

LONP1, an AAA+ mitochondrial protease, is implicated in protein quality control, but its precise role in this process remains poorly understood. In this study, we have investigated the role of human LONP1 in mitochondrial proteostasis and gene expression.
KEYWORDS: ISR, LONP1, mitochondria, mitochondrial gene expression, proteases, proteostasis, mtDNA
ABSTRACT
LONP1, an AAA+ mitochondrial protease, is implicated in protein quality control, but its precise role in this process remains poorly understood. In this study, we have investigated the role of human LONP1 in mitochondrial proteostasis and gene expression. Depletion of LONP1 resulted in partial loss of mitochondrial DNA (mtDNA) and a complete suppression of mitochondrial translation associated with impaired ribosome biogenesis. The levels of a distinct subset of mitochondrial matrix proteins (SSBP1, MTERFD3, FASTKD2, and CLPX) increased in the presence of a catalytically dead form of LONP1, suggesting that they are bona fide LONP1 substrates. Unexpectedly, the unprocessed forms of the same proteins also accumulated in an insoluble protein fraction. This subset of unprocessed matrix proteins (but not their mature forms) accumulated following depletion of the mitochondrial processing peptidase MPP, though all other MPP substrates investigated were processed normally. Prolonged depletion of LONP1 produced massive matrix protein aggregates, robustly activated the integrated stress response (ISR) pathway, and resulted in stabilization of PINK1, a mitophagy marker. These results demonstrate that LONP1 and MPPαβ are together required for the maturation of a subset of LONP1 client proteins and that LONP1 activity is essential for the maintenance of mitochondrial proteostasis and gene expression.
INTRODUCTION
The metazoan AAA+ proteases LONP1, CLPXP, and AFG3L2 are responsible for the protein quality control of the mitochondrial matrix by degrading misfolded or oxidized polypeptides that would otherwise accumulate with deleterious consequences for mitochondrial physiology (1). Mammalian LONP1 homo-oligomerizes to form a soluble hexameric ring in which the binding and degradation of substrates take place in an ATP-dependent manner (2–4). In addition to this proteolytic activity, it has been implicated in the maintenance of mitochondrial DNA (mtDNA).
In bacteria, LONP1 binds single- and double-stranded DNA (5) and plays a role in the degradation of proteins involved in DNA methylation and transcription (6, 7). Deletion of Pim1, the yeast homolog of LONP1, generates major mtDNA deletions, failure to process mitochondrially encoded mRNAs, and the accumulation of electrodense inclusion bodies in the mitochondrial matrix (8–10). In Drosophila melanogaster cells, LONP1 degrades mitochondrial transcription factor A (TFAM), a key player in the initiation of mitochondrial transcription and the major protein component of the mitochondrial nucleoid (11). In mammals, LONP1 is reported to bind only the heavy strand of the mtDNA, preferentially to G-rich sequences, and to the displacement loop (D-loop) (12), and it plays a role in the degradation of phosphorylated TFAM (13).
The majority of nucleus-encoded mitochondrial proteins are synthesized with an N-terminal mitochondrial targeting sequence (MTS) that is proteolytically cleaved to the mature form by the mitochondrial processing peptidase (MPP), a heterodimeric enzyme composed of an alpha (MPPα) and a beta (MPPβ) subunit. The catalytic activity of mitochondrial proteases is also required for the processing of a limited number of nucleus-encoded mitochondrial proteins and in the activation and/or regulation of retrograde signaling pathways. For instance, AFG3L2 is responsible for the maturation of the mitochondrial ribosomal protein MRPL32, enabling assembly of the mitochondrial ribosome (1). In the nematode Caenorhabditis elegans, the accumulation of damaged proteins triggers the mitochondrial unfolded protein response (mtUPR), which is initiated in the mitochondrial matrix by the proteolytic subunit of CLPXP, CLPP, which recognizes and degrades misfolded polypeptides. This step is followed by the activation of the bZip transcription factor ATFS-1, which accumulates in the nucleus and directs transcriptional upregulation of genes coding for molecular chaperones (14). It is not yet clear which components of this pathway are conserved in mammals, although ATF5, which can rescue an ATFS-1 deletion, has been proposed to drive a mammalian mtUPR (15). Several studies have shown that LONP1-depleted cells do not elicit this pathway (16–18); however, LONP1 was shown to degrade the RNase P subunit MRPP3A when mitochondrial proteostasis is impaired, decreasing mitochondrial translation and thus lessening the protein folding overload (19). A recent study in which mitochondrial proteostasis was disrupted failed to find evidence for a canonical mtUPR but rather showed that the response to mitochondrial stress was mediated through ATF4, which directed the expression of cytoprotective genes and a reprogramming of cellular metabolism, downstream of the integrated stress response (ISR) (17).
In this study, we investigated the role of LONP1 in the maintenance and expression of mtDNA and in mitochondrial protein homeostasis. We show that LONP1 is essential for the maintenance of mtDNA and ribosome biogenesis. We further identify a subset of mitochondrial proteins as LONP1 substrates and show that their maturation requires the activities of both LONP1 and the MPP. Loss of LONP1 activity resulted in massive accumulation of insoluble protein aggregates in the matrix, which triggers activation of the ISR.
RESULTS
Depletion of LONP1 impairs mtDNA expression.
To characterize the role of LONP1 in human mtDNA metabolism, we used small interfering RNA (siRNA) to transiently deplete it in immortalized human skin fibroblasts (LONP1-KD) and measured the effect on the levels of mtDNA and mtRNAs and on mitochondrial protein synthesis. The levels of mtDNA remained unchanged after 3 days of LONP1 depletion but were reduced by half after 6 days (Fig. 1A). There was a transient increase in the steady-state levels of all mitochondrial mRNAs tested (3 days), but after 6 days, all were substantially decreased (Fig. 1B). The synthesis of all 13 mtDNA-encoded polypeptides was decreased by more than 50% after 3 days of LONPI depletion, despite the lack of change in mtDNA and the increased levels of mitochondrial mRNAs. However, continued depletion of LONP1 completely abolished mitochondrial protein synthesis (Fig. 1C), a phenotype resembling that in [rho0] (no mtDNA) cell lines.
FIG 1.

Mitochondrial gene expression in LONP1 knockdown cell lines. (A) Southern blot analysis of control and LONP1 depletion. Specific probes were used for detection of the mitochondrial DNA and the cytosolic 18S rRNA. The percentages reflect the levels of mtDNA in both LONP1 knockdown cell lines after normalization with 18S rRNA. (B) Northern blot analysis of control and LONP1 knockdown for 3 and 6 days. Specific probes were hybridized for detection of the mRNA subunits of complex I (ND1 and ND5), complex IV (COX1 and COX2), complex III (cyt b), complex V (ATP6/8), and mitochondrial rRNA (12S and 16S). 18S cytosolic rRNA was used as loading control. (C) Mitochondrial translation experiments with control and LONP1-depleted cell lines (2, 3, and 6 days). Mitochondrially encoded polypeptides were pulse-labeled for 60 min with [35S]methionine and cysteine. The positions of the ND subunits of complex I (COX), subunits of complex IV (cyt b), subunit of complex III (ATP), and subunits of complex V are indicated on the right. (D) SDS-PAGE immunodetection of LONP1 and mitochondrial ribosomal subunits (MRPs) in control and LONP1-depleted cells (3 and 6 days). Supernatants from whole-cell extracts using n-dodecyl-β-d-maltoside (WCE-DDM) were used for this analysis. SDHA was used as a loading control. (E) Sucrose density gradients were done using mitochondria from control and LONP1-depleted cells (3 days), followed by immunodetection of LONP1 and MRPs. Immunoblots of MRPL44 and MRPS18B indicate the peaks for the small (28S) and large (39S) mitochondrial ribosomal subunits. “55S” indicates the mitochondrial monosome.
To investigate the molecular basis for the early protein synthesis defect associated with LONP1 depletion, we evaluated the assembly of the mitochondrial ribosomes. Immunoblot analysis showed a reduction in the steady-state levels of several mitochondrial ribosomal proteins (MRPs), including MRPL44, MRPS18B, and MRPL11 (Fig. 1D). Sucrose gradient sedimentation profiles showed a marked decrease in the fully assembled ribosome (55S) but no change in the sedimentation profile compared to the control (Fig. 1E). We conclude that LONP1 is required for the maintenance of mtDNA and for ribosome biogenesis.
MTERFD3 is a substrate of LONP1.
To search for LONP1-interacting proteins, we immunoprecipitated endogenous LONP1 and used mass spectrometry (MS) to identify coimmunoprecipitating proteins. This analysis revealed a complex network of interactors, including the mitochondrial proteases AFG3L2, HTRA2, CLPX, and YME1L1, the mitochondrial nucleoid components TFAM and SSBP1, several DNA/RNA-associated proteins, the entire MICOS complex, several OXPHOS subunits and assembly factors, 7 subunits of the TIM complex, and 11 mitochondrial ribosomal subunits of the large subunit (39S), including 5 that are part of the ribosome exit tunnel, MRPL17, MRPL22, MRPL23, MRPL39, and MRPL44 (see Fig. S1 in the supplemental material).
To identify putative LONP1 substrates, we generated a fibroblast line (LCD) that expressed a construct coding for a catalytically dead form of LONP1 containing an S855A substitution that disrupts the catalytic dyad of LONP1 responsible for protein degradation. This substitution should not interfere with substrate binding but would prevent subsequent proteolysis (20). Site-directed mutagenesis was used to produce two synonymous mutations in the LONP1-siRNA target sequence, allowing us to specifically deplete endogenous LONP1 in cells expressing the inactive LONP1 (Fig. 2A). Immunoblot analyses of whole-cell extracts from control, LCD, and LONP1-depleted cells (LONP1-KD-3D) and LCD depletion of LONP1 (LCD+LONP1-KD-3D) showed an increase in the steady-state level of LONP1 in the LCD cell line compared to the control but similar levels of expression after endogenous LONP1 was depleted (LCD+LONP1-KD-3D), confirming that we were able to express the catalytically dead LONP1 and effectively deplete endogenous LONP1 (Fig. 2B). We next examined the steady-state levels of MTERFD3, a protein involved in mitochondrial transcription, as a candidate substrate of LONP1 that might account for the biochemical phenotype observed in fibroblasts following LONP1 depletion. MTERFD3 codes for a soluble protein of 43 kDa containing a mitochondrial targeting sequence (MTS) that is cleaved after translocation into the mitochondrial matrix, generating a mature 39-kDa protein (21). A unique band of 39 kDa was detected in control and LCD cell lines; however, after depletion of wild-type LONP1, mature MTERF3 was increased and a second band of 43 kDa appeared (Fig. 2B). To test whether the solubility of MTERFD3 was affected by loss of LONP1 activity, we measured the steady-state level in the soluble fraction and found that it was reduced to nearly undetectable levels in the absence of LONP1 (Fig. 2B). These results show that loss of LONP1 affects the steady-state level, processing, and solubility of MTERFD3.
FIG 2.

Searching for LONP1 substrates. (A) The LCD cell line overexpresses a LONP1 construct containing two nonsynonymous substitutions that generates an S855A mutation in the P domain and two other synonymous mutations in the binding sequence of LONP1 siRNA that prevents its knockdown. (B) Immunoblot analysis of whole-cell extracts with sonication (WCE-sonication) of indicated proteins in control cells, LCD cells, cells in which LONP1 was knocked down for 3 days (LONP1-KD-3D), and LCD cells in which LONP1 was knocked down for 3 days (LCD+LONP1-KD-3D). SDHA is shown as a loading control. Supernatants from whole-cell extracts with n-dodecyl-β-d-maltoside (WCE-DDM) were used for SDS-PAGE immunodetection of the indicated proteins in control and LONP1 knockdown for 3 and 6 days. SDHA was used as a loading control.
LONP1 is necessary for the cleavage of the mitochondrial targeting sequences for a subset of mitochondrial proteins.
We next investigated the generality of the result obtained with MTERFD3 by determining the steady-state levels and solubility profiles of several mitochondrial proteins in control and LCD LONP1-depleted cells (LCD+LONP1-KD-3D) after alkaline carbonate extraction (Fig. 3A). We examined mitochondrial proteins covering a broad range of functions, for which we had reliable antibodies (Fig. 3A).
FIG 3.

Solubilization profile of mitochondrial proteins in LONP1-depleted cell lines. (A) Mitochondria from control and LCD cells in which LONP1 was depleted for 3 days (LCD+LONP1-KD-3D) were treated with alkaline carbonate (Na2CO3). Input (IN), supernatant (SN), and pellet (PE) fractions were analyzed by SDS-PAGE. (B) Whole-cell extracts were obtained by sonication and used for SDS-PAGE immunodetection of the indicated proteins in control and cell lines in which LONP1 was depleted for 1, 2, 3, and 4 days. Porin was used as a loading control.
The proteins could be classified into four different groups. Several, like AFG3L2 and TFAM, were agnostic to the loss of LONP1 activity. A few, including the methyltransferase TFB1M, which is responsible for posttranscriptional modification of the 12S rRNA (22), were reduced, without a change in solubility. Several others, including the RNA granule proteins GRSF1 and DHX30, showed an increase in the insoluble pellet fraction. MRPL11, a structural subunit of the large ribosomal subunit (39S), was detected in the control cell line roughly equally in the supernatant and pellet fractions but accumulated only in the pellet in LONP1-depleted cells. Finally, a fourth group contained proteins that all had an apparently unprocessed mitochondrial targeting sequence, including MTERFD3, FASTKD2, an RNA granule protein that interacts with the large 16S rRNA (23, 24), the protease chaperone CLPX, and the single-stranded DNA binding protein SSBP1.
To further characterize the processing impairment observed in SSBP1, MTERFD3, FASTKD2, and CLPX, we investigated the accumulation of these proteins over 1 to 4 days of LONP1 depletion (Fig. 3B). The loss of LONP1 correlated with increases in both the unprocessed and mature forms of these proteins, suggesting that LONP1 is necessary for both their maturation and degradation. CLPX oligomerizes with CLPP to form a mitochondrial matrix protease, and it is conceivable that its functional depletion could be the cause of the processing defect observed in LONP1-depleted cells; however, SSBP1 and MTERFD3 were correctly processed in CLPX-silenced cells (Fig. 4A).
FIG 4.

Analysis of LONP1 substrates. (A) Immunoblot analysis of whole-cell extracts obtained by sonication from control and CLPX-depleted cells (6 days). Immunoblots show the steady-state levels of the indicated proteins. Actin was used as a loading control. (B) Mitochondria from control and LONP1-depleted cells (3 days) were treated with increasing concentrations of proteinase K as indicated. MFN2, SCO1, and MRPL11 immunoblots are shown as reference controls for the outer mitochondrial membrane, the intermembrane space, and the mitochondrial matrix, respectively.
To confirm the identity of the putatively unprocessed precursor forms of the above-listed proteins (Fig. 3), we took advantage of the changes in protein solubility after LONP1 depletion. Purified mitochondria from control and LONP1-depleted cells (LONP1-KD-4D) were extracted with n-dodecyl-β-d-maltoside (DDM) and the pellets were solubilized with surfactant, run on SDS-PAGE gels, and silver stained. This showed a generalized increase in protein levels in the insoluble fraction of LONP1-depleted cells compared to the control (Fig. 5A). Bands corresponding to FASTKD2 (∼79 kDa), CLPX (∼70 kDa), and MTERFD3 (∼43 kDa) were excised from the gel (Fig. 5B) or analyzed in solution (SSBP1) by mass spectrometry in control and LONP-depleted cells. As shown in Fig. 6, peptides corresponding to sequences in the MTS of FASTKD2, CLPX, and SSBP1 were substantially enriched in the LONPI-depleted samples relative to the controls. MTERFD3 is not highly expressed in human fibroblasts, and we could not detect peptides from the MTS by mass spectrometry analysis.
FIG 5.

Analysis of insoluble protein fraction in LONP1-depleted cells. (A) Silver-stained gels obtained from the insoluble fraction of mitochondria from control and LONP1-KD-4D cell lines. (B) Silver-stained gels of proteins obtained from the insoluble fraction of mitochondria from control and LONP1-KD-6D cell lines. Bands of interest were excised from the silver-stained SDS-PAGE gel, digested with trypsin or glu-c, and analyzed by mass spectrometry (MS). (C) Analysis of the predicted MTS sequence (MitoProt II v1.101) for the indicated proteins was done using MS data obtained from the mitochondrial pellet fraction of control and LONP1-KD-4D cell lines. The shading indicates the two tryptic peptides identified within MRPL43 MTS. N/A, not available.
FIG 6.
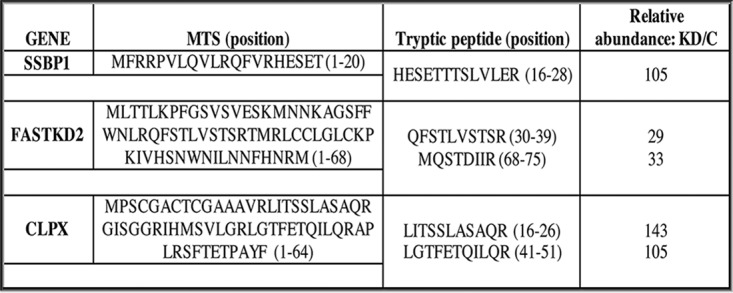
Identification of the MTS of SSBP1, FASTKD2, and CLPX. Mitochondria from control and LONP-depleted cells were treated with DDM (n-dodecyl-β-d-maltoside). The pellet fraction was further extracted in a urea solution and ProteaseMAX surfactant and analyzed in solution or by SDS-PAGE. Bands of interest were gel excised from silver-stained gels and analyzed by mass spectrometry (FASTKD2 and CLPX). The relative abundances of all peptides were calculated from the peak area of reconstructed ion chromatograms in control and LONP1-depleted cells and expressed as a ratio of the knockdown over control (KD/C).
To eliminate the possibility that the processing defect observed in the above-listed proteins was the result of deficient translocation of the precursor form to the mitochondrial matrix, we performed a proteinase K (PK) protection assay using purified mitochondria from control and LONP1-depleted cells (Fig. 4B). This experiment showed that both the precursor and mature forms of CLPX, MTERFD3, and SSBP1 were protected from degradation at high PK concentrations, similar to the matrix protein MRPL11.
LONP1 and MPP are required for processing the MTS of a common subset of mitochondrial proteins.
To investigate the protein composition of the insoluble fraction of LONP1-depleted cells, we suppressed LONP1 for 4 days and analyzed the pellet fraction of purified mitochondria by mass spectrometry after DDM extraction and solubilization with surfactant (Fig. 5; see also Fig. S2 in the supplemental material). LONP1 depletion led to the accumulation of a very specific insoluble fraction that included the above-mentioned proteins, whose maturation is impaired by LONP1 depletion, and proteins involved in mitochondrial translation and RNA metabolism, such as PNPT1, ELAC2, MTERFD1, ERAL1, POLRMT, NOA1, TFB2M, NOP9, GUF1, PUS1, TRUB2, TRMT10C, PTCD3, TFB1M, and GFM1. We also identified 43 mitochondrial ribosomal subunits of both the small (28S) and large (39S) subunits. Components of the MICOS and TIM complexes were not enriched in the pellet fraction of LONP1-depleted cells compared to the controls. We further examined the MS profiles of some of the proteins accumulated in LONP1-depleted cells to determine whether failure to process the MTS was a common feature. Of the 23 MRP proteins analyzed, we could identify only peptides belonging to the predicted MTS in MRPL43 (Fig. 5C).
PMPCB, the beta subunit of the MPP (MPPβ), was also identified in the insoluble fraction of LONP1-depleted cells, suggesting that the processing defect in SSBP1, MTERFD3, FASTKD2, and CLPX could be the result of an inactive MPP. To validate the mass spectrometry analysis, we used DDM to solubilize proteins in control and LONP1-depleted cells (Fig. 7A). Immunoblot analysis confirmed that the MPPβ subunit accumulates in the pellet fraction at 3 and 4 days of LONP1 knockdown; however, the amount of MPPβ present in the soluble fraction remained comparable to the control, suggesting that the depletion of LONP1 is primarily responsible for the processing defects. The MPPα subunit was detectable only in the soluble fraction under every condition, and we did not observe any changes in the processing or solubility profile of the malate dehydrogenase MDH2, one of the validated substrates of the MPP (25) (Fig. 7A).
FIG 7.

Analysis of MPP-depleted cell lines. (A) Input, supernatant, and pellet fractions from whole-cell extracts treated with n-dodecyl-β-d-maltoside were used for SDS-PAGE immunodetection of the indicated proteins in control and LONP1-depleted cells (3 and 4 days). (B and C) Immunoblot analysis of whole-cell extracts obtained by sonication from control cells, MPPα-depleted cells (MPPα-KD), and MPPβ-depleted cells (MPPβ-KD) for 2, 3, and 4 days. Immunoblots show the steady-state levels of the indicated proteins. Porin was used as a loading control.
To further evaluate the role of the MPP in the processing of mitochondrial proteins, we used specific siRNA constructs, MPPα-KD and MPPβ-KD, to deplete the MPPα and MPPβ subunits in a fibroblast line. Control and depleted cells were analyzed by SDS-PAGE (Fig. 7B). All proteins analyzed, including MDH2, were properly processed except for SSBP1.
Next, we investigated if the same four proteins that showed a processing defect in LONP1-depleted cells were also affected in MPPα-KD and MPPβ-KD depletion cell lines (Fig. 7C). Immunoblot analysis of CLPX, FASTKD2, MTERFD3, and SSBP1 confirmed that their processing was impaired in both MPPα- and MPPβ-depleted cell lines; however, in marked contrast to the results obtained with LONP1-depleted cells, MPP depletion resulted in accumulation of only the precursor and not the mature form of the proteins.
To investigate if LONP1 and the MPP are physically associated, we performed second-dimension denaturing gel electrophoresis experiments (2D-SDS-PAGE) in control fibroblasts (Fig. 8A). This experiment showed that LONP1, which is a 110-kDa protein, migrates at an apparent molecular mass of 210 kDa, with a small amount smearing up to 980 kDa. MPPα and MPPβ were present at their corresponding monomeric molecular masses of 56 and 53 kDa; however, the MPP beta subunit also accumulated at two discrete higher-molecular-weight complexes of about ∼630 and 830 kDa, suggesting that a fraction of LONP1 and MPPβ could be part of the same higher-molecular-weight complex. To investigate a potential interaction between LONP1 and MPPβ, we immunoprecipitated the MPPβ subunit using purified mitochondria. Immunoblot analysis showed that LONP1 was present in the elution fraction of MPPβ but not in the control (Fig. 8B). Mass spectrometry analysis of MPPβ immunoprecipitation confirmed the presence of LONP1 in the elution fraction (see Fig. S3 in the supplemental material).
FIG 8.
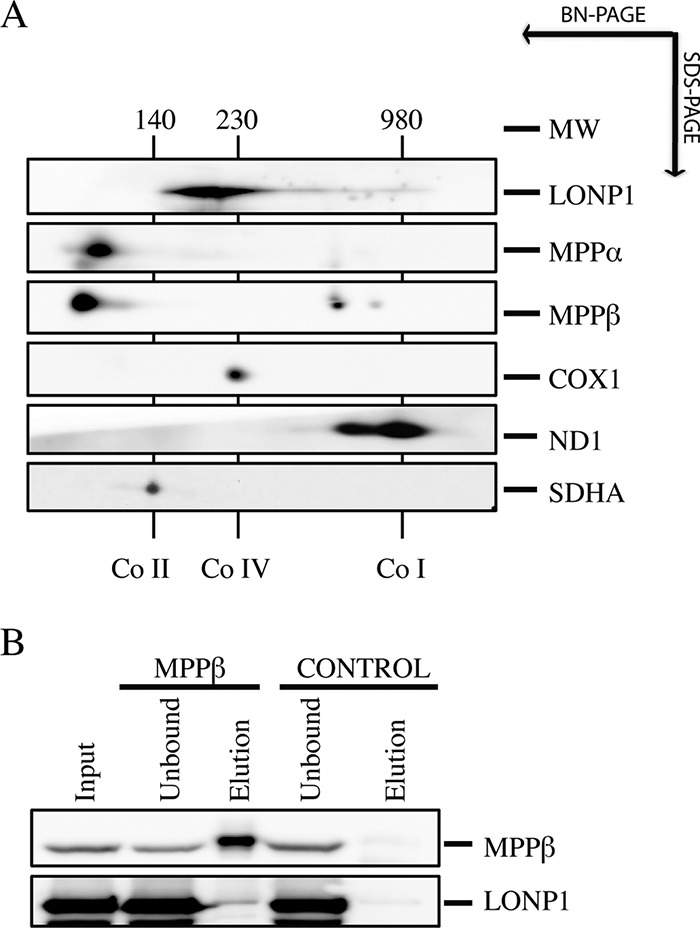
Analysis of MPP and LONP1 interaction. (A) 2D-SDS-PAGE immunodetection of LONP1 and MPP in a fibroblast control line. Complex I (ND1), complex IV (COX1), and complex II (SDHA) were identified with subunit-specific antibodies (right). Numbers indicate the molecular weights of fully assembled OXPHOS complexes. (B) The MPPβ immunoprecipitation experiment was done with control mitochondria. SDS-PAGE analysis of the indicated fractions showed steady-state levels of MPPβ and LONP1.
Depletion of LONP1 results in a massive accumulation of protein aggregates in the mitochondrial matrix.
Biochemical analysis demonstrated that the solubility profile of a subset of mitochondrial proteins was altered in LONP1-depleted cells. To further study this phenomenon, we analyzed LONP1-depleted cells by electron microscopy (Fig. 9). Analysis of thin sections of a control cell line showed round or ovoid mitochondria with defined cristae. After 3 days of LONP1 depletion, the cells showed a marked increase in early phagosome structures, few lysosomes, and crista-depleted mitochondria containing electrodense aggregates. By 6 days of depletion we identified late phagosome structures, mitochondria presenting electrodense aggregates, a more severe disruption of the cristae, and abundant cleared regions within the cytosol likely representing degraded mitochondria.
FIG 9.

TEM analysis of LONP1-depleted cell lines. Electron micrographs of Epon-treated fibroblast sections from control and LONP1-depleted cells (3 and 6 days) are shown. mt, mitochondria; ly, lysosome; e-pha, early phagosome; l-pha, late phagosome.
LONP1 depletion triggers an ISR.
It is thought that when the mitochondrial protein quality control system is not sufficient to counteract the accumulation of defective polypeptides, a retrograde transcriptional pathway, the mitochondrial unfolded protein response (mtUPR), is activated to promote the synthesis of nucleus-encoded mitochondrial chaperones (15, 19, 26). To investigate activation of the mtUPR in the context of LONP1 depletion, we knocked down LONP1 for 2 to 6 days and analyzed the protein steady-state levels of the CLPP protease and the chaperones HSP10, HSPA9, and HSP60 (Fig. 10A). Except for CLPP, which increased slightly, the levels of all chaperones investigated remained relatively unchanged, even after 6 days of LONP1 depletion. We next decided to analyze the mRNA levels of the mtUPR transcriptional activator ATF5 and several downstream components of the pathway by quantitative reverse transcription-PCR (qRT-PCR) (Fig. 10B). This analysis showed modest, but significant, increases in CLPP and HSPA9 after 4 days of LONP1 depletion; however, the transcript levels of ATF5 remained unchanged. In contrast, quantitative mRNA analysis of some of the components of the integrated stress response (ISR) machinery showed that ATF4, ASNS, and CHAC1 were all markedly upregulated after 4 days of LONP1 suppression. These results demonstrate that the most prominent response to the loss of LONP1 activity is activation of the ISR pathway (Fig. 10B).
FIG 10.

Analysis of stress responsive and mitophagy markers in LONP1-depleted cell lines. (A) Whole-cell extracts were obtained by sonication from control and LONP1-depleted cells (2 to 4 days) and used for SDS-PAGE and immunodetection of the indicated proteins. Porin was used as a loading control. (B) qRT-PCR analysis of the indicated markers of mtUPR (left) and ISR (right) was performed with control and LONP1-KD cell lines (n = 3). Data are means ± SEMs. ****, P < 0.0001; **, P < 0.01; *, P < 0.05. (C) Whole-cell extracts were obtained by sonication from control and LONP1-depleted cells (1, 2, and 4 days) and used for SDS-PAGE and immunodetection of the indicated proteins. Porin was used as a loading control. (D) Whole-cell extracts were obtained by sonication from control and LONP1-depleted cells (1 to 4 days) and used for SDS-PAGE and immunodetection of the indicated proteins. SDHA was used as a loading control.
To investigate if mitophagy was also triggered in LONP1-depleted cell lines, we analyzed the steady-state level of the mitochondrial autophagy marker PINK1, which was highly enriched in the LONP1-KD-4D cell line (Fig. 10C). PINK1 is a mitochondrial Ser/Thr kinase (27) that upon mitochondrial depolarization is stabilized in the outer mitochondrial membrane, where it phosphorylates ubiquitin to activate the cytosolic E3 ubiquitin ligase parkin, promoting recruitment of parkin to mitochondria; this allows mitochondrial ubiquitination, recruitment of autophagy receptors, and subsequent mitophagy (27, 28). To further investigate the degradation pathway of damaged mitochondria after LONP1 depletion, we generated a LONP1 time course depletion followed by immunoblot analysis of whole-cell extracts obtained by sonication (WCE-sonication) (Fig. 10D). Immublotting of the autophagy marker LC3 revealed that the steady-state levels of its processed and lipidated form, LC3-II, which localizes selectively to forming and newly formed autophagosomes, increased after the first day of LONP1 knockdown. However, the autophagy receptor P62 (sequestosome 1), a ubiquitin-binding scaffold protein that directly binds LC3 and that has also been suggested to link ubiquitinated proteins to the autophagic machinery, enabling their degradation in the lysosome (29), remained unaffected.
DISCUSSION
Although LONP1 has long been thought of as a protease responsible for the turnover of unfolded or oxidized proteins, this study demonstrated that it also plays an important role in the maturation mitochondrial proteins that are crucial for expression of the mitochondrial genome. We show that a decrease in mitochondrial protein synthesis, resulting from impaired ribosome biogenesis, is an early phenotype associated with loss of LONP1 activity. The maturation and solubility of FASTKD2, which interacts with the 16S rRNA and plays an essential role in mitochondrial ribosome biogenesis (23, 24), are impaired when LONP1 is depleted, and this could account for the protein synthesis defect. The core nucleoid protein SSBP1 is similarly affected, providing a plausible explanation for the defect in mtDNA maintenance.
Although LONP1 is a soluble protease of the mitochondrial matrix, immunoprecipitation experiments identified AFG3L2, YME1L1, and HTRA2 proteases, seven components of the TIM complex, and the entire MICOS complex as coimmunoprecipitating proteins, all of which are intimately associated with the inner mitochondrial membrane (IMM), suggesting that LONP1 localizes near the IMM. Additionally, five components of the ribosome exit tunnel (MRPL17, MRPL22, MRPL23, MRPL39, and MRPL44) coimmunoprecipitated with LONP1. These results suggest that LONP1 functions at the boundary of the IMM, interacting with a specific region of the mitochondrial ribosome. MS analysis of the pellet fraction after LONP1 depletion demonstrated that about half of the structural components of the mitochondrial ribosome accumulate in insoluble protein aggregates (see Fig. S2 in the supplemental material). Further evaluation of the MS data from 23 mitochondrial proteins that were found exclusively in the pellet of the LONP1-depleted cells showed that the ribosomal subunit MRPL43 contains peptides that are part of the predicted MTS, perhaps further contributing to the protein synthesis defect.
Depletion of the mitochondrial processing peptidase (MPP) results in impaired processing of the same subset of proteins as affected in LONP1 depletion but not in the accumulation of the mature proteins. Mass spectrometry and immunoblot analyses of the insoluble fraction in LONP1-depleted cells identified the MPPβ subunit of the MPP; however, the alpha subunit was never detected in this fraction. A small fraction of the total amount of LONP1 and MPPβ appeared to interact in a higher-molecular-weight complex of about 630 to 830 kDa. Similar to the phenotype of LONP1 depletion, the majority of the mitochondrial proteins analyzed following MPP depletion were processed normally, including the well-characterized substrate of the MPP MDH2. Recently, it was shown that depletion of the MPP in HEK293T cells did not affect the processing of other substrates such as UQCRFS1 (Rieske) (30, 31) or Sod2 (manganese superoxide dismutase [MnSOD]) (32).
Electron microscopic analysis showed that electrodense bodies amassed within the mitochondrial matrix of LONP1-depleted cells, a phenotype previously observed in yeast (Saccharomyces cerevisiae) and humans, likely reflecting protein aggregation (9, 33, 34). MS analysis of the insoluble fraction of LONP1-depleted cells confirmed that the electron-dense inclusion bodies of the matrix comprise a subset of mitochondrial proteins, mainly including most of the MRPs, and that the majority of these proteins were properly processed.
To evaluate the cellular response to the stress caused by the accumulation of protein aggregates in the mitochondrial matrix, we measured the accumulation of several markers of the mitochondrial unfolded protein response (mtUPR) and the integrated stress response (ISR) pathways. Depletion of LONP1 resulted in substantial increases in the transcript levels of several components of the ISR, viz., ATF4, ASNS, and CHAC1, which were previously shown to be upregulated in response to impaired mitochondrial proteostasis (26). Although mRNA levels of the transcriptional activator ATF5 did not accumulate in LONP1-depleted cells, the expression levels of some of the components of the mtUPR, like CLPP or HSPA9, increased modestly at the mRNA or protein level. This result is consistent with several studies in which suppression of LONP1 failed to robustly activate the canonical components of the mtUPR (17, 18). Modest depletion of the LONP1 homolog (Lon) in C. elegans similarly did not elicit an mtUPR (16). It has been previously shown that transcription of ATF5 is induced by both CHOP and ATF4 (35), and it is possible that the upregulation of CLPP and HSPA9 is connected to transient expression of ATF5 mediated by the sustained accumulation of ATF4.
LONP1-depleted cell lines exhibited a dramatic increase in the steady-state level of the mitophagy marker PINK1, which mediates parkin recruitment to fully depolarized mitochondria (27, 28). Consistently, the protein levels of the downstream autophagy marker LC3-II progressively accumulated with LONP1 depletion (Fig. 7), but the steady-state levels of P62 remained unaffected, suggesting that another autophagy receptor is responsible for the binding to the ubiquitinated cargo. Interestingly, LC3-II protein levels increased after 24 h of LONP1 depletion; however, the PINK1-parkin pathway was active only in LONP1-4D-KD cells. It is possible that the initial pathway of mitochondrial degradation in the context of LONP1 depletion is P62/parkin independent, as recently proposed by Lazarou et al. (36).
Together our results demonstrate that the human LONP1 protease is required for both the maturation and degradation of a subset of mitochondrial matrix proteins with key roles in the expression of the mitochondrial genome. Failure to process these substrates presumably prevents their proper folding, resulting in their accumulation in insoluble protein aggregates and ultimately triggering an ISR and mitophagy. Why a specific subset of mitochondrial matrix proteins requires LONP1 activity for proper processing remains an outstanding question. It is possible that the tertiary structures of immature SSBP1, MTERFD3, FASTKD2, and CLPX differ from the “canonical” features required for processing by MPP alone, but this requires further investigation.
MATERIALS AND METHODS
Cell culture.
Primary cell lines were established from subject skin fibroblasts and immortalized by transduction with a retroviral vector expressing the human papillomavirus 16 (HPV-16) E7 gene plus a retroviral vector expressing the catalytic component of human telomerase (htert) (37). Fibroblasts and 143B cells were grown at 37°C in an atmosphere of 5% CO2 in high-glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum.
Mitochondrial isolation.
Fibroblasts were resuspended in ice-cold SET buffer (250 mM sucrose–10 mM Tris-HCl–1 mM EDTA [pH 7.4]) supplemented with complete protease inhibitors (Roche) and homogenized with 10 passes through a prechilled, zero-clearance homogenizer (Kimble/Kontes). The homogenized cellular extract was then centrifuged twice for 10 min at 600 × g to obtain a postnuclear supernatant. Mitochondria were pelleted by centrifugation for 10 min at 10,000 × g and washed once in the same buffer.
LONP1 overexpression.
LONP1 cDNA was amplified by OneStep RT-PCR (Qiagen) and cloned into pBABE-Puro with Gateway cloning technology (Invitrogen). The accuracy of the clones was tested by automated DNA sequencing. Direct mutagenesis of LONP1 was done using the site-directed mutagenesis lightning kit (Stratagene). Retroviral constructs were transfected into the Phoenix packaging cell line using the HEPES-buffered saline (HBS)–Ca3(PO4)2 method. Control fibroblasts were infected 48 h later by exposure to virus-containing medium in the presence of 4 mg/ml of Polybrene as described previously (https://web.stanford.edu/group/nolan/_OldWebsite/index.html).
RNAi knockdown.
For the knockdown experiments with LONP1, CLPX, and MPP, stealth RNA interference (RNAi) duplex constructs were used as follows: LONP1, GGACGUCCUGGAAGAGACCAAUAUU; CLPX, AAUAUCUUCGCCUACAUAUCCAGCC; MPPαa, GCAUGUUCUCCAGGCUCUACCUCAA; MPPαb, GAGCCAAGACGCAGCUGACAUCAAU; MPPβa, GCAGCUUGAUGGUUCAACUCCAAUU; and MPPβb, CAGCUCACUUGUCAUGGCAAUCUUU.
siRNA constructs were designed through the BLOCK-iT RNAi Express website.
The desired cell lines were transfected following the Lipofectamine RNAiMAX protocol. Briefly, Lipofectamine and RNA constructs were diluted in Opti-MEM reduced-serum medium. BLOCK-iT Alexa Fluor red fluorescent oligonucleotide was used to assess the transfection efficiency (transfection performed in parallel with the other stealth constructs) and as a mock oligonucleotide transfection control (all reagents from Invitrogen).
Southern blotting.
DNA was isolated from siRNA-depleted LONP1 and control fibroblasts using the genElute mammalian genomic DNA miniprep kit (Sigma). Five micrograms of total DNA was PvuII digested overnight at 37°C. Samples were run on a 1% agarose–Tris-borate-EDTA (TBE) gel, followed by 20-min incubations at room temperature for depurination, denaturation, and neutralization. Next, the gel was transferred to a nylon membrane. mtDNA and 18S probes were labeled with [32P]dCTP (GE Healthcare) using the MegaPrime DNA labeling kit (GE Healthcare). Hybridization was conducted according to the manufacturer's manual using ExpressHyb hybridization solution (Clontech), and the radioactive signal was detected using the PhosphorImager system. Quantitative analysis was done using ImageQuant 5.2 software.
Northern blotting.
RNA was isolated from LONP1-depleted and control fibroblasts using an RNeasy kit (Qiagen). Ten micrograms of total RNA was separated on a denaturing morpholinepropanesulfonic acid (MOPS)-formaldehyde-agarose gel and transferred to a nylon membrane. ND1, ND5, COX1, COX2, ATP6/8, cytochrome b (cyt b), 12S, 16S, and 18S probes were labeled with [32P]dCTP (GE Healthcare) using a MegaPrime DNA labeling kit (GE Healthcare). Hybridization was conducted according to the manufacturer's manual using ExpressHyb hybridization solution (Clontech), and the radioactive signal was detected using the PhosphorImager system.
Pulse-labeling of mitochondrial translation products.
In vitro labeling of mitochondrial translation was performed as previously described (38). Briefly, cells were labeled for 60 min at 37°C in methionine- and cysteine-free DMEM containing 200 μCi/ml of [35S]methionine and 100 μg/ml of emetine and chased for 10 min in regular DMEM. Protein extraction was done in labeled cells by resuspension in loading buffer containing 93 mM Tris-HCl (pH 6.7), 7.5% glycerol, 1% SDS, 0.25 mg of bromophenol blue/ml, and 3% mercaptoethanol. Samples were sonicated for 5 s and run on 15 to 20% polyacrylamide gradient gels. The labeled mitochondrial translation products were detected by direct autoradiography on a PhosphorImager.
SDS-PAGE analysis.
Whole-cell extracts with n-dodecyl-β-d-maltoside (WCE-DDM) were prepared by solubilization in a 1:5 ratio with extraction buffer (1.5% DDM) for 30 min on ice, followed by centrifugation at 20,000 × g and 4°C for 20 min. The soluble fraction was mixed at a 1:1 ratio with 2× Laemmli sample buffer. The same protocol of extraction was used for the separation of the soluble (SN) and insoluble (PE) fractions of LONP1-KD and MPP-KD cells. Whole-cell extracts obtained by sonication (WCE-sonication) were prepared by resuspension of pelleted cells in loading buffer containing 93 mM Tris-HCl (pH 6.7), 7.5% glycerol, 1% SDS, 0.25 mg of bromophenol blue/ml, and 3% mercaptoethanol. Next, samples were sonicated for 5 s. WCE-DDM and WCE-sonication samples were loaded onto a 12% acrylamide-bisacrylamide (29:1) gel. Subsequent immunoblotting was done with the appropriate antibodies.
Antibodies.
The following antibodies were used: anti-LONP1 (kindly provided by Carolyn Suzuki), antiactin (MitoSciences), anti-succinate dehydrogenase subunit A (anti-SDHA) (Abcam), antiporin (EMD Calbiochem), anti-PHB1 (Abcam), anti-LRPPRC (in-house), anti-SLIRP (Abcam), anti-MRPL11 (Sigma-Aldrich), anti-MRPS18B (Proteintech), anti-MRPL44 (Sigma-Aldrich), anti-MTERFD3 (Sigma-Aldrich), anti-TFAM (a kind gift of Alexandra Trifunovic), anti-TFB1M (Sigma-Aldrich), anti-TFB2M (Abgent), anti-AFG3L2 (kindly provided by T. Langer), anti-GRSF1 (Sigma-Aldrich), anti-ATAD3A (Abcam), anti-SHMT2 (Abcam), anti-NDUFAF7 (in-house), anti-NOA1 (Sigma), anti-POLG (Abcam), anti-CLPX (Abcam), anti-SSBP1 (Novus Biologicals), anti-DHX30 (Abcam), anti-FASTKD2 (Santa Cruz), anti-MFN2 (Cell Signaling), anti-SCO1 (in-house), anti-MPPα (Sigma), anti-MPPβ (Proteintech), anti-MDH2 (Proteintech), anti-COX1 (Abcam), anti-ND1 (kindly provided by A. Lombes), anti-LC3 (Novus Biologicals), anti-P62 (BD Biosciences), anti-YME1L1 (Proteintech), anti-CLPP (Proteintech), anti-HSP10 (Santa Cruz), anti-HSPA9 (Proteintech), and anti-PINK1 (Novus Biologicals).
Sucrose density gradient.
Mitochondria were extracted in lysis buffer containing 260 mM sucrose, 100 mM KCl, 20 mM MgCl2, 10 mM Tris-Cl (pH 7.5), 1% Triton X-100, and 5 mM β-mercaptoethanol, supplemented with complete protease inhibitors without EDTA, on ice for 20 min. Lysates were centrifuged for 45 min at 9,400 × g at 4°C before being loaded on 1 ml of a 10 to 30% discontinuous sucrose gradient (50 mM Tris-Cl, 100 mM KCl, 10 mM MgCl2). Loaded gradients were centrifuged at 32,000 rpm for 130 min in a Beckman SW60-Ti rotor. After centrifugation, 14 fractions were collected and analyzed by SDS-PAGE.
Immunoprecipitation.
Mitochondria from control (LONP1 and MPPβ immunoprecipitations) cell lines were cross-linked with 1 mM disuccinimidyl glutarate (DSG) for 90 min on ice prior to protein extraction. The MPPβ immunoprecipitation experiment did not include cross-linking. One milligram of mitochondria was extracted on ice in 250 μl of lysis buffer (50 mM HEPES buffer [pH 7.6], 150 mM NaCl, 1% taurodeoxycholate), supplemented with complete protease inhibitors (Roche), for 45 min. The protein extract was centrifuged at 25,000 × g and 4°C for 40 min. The supernatant was then precleared overnight with naked magnetic Dynabeads. In parallel, the appropriate antibody (LONP1 or MPPβ) was cross-linked to Dynabeads according to the manufacturer's instructions. The incubation of the protein extract with antibody-coated beads was carried out overnight at 4°C. Bound protein was eluted with 400 μl of 0.1 M glycine (pH 2.5) with 0.5% DDM at 45°C, followed by trichloroacetic acid (TCA) precipitation. Elutions were trypsin digested and analyzed by mass spectrometry using an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany).
Sodium carbonate solubilization.
Purified mitochondria from control and LCD+LONP1-KD-3D fibroblasts were extracted with 100 mM Na2CO3 (pH 11.5) on ice for 30 min and centrifuged at 65,000 rpm to separate soluble and membrane fractions. The pellet fraction was washed twice with water prior to SDS-PAGE analysis. Subsequent immunoblotting was done with the appropriate antibodies.
Surfactant treatment and silver stain analysis.
Mitochondria from control, LONP1-KD-4D, and LONP1-KD-6D cells were resuspended in approximately a 1:5 ratio with extraction buffer (1.5% DDM) for 30 min on ice, followed by protein quantification by the Bradford assay. Equal amounts of protein from control and knockdown cell lines were centrifuged at 20,000 × g and 4°C for 20 min. The supernatant fraction was collected and the pellet was rinsed in acetone and resuspended in 8 M urea containing 0.2% ProteaseMAX surfactant. Samples were incubated at room temperature for 60 min, followed by centrifugation at 16,000 × g for 10 min. Next, supernatants were directly sent for mass spectrometry analysis and/or run by SDS-PAGE and silver stained. Gels were fixed in 50% methanol–10% acetic acid solution for 30 min, reduced in 0.02% sodium thiosulfate for 2 min, and stained with 0.2% silver nitrate for 30 min. Development was done with a solution containing 0.05% formalin, 3% sodium carbonate, and 0.0004% sodium thiosulfate. Bands of interest were gel excised, digested with trypsin or endoproteinase glu-c, and analyzed by mass spectrometry using an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany).
Mass spectrometry analysis.
The in-gel digestion protocol is based on results obtained previously (39). Bands of interest were gel excised and all volumes were adjusted according to the volume of gel pieces. Gel pieces were washed with water for 5 min and destained twice with destaining buffer (100 mM sodium thiosulfate, 30 mM potassium ferricyanide) for 15 min. This step was followed by a 5-min wash with 50 mM ammonium bicarbonate and then dehydration with acetonitrile. Next, reduction buffer (10 mM dithiothreitol [DTT], 100 mM ammonium bicarbonate) was added and incubated for 30 min at 40°C, followed by a 20-min incubation at 40°C in alkylation buffer (55 mM iodoacetamide, 100 mM ammonium bicarbonate). Gel pieces were dehydrated and washed at 40°C by adding acetonitrile for 5 min, then dried for 5 min at 40°C, and then rehydrated at 4°C for 40 min with trypsin solution (6 ng/μl of trypsin or glu-c [sequencing grade] from Promega, 25 mM ammonium bicarbonate). The concentration of trypsin or glu-c was kept low to reduce signal suppression effects and background originating from autolysis products when performing liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Protein digestion was performed at 58°C for 1 h and stopped with 1% formic acid–2% acetonitrile. The supernatant was transferred into a 96-well plate, and peptide extraction was performed with two 30-min extraction steps at room temperature using extraction buffer (1% formic acid, 50% acetonitrile). All peptide extracts were pooled into the 96-well plate and then completely dried in a vacuum centrifuge. The plate was sealed and stored at 20°C until LC-MS/MS analysis. Prior to LC-MS/MS, peptide extracts were resolubilized under agitation for 15 min in 11 μl of 0.2% formic acid and then centrifuged at 2,000 rpm for 1 min.
The LC column was a C18 reversed-phase column packed with a high-pressure packing cell. A 75-μm (inside diameter [i.d.]) Self-Pack PicoFrit fused silica capillary column (New Objective, Woburn, MA) 15 cm long was packed with the C18 Jupiter 5-μm 300-Å reverse-phase material (Phenomenex, Torrance, CA). This column was installed on an Easy-nLC II system (Proxeon Biosystems, Odense, Denmark) and coupled to the LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany) equipped with a Proxeon nanoelectrospray ion source. The buffers used for chromatography were 0.2% formic acid (buffer A) and 100% acetonitrile–0.2% formic acid (buffer B). During the first 12 min, 5 μl of sample was loaded onto the column at a flow rate of 600 nl/min, and subsequently, the gradient went from 2 to 80% buffer B in 60 min at a flow rate of 250 nl/min and then came back at 600 nl/min and 2% buffer B for 10 min. LC-MS/MS data acquisition was accomplished in positive-ion mode using an 11-scan-event cycle comprised of a full-scan MS for scan event 1 acquired in the Orbitrap. The mass resolution for MS was set to 60,000 (at m/z 400) and used to trigger the 10 additional MS/MS events acquired in parallel in the linear ion trap for the top 10 most intense ions. The mass-over-charge ratio range was from 360 to 2,000 for MS scanning, with a target value of 1,000,000 charges and a maximum injection time of 100 ms. To improve the mass accuracy, the lock mass option was enabled and decamethylcyclopentasiloxane (m/z 371.101233) was used for real-time internal mass calibration. The mass-over-charge ratio range for MS/MS scanning was from ∼1/3 of parent m/z ratio to 2,000, with a target value of 10,000 charges and a maximum injection time of 100 ms. The data-dependent scan events used a maximum ion fill time of 100 ms and 1 microscan. Target ions already selected for MS/MS were dynamically excluded for 25 s after two MS/MS acquisitions. Nanospray and S-lens voltages were set to 1.3 to 1.7 kV and 50 V, respectively. Capillary temperature was set to 225°C. MS/MS conditions were as follows: normalized collision energy, 35 V; activation q, 0.25; and activation time, 10 ms. The minimum signal required to trigger an MS/MS event was set to 2,000, and the isolation width for MS/MS scanning was set to 2. The peak list files were generated with extract_msn.exe (version 10 January 2011) using the following parameters: minimum mass set to 600 Da, maximum mass set to 6,000 Da, no grouping of MS/MS spectra, precursor charge set to auto, and minimum number of fragment ions set to 10. Protein database searching was performed with Mascot 2.3 (Matrix Science) against the human NCBInr protein database (version 18 July 2012). The mass tolerances for precursor and fragment ions were set to 10 ppm and 0.6 Da, respectively. Trypsin was used as the enzyme, allowing for up to 2 missed cleavages. Mono-, di-, and trimethylations of arginine were allowed as variable modifications, while carbamidomethylation was set as a fixed modification.
The Mascot cutoffs used for the MS analysis were based on the values shown in Table 1.
TABLE 1.
Mascot score valuesa
| Protein score | % false positive |
|---|---|
| >50 | <1 |
| 40–50 | 10 |
| 30–40 | 40 |
| 20–30 | 60 |
| <20 | >90 |
The values for false positives were used to determine the Mascot cutoff value presented in the analysis of the mass spectrometry data.
The relative abundances of peptides in Fig. 4 were estimated from the peak area of reconstructed ion chromatograms obtained with Qual Browser (Xcalibur; Thermo Fisher Scientific) using an m/z tolerance of 10 ppm.
Proteinase K protection assay.
Mitochondria from control and LONP1-depleted cell lines were resuspended in HIM (200 mM mannitol, 70 mM sucrose, 10 mM HEPES, and 1 mM EGTA, adjusted to pH 7.5 with KOH) buffer at a 1-mg/ml concentration containing the desired proteinase K concentration (see Fig. S3). Samples were incubated for 20 min on ice. Addition of 2 mM phenylmethylsulfanoxide (PMSF) for 20 min stopped the reaction. Samples were spun down for 20 min at 20,000 × g. The pellet fraction was resuspended in 50 μl of 1× Laemmli buffer, sonicated, and loaded onto a 12% acrylamide-bisacrylamide (29:1) gel. Subsequent immunoblotting was done with the appropriate antibodies.
2D-SDS-PAGE analysis.
Mitoplasts were prepared from fibroblasts as described previously (40) by treating the cells with 0.8 mg of digitonin/mg of protein. Mitoplasts were solubilized with 1% n-dodecyl-β-d-maltoside, and the solubilized proteins were used for electrophoresis. Blue native PAGE (BN-PAGE) (41) was used to separate the samples in the first dimension on 6 to 15% polyacrylamide gradients. For analysis of the second dimension, strips of the first-dimension gel were incubated for 30 min in 1% SDS and 1% mercaptoethanol. The strip was then run on 10% tricine–SDS-PAGE gel. Individual structural subunits of complexes I and IV were detected by immunoblot analysis.
Electron microscopy.
Control and LONP1-depleted cell lines were incubated overnight at 4°C in fixation solution containing 2.5% glutaraldehyde, 0.1 M sodium cacodylate, and 4% sucrose. Next, samples were incubated in postfixative solution containing 1% osmium tetraoxide and 1.5% potassium ferrocyanide, followed by 15-min incubations in increasing concentrations of ethanol (30% to 100%) to dehydrate; next, samples were infiltrated for 30 min with Epon-ethanol at 1:1, at 1:3, and finally with 100% Epon. Samples were embedded in fresh Epon for 48 h at 60°C, cut, and analyzed by transmission electron microscopy (TEM).
Quantitative RT-PCR.
RNA was purified from control and LONP1 siRNA knockdown fibroblasts (MCH64) using an RNeasy Plus kit (Qiagen). cDNA was obtained from 2 μg of purified RNA using the a high-capacity cDNA reverse transcription kit (4368814) from Applied Biosystems. qPCR was performed using the KAPA SYBR FAST qPCR master mix and run on a Quant Studio 6 Flex system. qPCR analysis was done by absolute quantification/2nd derivative of three independent biological replicates, each performed in triplicate. The statistical analysis of mRNA transcript abundance was done after normalization with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The statistics software GraphPad Prism 6 was used to perform a two-way analysis of variance (ANOVA) with Bonferroni multiple comparisons. Error bars in figures represent standard errors of the means.
Primers sequence used for qRT-PCR analysis were as follows: ATF4-F, CAGCAAGGAGGATGCCTTCT; ATF4-R, CCAACAGGGCATCCAAGTC; CHAC1-F, GTGGTGACGCTCCTTGAAGA; CHAC1-R, TTCAGGGCCTTGCTTACCTG; ASNS-F, GATGAACTTACGCAGGGTTACA; ASNS-R, CACTCTCCTCCTCGGCTTT; PCK2-F, AAACCCTGGAAACCTGGTG; PCK2-R, CAATGGGGACACCCTCTG; ATF5-F, GAGCCCCTGGCAGGTGAT; ATF5-R, CAGAGGGAGGAGAGCTGTGAA; CLPP-F, AAGCACACCAAACAGAGCCT; CLPP-R, AAGATGCCAAACTCCTGGG; HSPA9-F, TGGTGAGCGACTTGTTGGAAT; HSPA9-R, ATTGGAGGCACGGACAATTTT; HSPD1-F, ACTCGGAGGCGGAAGAAA; HSPD1-R, TGTGGGTAACCGAAGCATTT; GAPDH-F, AGGGTCATCATCTCTGCCCCCTC; and GAPDH-R, TGTGGTCATGAGTCCTTCCACGAT.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a CIHR grant to E.A.S. O.Z.R. was supported by fellowships from CONACyT (Consejo Nacional de Ciencia y Tecnología) (209378) and PBEEE (Fonds quebecois de la recherche sur la nature et les technologies) (140802).
We thank Woranontee Weraarpachai for reagents and support, Neil Webb for the generation of the LCD catalytically dead LONP1 construct, and Denis Faubert from IRCM for the generation of the mass spectrometry data and MS quantitative analysis.
O.Z.R. and E.A.S. conceived the project, designed the experiments, and wrote the manuscript. O.Z.R. executed all experiments and analyzed the data.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00412-17.
REFERENCES
- 1.Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. 2005. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Venkatesh S, Lee J, Singh K, Lee I, Suzuki CK. 2012. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim Biophys Acta 1823:56–66. doi: 10.1016/j.bbamcr.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duman RE, Lowe J. 2010. Crystal structures of Bacillus subtilis Lon protease. J Mol Biol 401:653–670. doi: 10.1016/j.jmb.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 4.Kereïche S, Kovacik L, Bednar J, Pevala V, Kunova N, Ondrovicova G, Bauer J, Ambro L, Bellova J, Kutejova E, Raska I. 2016. The N-terminal domain plays a crucial role in the structure of a full-length human mitochondrial Lon protease. Sci Rep 6:33631. doi: 10.1038/srep33631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu GK, Markovitz DM. 1998. The human LON protease binds to mitochondrial promoters in a single-stranded, site-specific, strand-specific manner. Biochemistry 37:1905–1909. doi: 10.1021/bi970928c. [DOI] [PubMed] [Google Scholar]
- 6.Wright R, Stephens C, Zweiger G, Shapiro L, Alley MR. 1996. Caulobacter Lon protease has a critical role in cell-cycle control of DNA methylation. Genes Dev 10:1532–1542. doi: 10.1101/gad.10.12.1532. [DOI] [PubMed] [Google Scholar]
- 7.Gottesman S, Gottesman M, Shaw JE, Pearson ML. 1981. Protein degradation in E. coli: the lon mutation and bacteriophage lambda N and cII protein stability. Cell 24:225–233. doi: 10.1016/0092-8674(81)90518-3. [DOI] [PubMed] [Google Scholar]
- 8.Van Dyck L, Langer T. 1999. ATP-dependent proteases controlling mitochondrial function in the yeast Saccharomyces cerevisiae. Cell Mol Life Sci 56:825–842. doi: 10.1007/s000180050029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki CK, Suda K, Wang N, Schatz G. 1994. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science 264:891. [DOI] [PubMed] [Google Scholar]
- 10.Bayot A, Gareil M, Rogowska-Wrzesinska A, Roepstorff P, Friguet B, Bulteau AL. 2010. Identification of novel oxidized protein substrates and physiological partners of the mitochondrial ATP-dependent Lon-like protease Pim1. J Biol Chem 285:11445–11457. doi: 10.1074/jbc.M109.065425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushima Y, Goto Y, Kaguni LS. 2010. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc Natl Acad Sci U S A 107:18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu B, Yadav S, Shah PG, Liu T, Tian B, Pukszta S, Villaluna N, Kutejova E, Newlon CS, Santos JH, Suzuki CK. 2007. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J Biol Chem 282:17363–17374. doi: 10.1074/jbc.M611540200. [DOI] [PubMed] [Google Scholar]
- 13.Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S, Bogenhagen DF, Temiakov D, Suzuki CK. 2013. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol Cell 49:121–132. doi: 10.1016/j.molcel.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. 2007. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 15.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. 2016. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26:2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. 2012. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, Gygi SP, Auwerx J. 31 May 2017. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayot A, Gareil M, Chavatte L, Hamon MP, L'Hermitte-Stead C, Beaumatin F, Priault M, Rustin P, Lombes A, Friguet B, Bulteau AL. 2014. Effect of Lon protease knockdown on mitochondrial function in HeLa cells. Biochimie 100:38–47. doi: 10.1016/j.biochi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Münch C, Harper JW. 2016. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534:710–713. doi: 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Lu B, Lee I, Ondrovicova G, Kutejova E, Suzuki CK. 2004. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J Biol Chem 279:13902–13910. doi: 10.1074/jbc.M309642200. [DOI] [PubMed] [Google Scholar]
- 21.Pellegrini M, Asin-Cayuela J, Erdjument-Bromage H, Tempst P, Larsson NG, Gustafsson CM. 2009. MTERF2 is a nucleoid component in mammalian mitochondria. Biochim Biophys Acta 1787:296–302. doi: 10.1016/j.bbabio.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Cotney J, McKay SE, Shadel GS. 2009. Elucidation of separate, but collaborative functions of the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 in mitochondrial biogenesis reveals new insight into maternally inherited deafness. Hum Mol Genet 18:2670–2682. doi: 10.1093/hmg/ddp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonicka H, Shoubridge EA. 2015. Mitochondrial RNA granules are centres for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep 10(6):P920–932. doi: 10.1016/j.celrep.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 24.Popow J, Alleaume AM, Curk T, Schwarzl T, Sauer S, Hentze MW. 2015. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 21:1873–1884. doi: 10.1261/rna.052365.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogishima T, Niidome T, Shimokata K, Kitada S, Ito A. 1995. Analysis of elements in the substrate required for processing by mitochondrial processing peptidase. J Biol Chem 270:30322–30326. doi: 10.1074/jbc.270.51.30322. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. 2002. A mitochondrial specific stress response in mammalian cells. EMBO J 21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. 2010. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjørkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. 2009. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 30.Nett JH, Schagger H, Trumpower BL. 1998. Processing of the presequence of the Schizosaccharomyces pombe Rieske iron-sulfur protein occurs in a single step and can be converted to two-step processing by mutation of a single proline to serine in the presequence. J Biol Chem 273:8652–8658. [DOI] [PubMed] [Google Scholar]
- 31.Nett JH, Trumpower BL. 1996. Dissociation of import of the Rieske iron-sulfur protein into Saccharomyces cerevisiae mitochondria from proteolytic processing of the presequence. J Biol Chem 271:26713–26716. [DOI] [PubMed] [Google Scholar]
- 32.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. 2012. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guha S, Lopez-Maury L, Shaw M, Bahler J, Norbury CJ, Agashe VR. 2011. Transcriptional and cellular responses to defective mitochondrial proteolysis in fission yeast. J Mol Biol 408:222–237. doi: 10.1016/j.jmb.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, Morse KM, Metcalfe HM, Skalska J, Andreeff M, Brookes PS, Suzuki CK. 2012. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 119:3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC. 2013. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell 24:2477–2490. doi: 10.1091/mbc.E13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lochmüller H, Johns T, Shoubridge EA. 1999. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp Cell Res 248:186–193. [DOI] [PubMed] [Google Scholar]
- 38.Sasarman F, Shoubridge EA. 2012. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol Biol 837:207–217. doi: 10.1007/978-1-61779-504-6_14. [DOI] [PubMed] [Google Scholar]
- 39.Havlis J, Thomas H, Sebela M, Shevchenko A. 2003. Fast-response proteomics by accelerated in-gel digestion of proteins. Anal Chem 75:1300–1306. doi: 10.1021/ac026136s. [DOI] [PubMed] [Google Scholar]
- 40.Klement P, Nijtmans LG, Van den Bogert C, Houstek J. 1995. Analysis of oxidative phosphorylation complexes in cultured human fibroblasts and amniocytes by blue-native-electrophoresis using mitoplasts isolated with the help of digitonin. Anal Biochem 231:218–224. [DOI] [PubMed] [Google Scholar]
- 41.Schägger H, von Jagow G. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199:223–231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.