete absence of reads from the deleted region in MYOD−/− cells compared to wild-type (WT) cells (Fig. 2C). The homozygous deletion was associated with the complete absence of MyoD protein in the cells, confirmed by antibodies recognizing an epitope in the C terminus of the protein (Fig. 2D).
FIG 2.

MYOD knockout decreases muscle differentiation in vitro. (A) Deletion of MYOD genomic sequence causing MyoD protein deletion. The triangles indicate the primers used for genotyping. The sequence across the deletion junction is shown and was confirmed by sequencing the genotyping PCR product from the genomic DNA of MYOD−/− cells. (B) PCR products with the genotyping primers on genomic DNA confirmed MYOD sequence deletion in MYOD−/− cells. The products were sequenced to confirm the deletion junction shown in panel A. The complete absence of a WT genotype band in the MYOD−/− cells confirmed that no WT allele was left. (C) RNA-Seq confirmed deletion of all alleles of MyoD in the MYOD−/− cells. A Sashimi plot of the RNA-Seq reads shows that the deleted region (shaded) in exon 1 of the MYOD gene is missing in MYOD−/− cell RNA. (D) Western blot analysis confirms the absence of MyoD protein in MYOD−/− cells. Tubulin served as a loading control. (E) Immunofluorescence analysis of fixed cells 3 days after differentiation (DM3). The cells were immunostained with antibodies against MyoD and MHC. DAPI (4′,6-diamidino-2-phenylindole) was used to visualize nuclei. (F to H) qRT-PCR analysis of proliferating (GM) and differentiating (DM3) C2C12 cells that were WT or miR-1a-1−/−. Levels of MYOD, MYOGENIN, and MYH3 mRNAs normalized to GAPDH are shown relative to that in proliferating WT cells (WT GM). (I to L) qRT-PCR analysis of proliferating (GM) and differentiating (DM3) cells that were WT or MYOD−/−. Levels of the indicated RNAs normalized to GAPDH are shown relative to that in proliferating WT cells (WT GM). The values represent three biological replicates and are presented as means and SEM. Statistical significance was calculated using the Wilcoxon-Mann-Whitney test. *, P < 0.05.
To ensure that nonspecific effects of CRISPR-Cas9 editing or clonal selection did not impair differentiation, we also engineered a C2C12 cell with homozygous deletion of miR1a-1. This microRNA is not expected to be essential for muscle differentiation because of the presence in skeletal muscle of two other microRNAs from the same sequence family, miR-206 and miR1a-2. The miR1a-1−/− cells differentiated in DM3 and induced the RNAs of three myogenic factors, MYOD, MYOGENIN, and MYH3, almost as efficiently as WT cells (Fig. 2F to H). Thus, CRISPR-Cas9 editing or clonal selection does not impair C2C12 cell differentiation.
In contrast, the MYOD−/− cells differentiated poorly. WT cells showed the expected induction of specific myogenic transcripts after differentiation: MYOD (Fig. 2I), MUNC (Fig. 2J), MYOGENIN, and MYH3 (Fig. 2K and L). In contrast, MYOD−/− cells with part of the MYOD transcript deleted (Fig. 2I) had low expression of MUNC (Fig. 2J) and nearly 100-fold less induction of MYOGENIN or MYH3 RNA than WT cells (Fig. 2K and L). Note that because of the nearly undetectable levels of MYOGENIN or MYH3 mRNA in C2C12 cells in GM, there is great variation in the high threshold cycle values (number of qPCR cycles after which the product becomes detectable) of these two transcripts in GM from experiment to experiment. Therefore, the fold induction in DM relative to this basal level varies greatly from experiment to experiment, even in WT cells, e.g., 14-fold versus 1,000-fold for MYOGENIN (Fig. 2G versus K) and 6-fold versus 30-fold for MYH3 (Fig. 2H versus L). This is why the fold induction during differentiation shown in the figures should not be compared between experiments but should always be interpreted relative to control cells included in each experiment. Thus, we conclude that the miR1a-1−/− cells are almost as good as WT cells at inducing the myogenic transcripts, while the MYOD−/− cells are 100-fold weaker than WT cells at inducing the same transcripts.
Consistent with this, the MYOD−/− cells lacked MyoD and myosin heavy chain (MHC) proteins by immunofluorescence assay (the background signal is due to incomplete cutoff by the filter) in DM3 (Fig. 2E). These results agree with previous reports that MYOD is essential for myogenesis in vitro and confirm that we successfully deleted MYOD in the C2C12 cells.
MUNC knockout disrupts myogenesis, which is rescued by overexpression of MyoD.
In parallel, we generated MUNC−/− C2C12 clones (Fig. 3A). The deletion of MUNC by CRISPR-Cas9 engineering was confirmed by PCR of genomic DNA (Fig. 3B) and Sanger sequencing of the PCR products (Fig. 3A). To confirm deletion of MUNC sequence, we performed Southern blotting of genomic DNA digested with BspHI enzyme. The digestion sites are labeled in Fig. 3A. WT cells produced a 9-kb DNA band, and MUNC−/− cells produced an 8-kb band, confirming full deletion of both alleles of MUNC (Fig. 3C). MUNC−/− cells were disabled in differentiation: MYOD, MYOGENIN, and MYH3 RNAs were decreased at least 5-fold compared to WT DM3 cells (Fig. 3D, E, and F). To examine whether the induction of the two RNAs was rescued by addition of MyoD, we overexpressed MyoD using a doxycycline-inducible MyoD-expressing lentivirus vector (Fig. 3H). After 3 days of differentiation, this was sufficient to induce MYOGENIN and MYH3 RNAs (Fig. 3G, lane 3 versus lane 2). This was accompanied by the induction of myogenin and MHC proteins (Fig. 3H) and morphological differentiation.
FIG 3.

MUNC knockout decreases expression of MYOGENIN and MYH3 RNAs, which is rescued by overexpression of MYOD. (A) The segment of MUNC genomic sequence that was deleted. The triangles indicate target sites for genotyping primers. Sequencing of the genotyping PCR products confirmed the deletion junction shown below. Locations of BspH1 restriction sites and the MUNC probe for Southern blotting are shown relative to the MUNC TSS. (B) PCR products genotyping MUNC in WT and MUNC−/− cells. (C) Confirmation of MUNC deletion by Southern blotting hybridization of BspHI-digested genomic DNA. The sizes of DNA fragments that hybridize with the MUNC probe are consistent with predicted sizes of genomic DNA from WT and MUNC−/− cells. (D to F) qRT-PCR analysis of differentiating (DM3) WT cells or MUNC−/− cells. The levels of the indicated mRNAs were normalized to GAPDH and are shown relative to WT cells. The values represent three biological replicates and are presented as means and SEM. Statistical significance was calculated using the Wilcoxon-Mann-Whitney test. *, P < 0.05. (G) qRT-PCR of the indicated RNAs in WT and MUNC−/− cells after 3 days in DM. The RNA levels were normalized to GAPDH and expressed relative to the level in WT cells. All the cells were transduced with lentivirus containing MYOD. Ex. MYODON, lentiviral MYOD induced by doxycycline. (H) Western blotting for the indicated proteins in MUNC−/− cells with and without MyoD overexpression.
To ensure that the failure to differentiate seen in the knockout cells was not due to delayed kinetics of differentiation and to compare the two types of knockout cells with each other, we compared the differentiation efficiencies of WT, MUNC−/−, and MYOD−/− cells over 5 days by measuring mRNA levels of myogenic factors (Fig. 4A to D). WT cells, as expected, showed a progressive increase of MYOD (Fig. 4A), MUNC (Fig. 4B), MYOGENIN (Fig. 4C), and MYH3 (Fig. 4D) after 1, 3, and 5 days of differentiation. MUNC−/− cells did not show any induction of myogenic mRNAs. In MYOD−/− cells, levels of RNA markers were very low compared to WT cells but were slightly induced after 5 days of differentiation. Neither mutant was able to synthesize myogenin or MHC protein (Fig. 4E), suggesting that they do not differentiate much, even though MYOGENIN and MUNC RNAs were induced to low levels in the MYOD−/− cells. Immunostaining cells after 5 days of differentiation showed many myotubes containing MHC in WT cells and none in either of the mutants (Fig. 4F). These results confirm that deletion of MYOD or MUNC equally impairs muscle differentiation.
FIG 4.

The time course of differentiation confirms that MUNC−/− and MYOD−/− C2C12 cells do not differentiate in vitro. (A to D) qRT-PCR analysis of proliferating (GM) and differentiating (DM1, DM3, and DM5) cells that were WT for MYOD and MUNC and of MUNC−/− and MYOD−/− cells. Levels of the indicated RNAs were normalized to GAPDH and are shown relative to that in proliferating WT cells (WT GM). The values represent three biological replicates and are presented as means and SEM. (E) Western blot of proliferating (GM) and differentiating (DM1, DM3, and DM5) cells that were WT for MYOD and for MUNC and of MUNC−/− or MYOD−/− cells. Protein levels for MyoD, myogenin, and MHC were measured. Tubulin served as a loading control. An arrow indicates the specific band for MyoD protein. (F) Immunofluorescence analysis of fixed cells 5 days after differentiation (DM5). Cells were immunostained with antibodies against MHC. DAPI was used to visualize nuclei. DIC, differential interference contrast.
Stable overexpression of MUNC in MYOD−/− cells induces MYOGENIN and MYH3 transcripts and proteins in the complete absence of MyoD protein.
We have seen the induction of MYOGENIN and MYH3 RNAs in WT C2C12 cells by the overexpression of MUNC (13). In those experiments, MyoD protein was not induced any further, but it was still present, so we could not definitively say that MUNC induced these RNAs independently of MyoD. We could rule out any role of MyoD by stably overexpressing spliced MUNC in MYOD−/− C2C12 cells. Cells overexpressing MUNC (Fig. 5A) showed higher expression of MYOGENIN RNA in both GM (100-fold induction) and DM (10-fold induction) than control cells not overexpressing MUNC (Fig. 5B). MYH3 RNA was also increased by 10-fold in both GM and DM (Fig. 5C). Thus, the lncRNA MUNC is able to induce MYOGENIN and MYH3 RNAs in the complete absence of MyoD protein.
FIG 5.

Stable overexpression of MUNC in MYOD−/− cells induces MYOGENIN and MYH3 transcript levels independently of MyoD. (A to C) qRT-PCR analysis of RNAs from proliferating (GM) and differentiating (DM3) MYOD−/− cells stably transfected with vector expressing MUNC. Levels of the indicated RNAs were normalized to GAPDH and are shown relative to MYOD−/− proliferating cells (GM). +, overexpression of exogenous MUNC. The values represent three biological replicates and are presented as means and SEM. Statistical significance was calculated using the Wilcoxon-Mann-Whitney test. *, P < 0.05. (D) Western blot analysis of MHC protein levels in MYOD−/− cells overexpressing MUNC under DM3 conditions. Tubulin was used as a loading control.
In WT C2C12 cells, the induction of MYOGENIN or MYH3 RNA by MUNC was not accompanied by the induction of the two proteins (13). However, the situation was slightly different in the MYOD−/− cells (Fig. 5D). Even though myogenin protein was not induced under GM or DM conditions, MHC protein was slightly induced upon MUNC overexpression only under DM conditions. The MYOD−/− cells overexpressing MUNC did not show any morphological signs of differentiation. The induction of MHC protein by MUNC in MYOD−/− cells can be explained by the MYH3 RNA reaching a threshold value in DM for the resulting protein to be detectable. However, the lack of MyoD protein still prevents the induction of myogenin protein or morphological differentiation in DM.
MyoD and MUNC cooperate to induce MYOGENIN and MYH3 RNAs but fail to promote differentiation of MYOD−/− cells in DM.
The RNA-Seq results we describe below suggest some cooperation between MUNC and MyoD in inducing MYOGENIN and MYH3 RNAs. MUNC overexpression is better in WT cells than in MYOD−/− cells at inducing MYOGENIN (5-fold) and MYH3 (12-fold). We therefore wanted to test whether MyoD synergizes with MUNC for the induction of these two genes. First we tested the maximum extent to which MyoD protein restoration in MYOD−/− cells would induce MYOGENIN and MYH3 RNAs by lentivirus-mediated doxycycline-inducible overexpression of MyoD (Fig. 6A and D). Exogenous MyoD induced MYOGENIN RNA and protein and MYH3 RNA (Fig. 6B, C, and D).
FIG 6.

Expression of MYOD partially rescued the MYOD−/− cell phenotype but did not significantly stimulate the induction of MYOGENIN or MYH3 by MUNC. (A to C) qRT-PCR analysis of MYOD−/− cells with or without doxycycline-mediated overexpression of exogenous MYOD in proliferating (GM) and differentiating (DM1, DM3, and DM5) cells as indicated on the x axes. Levels of expression were measured for MYOD (A), MYOGENIN (B), and MYH3 (C) mRNAs and normalized to GAPDH and are shown relative to MYOD−/− cells without overexpressed MYOD in GM. (D) Western blot showing exogenous MyoD and myogenin proteins induced in MYOD−/− cells when exogenous MyoD protein is induced by doxycycline. Tubulin was used as a loading control. (E to H) qRT-PCR analysis of MYOD−/− cells stably overexpressing MUNC, transiently overexpressing exogenous MYOD, and differentiated for 2 days. Levels of expression were measured for MYOD (E), MUNC (F), MYOGENIN (G), and MYH3 (H) mRNAs. The data were normalized to the GAPDH expression level and are shown relative to control cells. The values represent three biological replicates and are presented as means and SEM. Statistical significance was calculated using a Wilcoxon-Mann-Whitney test. *, P < 0.05. (I) Western blot analysis showing induction of exogenous MyoD protein in MYOD−/− cells when transiently transfected with MYOD. Tubulin was used as a loading control.
In order to see cooperation between MyoD and MUNC for the induction of MYOGENIN and MYH3 RNAs, MYOD−/− cells stably overexpressing MUNC were transiently transfected with a plasmid vector expressing MYOD (Fig. 6E and I). Relative to control cells, MYOGENIN RNA was induced 3-fold by MyoD alone, 4-fold by MUNC alone, and 6-fold by both MyoD and MUNC (Fig. 6G). MYH3 RNA was similarly induced 3-fold by MyoD alone, 4.5-fold by MUNC alone, and 6-fold by both MyoD and MUNC (Fig. 6H). Although MUNC plus MyoD genes induced more RNA than either gene alone, the differences did not reach statistical significance in DM. Interestingly, in GM, where the basal levels of MYOGENIN and MYH3 RNAs are lower, we saw statistically significant additive stimulation of the two RNAs upon coexpression of MUNC and MYOD (not shown), but the levels of MYOGENIN and MYH3 RNAs did not reach the levels seen during normal differentiation, and there was no morphological differentiation. Thus, transient expression of MyoD, expressed from heterologous sites, had a weak additive effect with MUNC to induce more MYOGENIN or MYH3 RNAs, but it was not statistically significant and was insufficient to promote any morphological differentiation in MYOD−/− cells.
MUNC overexpression regulates genes both in cooperation with MyoD and in the complete absence of MyoD.
MUNC induced MYOGENIN and MYH3 even in MYOD−/− cells, suggesting that it can act independently of MyoD. However, MUNC also induced MYOD, suggesting that the two genes could cooperate with each other in regulating gene expression. To determine how many genes are regulated by MUNC independently of MyoD and how many in cooperation with MyoD, we examined the global RNA changes produced by MUNC overexpression in WT cells and MYOD−/− cells after 3 days of differentiation (DM3) (Fig. 7A and C). The Venn diagram in Fig. 7A shows that 3,678 genes were induced by MUNC only in WT cells but not in MYOD−/− cells, suggesting that there is a large fraction of genes that are induced by MUNC only in the presence of MyoD. This could be either because MyoD stimulates these genes and MUNC increases MyoD protein expression or because there is cooperation between MUNC and MyoD (or a MyoD-induced factor) at these promoters. There were 35 genes similar to MYOGENIN and MYH3 that were induced by MUNC in the presence or absence of MyoD and 157 genes that were induced by MUNC in MYOD−/− cells but not in WT cells. These two groups clearly show that MUNC can regulate the expression of 192 genes independently of MyoD protein.
FIG 7.

MUNC overexpression regulates many cellular genes in the complete absence of MyoD protein. (A) Venn diagram representing overlap of genes that are upregulated upon MUNC overexpression in WT or MYOD−/− cells at DM3. The scatter plots show how MUNC overexpression regulates (log2 fold change in cells overexpressing MUNC relative to control cells transfected with the empty vector) the three classes of genes in WT cells and MYOD−/− cells. (B) The 157 genes upregulated by MUNC only in MYOD−/− cells were examined to see if they were induced or repressed by MyoD. The plots represent log2 fold changes of genes on DM3 in WT versus MYOD−/− cells. The red and green dots represent genes that were induced or repressed in WT cells (induced or repressed by the presence of MyoD protein); P < 0.05. (C) Same as panel A, except for genes downregulated upon MUNC overexpression in WT or MYOD−/− cells. (D) Same as panel B, except for 173 genes from panel C that were downregulated by MUNC only in MYOD−/− cells.
The scatter plots in Fig. 7A show how individual genes in each of these three groups behave upon MUNC overexpression in WT cells and in MYOD−/− cells. The 35 genes that were induced in both types of cells (upper-right plot), were less induced in the absence of MyoD. The 3,678 genes that were induced by MUNC exclusively in WT cells (lower-left plot) were mostly unaffected in the MYOD−/− cells (log2 fold change from 0.2 to −0.2), though there were a few that were repressed by MUNC in the absence of MyoD. Surprisingly, of the 157 genes that were induced by MUNC exclusively in the MYOD−/− cells (lower-right plot), a large number were repressed by MUNC in WT cells, suggesting that the presence of MyoD reverses the direction of change produced by MUNC.
The last observation raises the possibility that MyoD induced by MUNC overexpression is responsible for the repression of the 157 genes. If that is the case, all 157 genes would be expressed less in WT cells than in MYOD−/− cells even without overexpressing MUNC (Fig. 7B). Out of these 157 genes, expression of only 43 was lower in WT cells than in MYOD−/− cells (P < 0.05), suggesting that they were repressed by MyoD and so could be repressed by MUNC through the induction of MyoD. However, in the absence of MyoD, MUNC induces these genes, providing further support for the hypothesis that MUNC regulates many genes completely independently of the MyoD protein. The 45 genes at the left end of the plot in Fig. 7B were induced by the presence of MyoD, so their repression by MUNC in WT cells cannot be explained by postulating an indirect effect through the induction of MyoD by MUNC.
Turning to genes repressed by MUNC under differentiating conditions (Fig. 7C), we found 4,021 genes that were repressed by MUNC only in the presence of MyoD. MUNC either represses these genes indirectly through the induction of MyoD or cooperates with MyoD (or some MyoD-induced factor) at their promoters. We analyzed chromatin immunoprecipitation sequencing (ChIP-seq) data available for MyoD protein in C2C12 cells to determine whether the repressed genes had MyoD binding sites near their transcription start sites (12). Forty-seven percent of the repressed genes were closest to (nearest neighbors to) a MyoD binding site. Thus, at least 53% of the genes repressed by MUNC are repressed indirectly by cooperation with some factor present when MyoD is present, but not MyoD itself. Looked at another way, only 47% of the 4,021 genes are repressed by MyoD alone in MyoD-converted mouse embryonic fibroblasts (MEFs) and so could be repressed by MUNC through the induction of MyoD (26). However, all these genes do not contain MyoD ChIP sites and so may be repressed indirectly by factors induced by MyoD. The data suggest that many genes are repressed by overexpressed MUNC in MyoD+ cells in direct or indirect cooperation with MyoD.
Twenty-six genes were repressed by MUNC in the presence or absence of MyoD, and 173 genes were repressed by MUNC only in the absence of MyoD, again showing evidence of MUNC activity independent of MyoD protein. The scatter plot in Fig. 7C, lower right, suggests that the 173 genes repressed by MUNC in MYOD−/− cells include many genes that are paradoxically upregulated by MUNC in WT cells. Among these, the plot in Fig. 7D identifies 6 genes that are induced by the presence of MyoD and so might be induced by MUNC in WT cells through the induction of MyoD. However, in the absence of MyoD, MUNC independently acts on the same genes and represses them. Figure 7D also identifies 88 genes that are repressed by MyoD (in a comparison of WT and MYOD−/− cells). These genes are repressed by MUNC in the absence of MyoD (they are among the 173 genes in Fig. 7C), and yet overexpression of MUNC in WT MYOD+ cells did not lead to their repression (Fig. 7C, lower right scatter plot), suggesting that MyoD and MUNC do not act additively on these promoters.
Collectively, these results suggest that MUNC and MyoD cooperate to regulate thousands of genes but that there are a few hundred genes that are regulated by MUNC in the complete absence of MyoD protein, consistent with our hypothesis that MUNC is not merely an eRNA whose only role is to induce MYOD transcription. Additionally, we observed a group of genes that were regulated by MyoD and MUNC in opposite directions, which also suggests independence of action. Finally, this is the first evidence that overexpressed MUNC can also repress thousands of cellular genes.
Confirmation of induction of genes by MUNC in MYOD−/− cells.
We selected 4 of the 35 genes besides MYOGENIN and MYH3 that are induced by MUNC in WT and MYOD−/− C2C12 cells (Fig. 7A) to confirm the induction by quantitative reverse transcription (qRT)-PCR in WT cells (Fig. 8A) and in MYOD−/− cells (Fig. 8B). We focused on genes whose products are functionally and structurally connected to skeletal muscle function: Tmem8c, a gene coding for Myomaker, a protein essential for fusion of embryonic and adult myoblasts; Mylpf, a gene coding for the regulatory light chain of striated muscle (27); Ablim3, encoding a protein that binds strongly to F-actin, suggesting its role as a scaffold for actin cytoskeleton signaling (28); and Tnnc1, a gene coding for troponin C, a part of the troponin complex, a structural complex responsible for muscle contraction (29). All four genes were induced by MUNC in WT and MYOD−/− C2C12 cells.
FIG 8.

Other myogenic genes and proteins are induced by MUNC in MYOD−/− cells. (A and B) qRT-PCR confirmation of genes upregulated upon MUNC overexpression. Shown is analysis of WT cells (A) and MYOD−/− cells (B) under differentiating conditions. The data were normalized to the GAPDH expression level and are shown relative to control cells (EV). The values represent three biological replicates and are presented as means and SEM. Statistical significance was calculated using the Wilcoxon-Mann-Whitney test. *, P < 0.05. (C) Western blots of protein products of genes analyzed in panels A and B.
We next checked the levels of protein products from Mylpf, Ablim3, and Tnnc1 (Fig. 8C). Tmem8c was not studied because there are no suitable commercial antibodies (Abs) available for immunoblotting. The MYOD−/− cells in DM expressed low levels of myosin light chain (Mylpf product) and troponin C1 (Tnnc1 product), allowing us to detect induction of these proteins when MUNC was overexpressed and induced the corresponding RNAs. We suggest that the levels of these proteins are regulated posttranscriptionally so that further protein induction is not seen when the protein levels are already high (as in WT cells with empty vector), even though the RNAs are induced by MUNC in WT cells.
MUNC regulates muscle-related genes in MYOD−/− cells.
We first tested the reproducibility of the gene expression changes seen with MUNC overexpression independently of MyoD. Hierarchical clustering of the differentially expressed genes in MYOD−/− C2C12 cells in both GM and DM3 showed that the pattern of gene expression changes was preserved in two independent experiments (Fig. 9A). Gene ontology (GO) terms that were enriched among the genes regulated by MUNC in DM3 in the MYOD−/− cells indicated that many of them are associated with skeletal muscle development and muscle structure (Fig. 9B). Fewer genes involved in skeletal muscle development and structure are regulated by MUNC in GM in MYOD−/− cells (GO term enrichment analysis for GM not shown). Therefore, DM likely induces factors independent of MyoD that cooperate with MUNC to regulate many myogenic genes.
FIG 9.

MUNC globally regulates many muscle-related genes in MYOD−/− cells. (A) Heat maps showing clustering of samples based on differentially regulated genes upon MUNC overexpression under proliferating conditions (GM) (left) and under differentiating conditions (DM) (right) in MYOD−/− cells. There were two biological replicates for each condition. Bootstrap values based on 1,000 repetitions are shown near the corresponding branches. (B) Top 30 significant gene ontology terms enriched in differentially expressed genes in DM upon MUNC overexpression in MYOD−/− cells. The arrowheads indicate gene terms related to skeletal muscle development and regeneration. (C) Enrichment plot from GSEA showing that the gene set involved in muscle contraction is enriched among differentially regulated genes upon MUNC overexpression in MYOD−/− cells in DM (P < 0.01). The table lists the top 10 genes contributing to enrichment scores for muscle contraction GO terms. (D) Venn diagrams representing overlap between differentially expressed genes upon differentiation of control cells (EV DM/EV GM) versus differentially expressed genes upon MUNC overexpression under proliferating conditions (MUNC GM/EV GM) in WT cells.
To determine the most significant molecular pathway regulated by MUNC in the absence of MyoD, we performed a gene set enrichment analysis (GSEA) on the genes differentially regulated upon MUNC overexpression in MYOD−/− cells in DM3. The plot in Fig. 9C shows significant enrichment of genes involved in muscle contraction among the genes induced by MUNC. The table below the plot lists the top 10 genes contributing to the enrichment score for muscle contraction GO terms, which are mainly muscle structure protein-coding genes.
As discussed above, the MyoD-independent activity of MUNC is more myogenic in DM than in GM, but we wanted to test whether the global change in gene expression induced by MUNC in WT C2C12 cells in GM is similar to that seen when the same cells undergo differentiation in DM. A total of 1,982 genes were induced and 1,733 genes were repressed by MUNC in WT cells growing in GM. When these genes were compared with the genes that were induced or repressed upon differentiation of WT C2C12 cells, a highly significant number of genes were found to overlap (Fig. 9D). This result suggests that MUNC overexpression alone in GM is able to push C2C12 cells in the direction of myogenic differentiation, although of course, MUNC overexpression alone is not as potent as the differentiation induced by moving cells from GM to DM.
DISCUSSION
The first question this paper answers is whether MUNC is an lncRNA that has functions independent of acting as an eRNA for MYOD (Fig. 10). Recent reports suggest that long noncoding RNAs derived from enhancer loci directly regulate the expression level of neighboring genes by a cis-acting mechanism (2). p53-bound enhancer regions produce eRNAs that regulate the transcription of adjacent genes, as shown by reporter assays and RNA Pol II ChIP assay (30). Additional examples are activating noncoding RNAs (ncRNAs), ncRNA-a3 and ncRNA-a7, whose depletion decreases RNA Pol II abundance at adjacent genes, as well as the recruitment of Mediator to the adjoining promoter (46). Estrogen receptor alpha (ERα)-inducible enhancer RNAs are functionally important for the expression of their target genes and are crucial for proper chromatin looping between enhancer loci and target gene bodies, which facilitates interactions between chromatin modifiers and transcription machinery (31). It was suggested that MUNC, coded by DRR genomic sequence, acts primarily as an enhancer RNA (12), inducing transcription of MYOD, but also induced MYOGENIN in trans (perhaps through the induction of MYOD). We now present data showing that MUNC positively regulates different myogenic genes, not only MYOD, and that it has many target genes that are regulated by MUNC overexpression in the complete absence of MyoD protein. The fact that specific sequence and structural elements of MUNC are necessary for the induction of MYOD, MYOGENIN, or MYH3 argues that the mere act of transcription or splicing of MUNC is not sufficient for its activity, as has been suggested for eRNAs (2). In addition, the structure-function studies show that even in WT cells, different parts of MUNC stimulate MYOD, MYOGENIN, and MYH3 RNAs to different extents that are not correlated with each other, something that would have been expected if all of MUNC's actions were through the induction of MYOD RNA and protein. These results suggest that MUNC is both a classical eRNA that induces transcription of the adjoining MYOD RNA and also a trans-acting lncRNA that has actions independent of MYOD induction.
FIG 10.
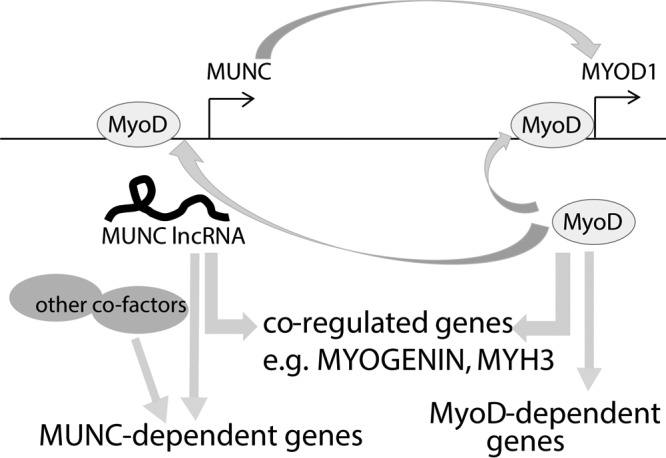
Schematic showing that MUNC and MYOD positively regulate each other and coregulate many genes but also regulate many genes independently of each other.
This result raises the possibility that there are other eRNAs that also act as lncRNAs. So far, reports suggest that eRNAs are not spliced, that transcription from the enhancer region is bidirectional, and that transcriptionally active enhancers are tagged with H3K4me1 rather than H3K4me3 marks. Enhancer RNAs are also usually much shorter than lncRNAs (32). We know from this report and our previous study (13) that MUNC is spliced, that the predominant stable transcript at the DRR locus is in the direction of MUNC, and that the DRR genomic locus during muscle differentiation acquires H3K4me3 marks. We hypothesize that eRNAs with similar features may have dual actions as an eRNA (enhancing the transcription of the adjoining gene) and as an lncRNA, which executes functions independent of its nearby neighbor.
The next question is whether MUNC lncRNA acts through the expression of an encoded micropeptide. There are growing reports that some lncRNAs code for functional micropeptides of even 30 amino acids. The most recent examples are micropeptides described by Olson and colleagues, which by interaction with SERCA regulate calcium signaling in muscle (33, 34) and nonmuscle (35) cells. Additionally, it was shown that one genomic locus may produce both a functional micropeptide, MLN, and a functional lncRNA, linc-RAM, working independently of each other (22). Spliced MUNC transcript could code for three such micropeptides unrelated in sequence to each other (underlined in red in Fig. 1A). However, the structure-function studies on MUNC rule out the possibility that MUNC's lncRNA-like function is due to any of the three putative micro-open reading frames (ORFs) in MUNC and suggest instead that the sequence and the folding of the RNA fragments are important for their function.
Both MUNC and MyoD are promyogenic factors, raising the question of whether they are additive with each other and whether they ever act in opposite directions. Our results suggest that, indeed, MUNC and MyoD cooperate to regulate many genes. However, there is a clear subset of genes that are regulated by MUNC in the complete absence of MyoD protein. Additionally, we observed a group of genes that are regulated by MyoD and MUNC in opposite directions, which suggests that the two factors may work in some pathways as antagonists.
The lack of MUNC or MyoD disables differentiation in vitro. The weak induction of some myogenic transcripts, like MYOGENIN and MYH3, when MYOD−/− cells are moved to DM (Fig. 2 and 4) is very slight relative to what is seen with WT cells. Overexpression of MUNC in MYOD−/− cells in DM induces the MYOGENIN RNA 4- to 10-fold (Fig. 5B and 6G), while overexpression of MyoD in the same cells induces MYOGENIN RNA 50,000-fold (Fig. 6B, DM3), suggesting that overexpressed MUNC cannot completely compensate for the absence of MyoD. Conversely, overexpression of MyoD in MUNC−/− cells stimulated MYOGENIN and MYH3 RNAs and proteins quite effectively (Fig. 3H to I). These results suggest that at loci like MYOGENIN, MUNC can partly compensate for lack of MyoD and vice versa, consistent with independent modes of action of MUNC and MyoD.
When MUNC was expressed stably and MyoD was expressed transiently together in the MYOD−/− cells, there was weak additive induction of MYOGENIN or MYH3 RNA in DM (and more so in GM). This was insufficient to allow differentiation of the cells. Even when MyoD protein was expressed at a high level in MYOD−/− cells (Fig. 6A to D), we saw induction of MYOGENIN RNA and protein, but not enough to permit differentiation.
In our previous report (13), overexpression of MUNC induced the expression of three genes, MYOD, MYOGENIN, and MYH3, so we focused on MUNC as a positive factor for gene expression. The genomewide analysis of genes regulated by MUNC in WT cells and in MYOD−/− cells presents a more complicated picture where in both types of cells MUNC induces and represses a large number of genes. MyoD, similarly, was initially thought to be a transcriptional factor that positively regulated expression of its target genes. However, it has since been recognized that MyoD also plays a role as a repressor of transcription, in cooperation with histone deacetylase 1 (HDAC1). For example, in proliferating myoblasts, MyoD binds to the promoter region of MYOGENIN to recruit HDAC1 and to suppress transcription (36). After serum withdrawal, MyoD changes its interaction partners to P/CAF and activates transcription of MYOGENIN (36). Another study showed that MyoD can repress c-Jun-mediated activation of genes linked to an AP-1 site in C2 cells (37). Thus, MyoD may repress specific gene promoters and MUNC may cooperate with such repression. It has also been proposed that MyoD can interact with chromatin-looping proteins, such as CTCF, to disrupt repressive loops, thus inducing transcription from specific genomic regions (38). Thus, there are different, independent mechanisms by which MyoD regulates its targets. Similarly, we propose that MUNC interacts with different cellular factors to induce or repress different targets and that the induction and repression functions are sometimes MyoD dependent and sometimes not.
Although one important conclusion of this paper is that MUNC can act independently of MyoD and sometimes in the opposite direction to MyoD, it is clear that there are many functional interactions between the two promyogenic factors. For example, MyoD promotes the transcription of MUNC (as evidenced by the decrease of MUNC in the MYOD−/− cells), and MUNC promotes the expression of MyoD. In addition, there are many genes that are regulated in the same direction by MUNC (in the presence or absence of MyoD) and by MyoD. Our future goal is to describe how MUNC and MyoD cooperate on the genes that they both induce or repress. Although we have failed to detect any direct physical interaction between MyoD and MUNC, we cannot yet rule out this possibility. Transient and weak interactions between MyoD and MUNC may be functionally important but difficult to show. In addition, MyoD interacts with numerous proteins to build whole complexes that regulate the expression of target genes, and MUNC may interact with and activate another protein from such a complex or may function as a scaffold, helping to maintain stability of interaction between transcriptional factors and chromatin remodelers.
A related goal is to describe how MUNC acts on many genes independently of MyoD (Fig. 10). We have to identify cellular proteins that interact with MUNC independently of MyoD. The MUNC-overexpressing MYOD−/− cells will be very important for such a search. As a nuclear transcript, MUNC may interact with chromatin modifiers, transcription factors, or repressors on the chromatin. Thus, we plan to examine whether we can identify specific genomic sites at which MUNC associates with the chromatin or alters the chromatin landscape without stable association with the chromatin.
An important possibility is that the MyoD-related proteins Myf5, myogenin, and MRF4 act as cofactors for MUNC. MyoD and Myf5 play redundant roles in skeletal muscle differentiation: mice with deletion of either gene remain alive and healthy, but double-knockout pups die shortly after birth (39). Studies on double-knockout mice showed that each of the factors is essential for proper development of different parts of the musculature (40). Expression of these transcription factors during development is temporally regulated: MYF5 transcript is evident at 7.5 days postcoitum (dpc) MYOGENIN at 8.5 dpc, and MYOD at 9.5 dpc (41). In our previous study, MUNC was induced between days 11 and 15 of embryonic development, which suggested a role at later points in development, when Myf5, myogenin, and MyoD are all present. We also showed that MUNC is induced during differentiation of myoblasts, with its abundance being very low in undifferentiated C2C12 cells in proliferating medium (13). One argument against Myf5 being a cofactor for MUNC is that Myf5 does not induce myogenic gene transcription as robustly as MyoD (26) or MUNC (Fig. 7A). MUNC could also work with myogenin, another transcription factor, which is strongly induced during differentiation and whose expression is itself MUNC dependent. Identifying the cofactors that assist MUNC activity will be another important area of future research.
MATERIALS AND METHODS
Cell culture.
The C2C12 mouse myoblast cell line was supplied by the American Type Culture Collection (ATCC). C2C12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM)–high-glucose medium (GE Healthcare Life Sciences Co.) with 10% fetal bovine serum (FBS) (Life Technologies Co.); when differentiating, serum was switched to 2% horse serum (GE Healthcare Life Sciences Co.)
Knockout strategy.
CRISPR protocol with minor changes was followed to achieve deletion of a part of the MYOD gene (25). Briefly, single guide RNAs (sgRNAs) were designed using the CRISPR DESIGN tool (http://crispr.mit.edu/). Cells were cotransfected with vectors coding for Cas9 (the vectors were obtained from Addgene [no. 41815]), and the sgRNAs were cloned into gRNA_GFP-T2 (a vector obtained from Addgene [no. 41820]) and a spiking vector coding for a resistance gene. After 24 to 48 h, the cells were treated with puromycin (concentration = 2 μg/ml), and resistant cells were seeded to 96-well plates using a single-cell dilution method. Growing clones were examined for the desired deletion by PCR on extracted genomic DNA (Quick Extract DNA extraction solution; Epicentre Co.), and candidates with complete loss of the WT PCR product (homozygous deletion) were screened by immunoblotting for MyoD protein.
Stable overexpression of MUNC in C2C12 cells.
PCR-amplified sequence of genomic MUNC (PCR using C2C12 genomic DNA) or of spliced MUNC (PCR using cDNA from DM3 C2C12 cells) was cloned into the pLPCX vector by ligation. The constructs were linearized and introduced into the C2C12 cells (XtremeGene transfection reagent; Roche). After 24 h, pools of stably transfected cells were selected with puromycin (concentration = 2 μg/ml). Vectors coding for mutant forms of MUNC were generated similarly, using genomic DNA or DM3 cDNA as necessary.
To generate reagents for MUNC overexpression in MYOD−/− cells, the insert was cloned into the pLHCX vector by ligation. The construct was linearized and introduced into the cells (XtremeGene transfection reagent; Roche). After 48 h, pools of stably transfected cells were selected with hygromycin (concentration = 300 μg/ml).
Estimation of the proportions of spliced and unspliced MUNC in C2C12 cells transfected with genomic sequence of MUNC.
To estimate the proportions of spliced and unspliced MUNC, we performed RNA-Seq from C2C12 cells stably transfected with genomic MUNC. We counted the reads overlapping three 30-base junctions made of 15 bases from each side of the exon 1-intron, exon 1-exon 2, and intron-exon 2 boundaries. The exon 1-exon 2 junction gave us an estimate of spliced MUNC, and the mean count of exon 1-intron and intron-exon 2 junctions gave an estimate for unspliced MUNC. The ratios of unspliced to spliced MUNC were 120:1 in WT C2C12 cells and 2:1 in MYOD−/− C2C12 cells.
Prediction of RNA structures.
MUNC fragment structures were predicted using the Forna prediction tool (21).
Transient overexpression of MYOD in C2C12 cells.
Cells were seeded on 6-well plates and after 12 h were transfected with vector coding for MYOD. The medium was changed 12 h posttransfection to differentiation medium, and cells were harvested 2 days later.
Stable overexpression of inducible MYOD in WT, MUNC−/−, and MYOD−/− C2C12 cells.
PCR-amplified sequence of the MYOD ORF (PCR using C2C12 cDNA) was cloned by ligation into the pCW-Cas9 (Addgene; no. 50661) vector upon Cas9 removal by enzymatic digestion with BamHI and NheI. The vector was packed in the virus using psPAX2 (Addgene; no. 12260) and pMD2.G (Addgene; no. 12259) in 293T cells. WT, MUNC−/−, and MYOD−/− C2C12 cells were transduced with the filtered supernatant containing virus. After 24 h, the cells were treated with puromycin (C = 2 μg/ml).
MYOD expression was induced in MUNC−/− and MYOD−/− C2C12 cells before differentiation using doxycycline (concentration = 1 μg/ml). The samples were collected under proliferation (GM) and differentiation (DM1, DM3, and DM5) conditions.
RNA analysis by qRT-PCR.
RNA was isolated by TRIzol extraction or using an RNeasy minikit (Qiagen), and RNA samples were treated with RQ1 RNase-free DNase (Promega Co.) to eliminate potential DNA contamination of samples. cDNA synthesis was performed using a Superscript III RT cDNA synthesis kit (Life Technologies Co.) with random-hexamer and oligo(dT) priming. After cDNA synthesis, quantitative PCR (qPCR) was performed with Applied Biosystems 7500 real-time PCR systems using Power SYBR green master mix (ThermoFisher Scientific) or a SensiFast SYBR Hi-Rox kit (Bioline). All the primers used in this study are listed in Table 1.
TABLE 1.
Primers used in this study

Western blotting.
Cells were lysed in IPH buffer (50 mM Tris-Cl, 0.5% NP-40, 50 mM EDTA), run on a 10% polyacrylamide SDS-PAGE gel, and transferred to nitrocellulose membranes. The membranes were blocked for 30 min in 5% milk containing phosphate-buffered saline with Tween 20 (PBS-T) and incubated overnight with primary antibody in 1% milk. Secondary-antibody incubation was carried out for 1 h after washing and at 1:4,000 dilution before washing and incubation with Millipore Immobilon horseradish peroxidase (HRP) substrate. Antibodies were used as follows: MyoD1 (sc-12732; Santa Cruz Co.), MHC (MF-20; Developmental Studies Hybridoma Bank, University of Iowa), Ablim3 (sc-398575; Santa Cruz Co.), Mylpf (16052-1-AP; Proteintech Co.), and Tnnc1 (13504-1-AP; Proteintech Co.).
Southern blotting.
Ten micrograms of genomic DNA was digested with a restriction enzyme and electrophoresed in a 0.8% agarose gel. The DNA was transferred to a Nitran SuperCharge membrane (Schleicher & Schuell) using alkaline denaturing conditions. The membrane was hybridized with a DNA probe labeled with a random-primer DNA-labeling kit (TaKaRa) using [32P]dCTP. The probe was amplified from genomic DNA with MUNC probe forward and MUNC probe reverse primers (listed in Table 1).
Immunofluorescence assay.
Cells were plated on glass coverslips and collected in growth medium or after 3 days of differentiation. The coverslips were fixed with 4% formaldehyde in PBS for 10 min, permeabilized in 0.5% Triton X-100 in PBS, and blocked in 5% goat serum. The coverslips were incubated at room temperature with primary antibody for 1 h and Alexa Fluor 488- or 549-conjugated secondary antibody for 1 h, with three PBS washes following each antibody incubation. The coverslips were then mounted with Vectashield mounting solution (Vector Laboratories). The antibodies used were anti-MyoD C-20 antibody (Santa Cruz Laboratories) and anti-myosin heavy chain M4276 antibody (Sigma). The antibodies were diluted 1:200 in 5% goat serum containing PBS.
Microscopy.
Images were captured using a Nikon Microphot SA upright microscope equipped with a Nikon NFX35 camera using SPOT imaging software (Diagnostic Instruments Inc.) and a Nikon PlanApo 60× oil objective lens. Fluorescence images were acquired on the same day using the same exposure times, gamma, and gain between samples. Images were enhanced for brightness and contrast to the same extent within Adobe Photoshop software.
RNA-Seq library preparation.
RNA samples were isolated from proliferating or differentiating cells using an RNeasy minikit (Qiagen Co.). One microgram of RNA was enriched for poly(A)-tailed mRNA molecules using a NEBNext Poly(A) mRNA Magnetic Isolation Module, and RNA-Seq libraries were made using NEBNext Ultra Directional RNA library prep kit for Illumina (NEB Co.) according to the manufacturer's protocol. Pooled libraries were sequenced using a paired-end protocol on the Illumina platform, using a NextSeq 500 instrument in the Biomolecular Analysis Facility, University of Virginia School of Medicine.
RNA-Seq analysis.
We obtained ≥40 million paired-end 75-bp-long reads for WT and MYOD knockout (MYOD−/−) conditions. The WT control cell line, the WT cell line overexpressing MUNC, the MYOD−/− cell line, and the MYOD−/− cell line overexpressing MUNC were grown in GM conditions and harvested at ~80% confluence. To achieve differentiated samples (DM) at ~90% confluence of cells, medium was changed to differentiation medium, and cells were harvested after 3 days. Paired-end reads were obtained from the two biological replicates with EV and MUNC overexpression in both GM and DM in WT and MYOD−/− C2C12 cell lines. Transcripts for mm10 RefSeq genes were downloaded from the UCSC table browser (http://genome.ucsc.edu). We used the default settings of Kallisto (42) to build an index for the downloaded 35,818 transcript sequences and then quantified the abundance of each transcript from the paired-end reads (42). We used the DESeq2 package in R for differential expression analysis of the quantified data obtained from Kallisto (43). A P value (obtained by DESeq2) cutoff of 0.05 was used to define differentially expressed genes. Gene Trail (44) and GSEA (45) were used for functional gene ontology term enrichment analysis and gene set enrichment analysis, respectively.
Accession number(s).
All RNA-Seq library data files are available under GEO accession number GSE99258.
ACKNOWLEDGMENTS
We thank all the members of the Dutta laboratory for helpful discussions, especially Etsuko Shibata for technical guidance regarding the CRISPR-Cas9 method.
This work was supported by a grant from the NIH (NIAMS), R01 AR067712, to A.D. M.A.C. was partly supported by a Wagner Fellowship from the University of Virginia during the study. M.K. was partly supported by a DOD award (PC151085).
M.A.C. performed structure-function studies. R.K.P. analyzed the structure predictions. M.A.C. designed CRISPR-Cas9 for MYOD and MUNC KO and obtained the knockout cells with help from R.K.P. R.K.P. designed CRISPR-Cas9 for miR-1a-1 KO and obtained the knockout cells with help from E.S. M.A.C. performed immunofluorescence experiments. M.A.C., R.K.P., and E.S. performed differentiation assays and qPCR analyses. M.A.C. and R.K.P. performed Western blot analyses. Y.S. performed Southern blotting experiments. M.A.C. designed MUNC stable-overexpression studies. R.K.P. designed MYOD stable-overexpression studies. M.A.C. performed the RNA-Seq experiment with help from R.K.P. M.K. performed analysis related to Fig. 7 and 9. M.A.C. confirmed RNA-Seq results. M.A.C. wrote the manuscript with help from A.D., M.K., and R.K.P.
REFERENCES
- 1.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. 2010. Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engreitz AJM, Haines JE, Munson G, Chen J, Elizabeth M, Kane M, Mcdonel PE, Guttman M, Lander ES. 2016. Neighborhood regulation by lncRNA promoters, transcription, and splicing. Nature 539:1–15. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun T, Gautel M. 2011. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol 12:349–361. doi: 10.1038/nrm3118. [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama S, Asahara H. 2011. The myogenic transcriptional network. Cell Mol Life Sci 68:1843–1849. doi: 10.1007/s00018-011-0629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berkes CA, Tapscott SJ. 2005. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16:585–596. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Tapscott SJ, Lassar AB, Weintraub H. 1992. A novel myoblast enhancer element mediates MyoD transcription. Mol Cell Biol 12:4994–5003. doi: 10.1128/MCB.12.11.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asakura A, Lyons GE, Tapscott SJ. 1995. The regulation of MyoD gene-expression; conserved elements mediate expression in embryonic axial muscle. Dev Biol 171:386–398. doi: 10.1006/dbio.1995.1290. [DOI] [PubMed] [Google Scholar]
- 8.Goldhamer DJ, Faerman A, Shani M, Emerson CP. 1992. Regulatory elements that control the lineage-specific expression of myoD. Science 256:538–542. doi: 10.1126/science.1315077. [DOI] [PubMed] [Google Scholar]
- 9.Chen JCJ, Ramachandran R, Goldhamer DJ. 2002. Essential and redundant functions of the MyoD distal regulatory region revealed by targeted mutagenesis. Dev Biol 245:213–223. doi: 10.1006/dbio.2002.0638. [DOI] [PubMed] [Google Scholar]
- 10.L'honore A, Lamb NJ, Vandromme M, Turowski P, Carnac G, Fernandez A. 2003. MyoD distal regulatory region contains an SRF binding CArG element required for MyoD expression in skeletal myoblasts and during muscle regeneration. Mol Biol Cell 14:2151–2162. doi: 10.1091/mbc.e02-07-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JCJ, Love CM, Goldhamer DJ. 2001. Two upstream enhancers collaborate to regulate the spatial patterning and timing of MyoD transcription during mouse development. Dev Dyn 221:274–288. doi: 10.1002/dvdy.1138. [DOI] [PubMed] [Google Scholar]
- 12.Mousavi K, Zare H, Dell'Orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, Hager G, Sartorelli V. 2013. ERNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 51:606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller AC, Cichewicz MA, Dey BK, Layer R, Reon BJ, Gagan JR, Dutta A. 2015. MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol Cell Biol 35:498–513. doi: 10.1128/MCB.01079-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballarino M, Morlando M, Fatica A, Bozzoni I. 2016. Non-coding RNAs in muscle differentiation and musculoskeletal disease. J Clin Invest 126:2021–2030. doi: 10.1172/JCI84419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume D, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake J, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani T, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons P, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple C, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y, FANTOM Consortium, RIKEN Genome Exploration Research Group Phase I and II Team. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 16.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neguembor MV, Jothi M, Gabellini D. 2014. Long noncoding RNAs, emerging players in muscle differentiation and disease. Skelet Muscle 4:8. doi: 10.1186/2044-5040-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colley SM, Leedman PJ. 2011. Steroid receptor RNA activator—a nuclear receptor coregulator with multiple partners: insights and challenges. Biochimie 93:1966–1972. doi: 10.1016/j.biochi.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Zhao Y, Bao X, Zhu X, Kwok YK-Y, Sun K, Chen X, Huang Y, Jauch R, Esteban MA, Sun H, Wang H. 2015. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res 25:335–350. doi: 10.1038/cr.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D. 2012. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerpedjiev P, Hammer S, Hofacker IL. 2015. Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 31:3377–3379. doi: 10.1093/bioinformatics/btv372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu X, Zhang Y, Li T, Ma Z, Jia H, Chen Q, Zhao Y, Zhai L, Zhong R, Li C, Zou X, Meng J, Chen AK, Puri PL, Chen M, Zhu D. 2017. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat Commun 8:14016. doi: 10.1038/ncomms14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. 1996. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev 10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- 24.Rajan S, Dang HCP, Djambazian H, Zuzan H, Fedyshyn Y, Ketela T, Moffat J, Hudson TJ, Sladek R. 2012. Analysis of early C2C12 myogenesis identifies stably and differentially expressed transcriptional regulators whose knock-down inhibits myoblast differentiation. Physiol Genomics 44:183–197. doi: 10.1152/physiolgenomics.00093.2011. [DOI] [PubMed] [Google Scholar]
- 25.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conerly ML, Yao Z, Zhong JW, Groudine M, Tapscott SJ. 2016. Distinct activities of Myf5 and MyoD indicate separate roles in skeletal muscle lineage specification and differentiation. Dev Cell 36:375–385. doi: 10.1016/j.devcel.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Szczesna-Cordary D, Craig R, Diaz-Perez Z, Guzman G, Miller T, Potter JD. 2007. Fast skeletal muscle regulatory light chain is required for fast and slow skeletal muscle development. FASEB J 21:2205–2214. doi: 10.1096/fj.06-7538com. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda M, Yamashita JK, Tsukita S, Furuse M. 2010. AbLIM3 is a novel component of adherens junctions with actin-binding activity. Eur J Cell Biol 89:807–816. doi: 10.1016/j.ejcb.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Perry SV. 1985. Properties of the muscle proteins—a comparative approach. J Exp Biol 115:31–42. [DOI] [PubMed] [Google Scholar]
- 30.Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Vrielink JAFO, Elkon R, Melo SA, Léveillé N, Kalluri R, de Laat W, Agami R. 2013. ERNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 31.Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen Y, Merkurjev D, Zhang J, Ohgi K, Song X, Kim H, Glass CK, Rosenfeld MG. 2013. Functional importance of eRNAs for estrogen-dependent transcriptional activation events. Nature 498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim TK, Hemberg M, Gray JM. 2015. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb Perspect Biol 7:a018622. doi: 10.1101/cshperspect.a018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN. 2015. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160:595–606. doi: 10.1016/j.cell.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R, Olson EN. 2016. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351:271–275. doi: 10.1126/science.aad4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, Olson EN. 2016. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci Signal 9:ra119. doi: 10.1126/scisignal.aaj1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mal A, Harter ML. 2003. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Natl Acad Sci U S A 100:1735–1739. doi: 10.1073/pnas.0437843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, Verma IM. 1992. Functional antagonism between c-Jun and MyoD proteins: A direct physical association. Cell 68:507–519. doi: 10.1016/0092-8674(92)90187-H. [DOI] [PubMed] [Google Scholar]
- 38.Battistelli C, Busanello A, Maione R. 2014. Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J Cell Sci 127:3757–3767. doi: 10.1242/jcs.149427. [DOI] [PubMed] [Google Scholar]
- 39.Rudnicki MA, Schnegelsberg PNJ, Stead RH, Braun T, Arnold HH, Jaenisch R. 1993. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75:1351–1359. doi: 10.1016/0092-8674(93)90621-V. [DOI] [PubMed] [Google Scholar]
- 40.Kablar B, Krastel K, Ying C, Asakura A, Tapscott SJ, Rudnicki M. 1997. MyoD and Myf-5 differentially regulate the development of limb versus trunk skeletal muscle. Development 124:4729–4738. [DOI] [PubMed] [Google Scholar]
- 41.Hannon K, Smith CK, Bales KR, Santerre RF. 1992. Temporal and quantitative analysis of myogenic regulatory and growth factor gene expression in the developing mouse embryo. Dev Biol 151:137–144. doi: 10.1016/0012-1606(92)90221-2. [DOI] [PubMed] [Google Scholar]
- 42.Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller A, Backes C, Al-Awadhi M, Gerasch A, Küntzer J, Kohlbacher O, Kaufmann M, Lenhof H-P. 2008. GeneTrailExpress: a web-based pipeline for the statistical evaluation of microarray experiments. BMC Bioinformatics 9:552. doi: 10.1186/1471-2105-9-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette M, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai F, Orom U, Cesaroni M, Beringer M, Taatjes DJ, Blobel G, Shiekhattar R. 2013. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]