

Abstract
Background
Inherited unconjugated hyperbilirubinemia is caused by variants in the gene UGT1A1 leading to Gilbert’s syndrome and Crigler-Najjar syndrome types I and II. These syndromes are differentiated on the basis of UGT1A1 residual enzymatic activity and its affected bilirubin levels and responsiveness to phenobarbital treatment.
Case presentation
In this report, we present a boy with Crigler-Najjar syndrome type II with high unconjugated bilirubin levels that decreased after phenobarbital treatment but increased in adolescence. Four different UGT1A1 gene variants have been identified for this patient, of which one is novel (g.11895_11898del) most likely confirming diagnose molecularly.
Conclusions
The presented case highlights the challenges encountered with the interpretation of molecular data upon identification of multiple variants in one gene that are causing different degree reducing effect on enzyme activity leading to several clinical conditions.
Keywords: CNS-I, CNS-II, UGT1A1
Background
Inherited unconjugated hyperbilirubinemia is caused by pathogenic variants in the UGT1A1 gene and, depending on the bilirubin levels, is categorized as Gilbert’s syndrome (GS; OMIM: 143500), Crigler-Najjar syndrome type I (CNS-I; OMIM: 218800), or Crigler-Najjar syndrome type II (CNS-II; OMIM: 606785); the total bilirubin levels range from 17.1–102.6 μmol/L, 102.6–342 μmol/L, and 342–769.5 μmol/L, respectively [1]. When not appropriately treated, CNS-I is an early lethal condition characterized by kernicterus caused by the absence of active UDP-glucuronosyltransferase 1A1 (UGT1A1). In the case of CNS-II, UGT1A1 activity is greatly reduced [1].
There are numerous reports on UGT1A1 gene variants that are related to the aforementioned clinical conditions [2, 3]. The most common condition is GS which affects ~ 10% of Europeans. As there are cases where it is difficult to distinguish the disorders caused by pathogenic variants in the UGT1A1 gene, genetic investigations are becoming increasingly important [4].
The patient described here has unconjugated hyperbilirubinemia that is responsive to phenobarbital treatment and carries four genetic variations in the UGT1A1 gene, one of which is reported for the first time.
Case presentation
The proband is a 17-year-old Caucasian male who first presented at the neonatal stage with prolonged jaundice with an unconjugated bilirubin level of 400 μmol/L without hemolytic anemia data. Following an uneventful pregnancy, the patient was the first child of non-consanguineous Caucasian parents. Due to jaundice and elevated indirect bilirubin levels, he was diagnosed with GS soon after delivery as he had the A(TA)7TAA allele in homozygous state (identified by fragment analysis as previously described [5]). During his childhood years, the patient did well. He periodically underwent phenobarbital treatment for the reduction of bilirubin levels associated with jaundice; the mean bilirubin levels achieved as the result of these treatments were 100–200 μmol/L. The patient’s parents had no complaints until puberty.
After reaching puberty, the boy became severely jaundiced and his bilirubin levels increased to 300–350 μmol/L. However, there were no other complaints, he was doing well in school and his weight and height parameters were according to his age. The boy was referred to a hepatologist and multiple investigations were carried out. No pathological findings were observed in the laboratory analysis and hepatitis viral markers were negative. Furthermore, there was no evidence of other inborn errors of metabolism, i.e. organic acidurias and amino acidurias or changes in the fatty acid profile.
The results of abdominal ultrasound and abdominal MRI were unremarkable. Procurement of a percutaneous liver biopsy revealed no pathological findings. Based on isolated elevation of indirect bilirubin levels from standard laboratory investigations, reduced glutathione (0.89 mmol/l; reference value 1.12–1.216 mmol/l), a glucuronide level in 24-h urine of up to 102 mg/ml (reference value 430–600 mg/ml), phenobarbital responsiveness, and no evidence of kernicterus (as the child had no complaints about his health condition), CNS-II was suspected.
To confirm the CNS-II diagnosis, bidirectional sequencing of five exons and exon/intron boundaries of the gene UGT1A1 (OMIM: 191740) was performed using previously described primers [6] and a BigDye 3.0 kit (following the manufacturer’s protocol; Applied Biosystems, USA). The nomenclature of the identified variations was identified by using Mutalyzer (https://mutalyzer.nl/) and assessed sequentially against the Single Nucleotide Polymorphism Database (dbSNP; www.ncbi.nlm.nih.gov/SNP), Exome Aggregation Consortium (ExAC; exac.broadinstitute.org), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), and the UGT1A1 variant database [3]. A search was performed with MEDLINE if the variation was not found in any of the aforementioned databases. The biological significance of observed nucleotide changes located at splice sites was assessed using the Human Splicing Finder (HSF) 3.0 [7] and MutationTaster [8].
Four different variants in the UGT1A1 gene were identified in the patient: g.3664A > C (c.1352A > C, rs3755319); g.4963_4964TA[7] (c.-53_-52insTA, A(TA)7TAA, UGT1A1*28, rs8175347); g.5884G > T (c.864 + 5G > T, IVS1 + 5G > T); and g.11895_11898del (c.996+2_996+5del) (reference sequence NG_033238.1) .
In the ClinVar database, the variant g.4963_4964TA[7] is described as a variant affecting response to drug treatment. This is the most common variant identified in patients with GS. The variant g.3664A > C, as reported in the ClinVar database, causes transient familial neonatal hyperbilirubinemia (OMIM: 237900). The variant g.5884G > T located in the first intron has previously been reported in a patient with CNS-II [9], and from exome sequencing has been identified only in Europeans in five alleles (exac.broadinstitute.org). The second intronic variant g.11895_11898del (located in second intron two nucleotides after the second exon) is reported for the first time (sequence showed in the Fig. 1). Considering that one of the criteria for evaluating variant pathogenicity is its frequency in the healthy population, 90 healthy individuals from the Genome Database of the Latvian Population (LGDB), a government-funded biobank (the principle of LGDB has previously been reported [10]), were randomly selected and screened for the two lesser-described variants g.5884G > T and g.11895_11898del. The screened variants were not identified for any of the selected individuals. In order to identify variant segregation in the family, the parents were assessed for all the variants (Fig. 2). Both parents demonstrated normal-range levels of bilirubin from multiple measurements.
Fig. 1.

Electropherogram of the identified novel variant NG_033238.1: g.11895_11898del (c.996+2_996+5del) in case patient. In the first row showed reference sequence, in square given deleted nucleotides
Fig. 2.
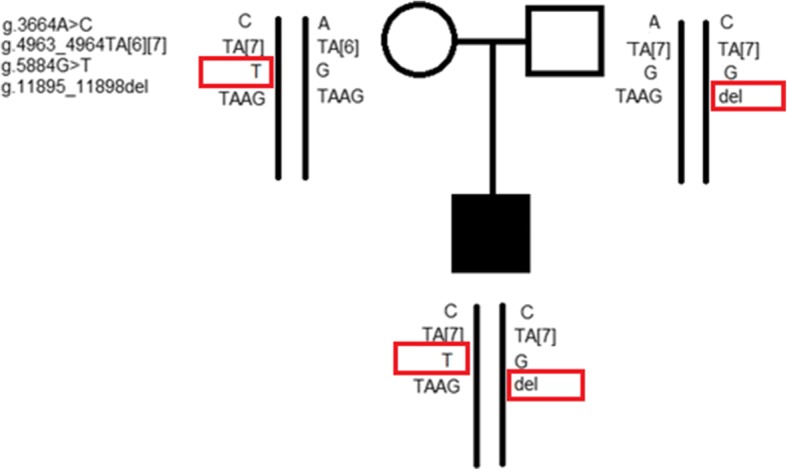
Pedigree of the family with Crigler-Najjar syndrome type II showing segregation of the identified genetic variants in the gene UGT1A1 (reference sequence NG_033238.1)
The intronic variants not mentioned in the ClinVar database were annotated according to ACMG standards and guidelines [11]. Both variants are classified as likely pathogenic (for variant g.5884G > T – fulfilled criteria PM2, PM3, PP3, PP4, PP5; for variant g.11895_11898del – fulfilled criteria PM2, PM3, PP3, PP4; Table 1).
Table 1.
Pathogenicity criteria for two variants not reported in the ClinVar database according to ACMG standards and guidelines [11]
| Evidence of pathogenicity | Fulfilled criteria with explanation | g.5884G > T | g.11895_11898del |
|---|---|---|---|
| Moderate evidence | PM2 – the prevalence of the variants in affected individuals is significantly increased compared with the prevalence in controls | 1) The variant was not found in 180 healthy Latvian control chromosomes 2) 0.00004119 (for Europeans in ExAC database) |
1) The variant was not found in 180 healthy Latvian control chromosomes 2) Not reporteda |
| PM3 – for recessive disorders, detected in trans with a pathogenic allele | Located in trans position with other allele (in our case likely pathogenic allele) | Located in trans position with other allele (in our case likely pathogenic allele) | |
| Supporting evidence | PP3 – multiple lines of computational evidence support a deleterious effect on the gene product | 1) HSF – predicted WT donor site broken ΔCT- -13.62%b (max entropy − 67.38%c) 2) Mutalyzer – variant located near to splice site 3) MutationTaster – disease causing (protein features (might be) affected; splice site change) |
1) HSF – predicted WT donor site broken ΔCT-55.3%b (max entropy − 284.9%c) 2) Mutalyzer - variant located in splice site 3) MutationTaster – disease causing (protein features (might be) affected; splice site change) |
| PP4 – patient’s phenotype or family history is highly specific for disease with single genetic etiology | Yes | Yes | |
| PP5 – reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation | Passuello et al., 2009 | None | |
| Total score | Likely pathogenic | Likely pathogenic |
aAs identified variant is indel frequency, should be evaluated with caution
b ΔCT – if difference between consensus value for wild type and mutated variantis below − 10% it is considered that splice site is broken [7]
cif maximal entropy is below − 30%, it is considered that splice site is broken [7]
Discussion and conclusions
Three inherited disorders leading to unconjugated hyperbilirubinemia have been identified. They are categorized depending on the bilirubin levels, phenobarbital responsiveness, and clinical picture.
We present here a 17-year-old male with CNS-II who has significant hyperbilirubinemia that decreases after phenobarbital intake and some UGT1A1 activity confirmed by glucuronide traces in his urine. He carries four genetic variants in the UGT1A1 gene; one reported for the first time. Phenobarbital responsiveness and the absence of kernicterus are described as criteria to discriminate CNS-I and CNS-II [1, 12]. However, atypical cases have also been reported. Duhamel et al. described a 25-year-old male with total bilirubin levels suggestive of CNS-I but without any neurological changes and phenobarbital unresponsiveness; no molecular investigations were carried out for that particular patient [13]. Chalasani et al. reported a CNS-II patient who developed kernicterus after laparoscopic surgery, leading to hematoma and resulting in a more significant increase in bilirubin production; the patient had three mutations: A(TA)7TAA in homozygous state and c.1391A > C in heterozygous state [14].
With regard to genotype/phenotype correlation in the case of variants of UGT1A1, Kadakol et al. proposed how to differentiate between CNS-I and CNS-II based upon molecular findings [15]. CNS-I results from genetic lesions that cause the premature truncation of UGT1A1, also caused by splice defects or substitution of critical amino acid residues, whereas CNS-II is caused by the substitution of one amino acid residue that markedly reduces without abolishing the catalytic activity of the enzyme [15]. Li et al. analyzed CNS-II patients in the Chinese population and discovered the existence of a genotype/phenotype correlation [16]. Patients homozygous for both variations G71R and Y486D had a bilirubin level > 200 μmol/L, but patients with total bilirubin levels ranging from 102.6 to 200 μmol/L carried a broader spectrum of UGT1A1 variants [16]. However, due to a different variant spectrum, these data are not attributable to Europeans.
Our described patient has four different molecular variants. This is the first reported case of four variants in one patient; previous reports have identified three variants in a single patient [17–19]. It is not clear how multiple variants in the UGT1A1 gene affect the clinical status. Kadakol et al. described four different gender and age families with identified multiple genetic variants in patients with unconjugated hyperbilirubinemia (> 100 μmol/L) and concluded based on the high frequency of UGT1A1 promoter variant A(TA)7TAA that further examination of the structural variants in the gene should be conducted for patients with hyperbilirubinemia as it could help find more severe phenotypes [15]. The variant A(TA)7TAA in homozygous state leads to 70% reduction in UGT1A1 expression compared to A(TA)6TAA carriers [20], and the presence of additional variants is likely to affect the enzyme activity even more [15].
Our patient has the rs3755319 variant that could explain his neonatal hyperbilirubinemia, the A(TA)7TAA variant that could contribute to his prolonged hyperbilirubinemia, and two other variants, one of which (c.864 + 5G > T) has been reported in a compound heterozygous state in pregnant women with variant c.1175C > T in patients with CNS-II [9]. The other variant (g.11895_11898del) is reported for the first time and according to the ACMG guidelines could be classified as a likely pathogenic variant (see Table 1). Unfortunately, no functional studies for the identified variants have been performed as UGT1A1 is expressed only in the liver [21] and there were no indications for a liver biopsy, no RNA sample was available and in vitro study as presented by Gupta et al. were not performed [22]. Splice site mutations in UGT1A1 were first reported in CNS-I [23], then reported in a subsequent mutation classification to be causing CNS-I [2]. However, although the total bilirubin levels of our patient in adolescence correspond to CNS-I, he is responsive to phenobarbital treatment and to date there are no signs of kernicterus, thus classifying as CNS-II. According to the ACMG guidelines, functional studies provide likely pathogenic criteria for the interpretation of the classification of unreported splice site variants. In the case of our patient, functional studies were not conducted. However, previous studies have shown that functional studies correlate well with pathogenicity prediction tools in case of UGT1A1 allelic variants [22], but to our knowledge there is no computational tool for analysing multiple allelic variant effect on one allele. In our study computation tools were used analysing effect of single allelic variant – here, MutationTaster and HSF predicted that the variation breaks the wild type splice site and as the variant is localized in the intron after exon 2, then it would affect all transcripts of the gene. Also based on family segregation analysis clinical symptoms could be caused by both variants g.11895_11898del and c.864 + 5G > T located in trans position, because for both parents one variant was identified but without remarkable changes in the bilirubin level.
The reported case demonstrates that it is not always possible to distinguish between CNS-I and CNS-II based on molecular findings and for patients with elevated unconjugated bilirubin level if confirming most common genotype for GS (homozygote for (A(TA)7TAA allele) should be performed full UGT1A1 sequencing if level of unconjugated bilirubin is atypically high Furthermore, wider opportunities present a greater challenge concerning interpretation, as encountered by our group when identifying an unreported variant.
Study limitation is missing functional studies of multiple variant effects on UGT1A1 expression level.
Acknowledgements
We acknowledge the Latvian Biomedical Research and Study Centre and the Genome Database of the Latvian Population for providing data and DNA samples.
Availability of data and materials
Available upon request (raw data of the sequencing results are available).
Abbreviations
- ACMG
American College of Medical Genetics
- CNS-I
Crigler-Najjar syndrome type I
- CNS-II
Crigler-Najjar syndrome type II
- CT
Consensus value
- dbSNP
Single Nucleotide Polymorphism Database
- ExAC
Exome Aggregation Consortium
- GS
Gilbert’s syndrome
- HSF
Human Splicing Finder
- LGDB
Genome Database of the Latvian Population
- MRI
Magnetic resonance imaging
- OMIM
Online Mendelian Inheritance in Man
- PM
Moderate evidence of pathogenicity
- PP
Supporting evidence of pathogenicity
- UGT1A1
UDP-glucuronosyltransferase 1A1
- WT
Wild type
Authors’ contributions
LG – wrote the manuscript, analyzed the molecular data. IP and GC – gastroenterologist and hepatologist, respectively, involved in the patient’s treatment process and management. DR – conducted the molecular study for the patient. MK – pediatrician involved in the patient’s enrolment in the study, provided the clinical description, and designed the investigation’s structure. All authors have read and approved the final version of manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from all involved individuals. The study was performed according to the Declaration of Helsinki and the protocol was approved by the Central Medical Ethics Committee of Latvia.
Consent for publication
The patient and both of his parents have given written informed consent for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Linda Gailite, Email: linda.gailite@rsu.lv.
Dmitrijs Rots, Email: dmitrijs.rots@rsu.lv.
Ieva Pukite, Email: ieva.pukite@rsu.lv.
Gunta Cernevska, Email: lnsr@latnet.lv.
Madara Kreile, Email: madara.kreile@rsu.lv.
References
- 1.Arias IM, Gartner LM, Cohen M, Ezzer JB, Levi AJ. Chronic nonhemolytic unconjugated hyperbilirubinemia with glucuronyl transferase deficiency. Clinical, biochemical, pharmacologic and genetic evidence for heterogeneity. Am J Med. 1969;47(3):395–409. doi: 10.1016/0002-9343(69)90224-1. [DOI] [PubMed] [Google Scholar]
- 2.Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat. 2000;16(4):297–306. doi: 10.1002/1098-1004(200010)16:4<297::AID-HUMU2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 3.Canu G, Minucci A, Zuppi C, Capoluongo E. Gilbert and Crigler Najjar syndromes: an update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells Mol Dis. 2013;50(4):273–280. doi: 10.1016/j.bcmd.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Maruo Y, Nakahara S, Yanagi T, Nomura A, Mimura Y, Matsui K, Sato H, Takeuchi Y. Genotype of UGT1A1 and phenotype correlation between Crigler-Najjar syndrome type II and Gilbert syndrome. J Gastroenterol Hepatol. 2016;31(2):403–408. doi: 10.1111/jgh.13071. [DOI] [PubMed] [Google Scholar]
- 5.Lin JP, O'Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, Yang S, Kronenberg F. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham heart study. Circulation. 2006;114(14):1476–1481. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 6.Costa E, Vieira E, Martins M, Saraiva J, Cancela E, Costa M, Bauerle R, Freitas T, Carvalho JR, Santos-Silva E, et al. Analysis of the UDP-glucuronosyltransferase gene in Portuguese patients with a clinical diagnosis of Gilbert and Crigler-Najjar syndromes. Blood Cells Mol Dis. 2006;36(1):91–97. doi: 10.1016/j.bcmd.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 9.Passuello V, Puhl AG, Wirth S, Steiner E, Skala C, Koelbl H, Kohlschmidt N. Pregnancy outcome in maternal Crigler-Najjar syndrome type II: a case report and systematic review of the literature. Fetal Diagn Ther. 2009;26(3):121–126. doi: 10.1159/000238122. [DOI] [PubMed] [Google Scholar]
- 10.Ignatovica V, Latkovskis G, Peculis R, Megnis K, Schioth HB, Vaivade I, Fridmanis D, Pirags V, Erglis A, Klovins J. Single nucleotide polymorphisms of the purinergic 1 receptor are not associated with myocardial infarction in a Latvian population. Mol Biol Rep. 2012;39(2):1917–1925. doi: 10.1007/s11033-011-0938-4. [DOI] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seppen J, Bosma PJ, Goldhoorn BG, Bakker CT, Chowdhury JR, Chowdhury NR, Jansen PL, Oude Elferink RP. Discrimination between Crigler-Najjar type I and II by expression of mutant bilirubin uridine diphosphate-glucuronosyltransferase. J Clin Invest. 1994;94(6):2385–2391. doi: 10.1172/JCI117604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duhamel G, Blanckaert N, Metreau JM, Preaux AM, Bouvry M, Fevery J, Berthelot P. An unusual case of Crigler-Najjar disease in the adult. Classification into types I and II revisited. J Hepatol. 1985;1(1):47–53. doi: 10.1016/S0168-8278(85)80067-2. [DOI] [PubMed] [Google Scholar]
- 14.Chalasani N, Chowdhury NR, Chowdhury JR, Boyer TD. Kernicterus in an adult who is heterozygous for Crigler-Najjar syndrome and homozygous for Gilbert-type genetic defect. Gastroenterology. 1997;112(6):2099–2103. doi: 10.1053/gast.1997.v112.pm9178703. [DOI] [PubMed] [Google Scholar]
- 15.Kadakol A, Sappal BS, Ghosh SS, Lowenheim M, Chowdhury A, Chowdhury S, Santra A, Arias IM, Chowdhury JR, Chowdhury NR. Interaction of coding region mutations and the Gilbert-type promoter abnormality of the UGT1A1 gene causes moderate degrees of unconjugated hyperbilirubinaemia and may lead to neonatal kernicterus. J Med Genet. 2001;38(4):244–249. doi: 10.1136/jmg.38.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Deng G, Tang Y, Mao Q. Spectrum of UGT1A1 variations in Chinese patients with Crigler-Najjar syndrome type II. PLoS One. 2015;10(5):e0126263. doi: 10.1371/journal.pone.0126263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tesapirat L, Nilyanimit P, Wanlapakorn N, Poovorawan Y. Compound heterozygosity of a novel exon 3 frameshift (p.R357P fs*24) mutation and Y486D mutation in exon 5 of the UGT1A1 gene in a Thai infant with Crigler-Najjar syndrome type 2. Genet Mol Res. 2015;14(2):3293–3299. doi: 10.4238/2015.April.13.8. [DOI] [PubMed] [Google Scholar]
- 18.D'Angelo R, Rinaldi C, Donato L, Nicocia G, Sidoti A. The combination of new missense mutation with [a(TA)7TAA] dinucleotide repeat in UGT1A1 gene promoter causes Gilbert's syndrome. Ann Clin Lab Sci. 2015;45(2):202–205. [PubMed] [Google Scholar]
- 19.Zheng B, Hu G, Yu J, Liu Z. Crigler-Najjar syndrome type II in a Chinese boy resulting from three mutations in the bilirubin uridine 5′-diphosphate-glucuronosyltransferase (UGT1A1) gene and a family genetic analysis. BMC Pediatr. 2014;14:267. doi: 10.1186/1471-2431-14-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosma PJ, Seppen J, Goldhoorn B, Bakker C, Oude Elferink RP, Chowdhury JR, Chowdhury NR, Jansen PL. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 1994;269(27):17960–17964. [PubMed] [Google Scholar]
- 21.Ritter JK, Kessler FK, Thompson MT, Grove AD, Auyeung DJ, Fisher RA. Expression and inducibility of the human bilirubin UDP-glucuronosyltransferase UGT1A1 in liver and cultured primary hepatocytes: evidence for both genetic and environmental influences. Hepatology. 1999;30(2):476–484. doi: 10.1002/hep.510300205. [DOI] [PubMed] [Google Scholar]
- 22.Gupta N, Benjamin M, Kar A, Munjal SD, Sarangi AN, Dalal A, Aggarwal R. Identification of promotor and Exonic variations, and functional characterization of a splice site mutation in Indian patients with unconjugated hyperbilirubinemia. PLoS One. 2015;10(12):e0145967. doi: 10.1371/journal.pone.0145967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gantla S, Bakker CT, Deocharan B, Thummala NR, Zweiner J, Sinaasappel M, Roy Chowdhury J, Bosma PJ, Roy Chowdhury N. Splice-site mutations: a novel genetic mechanism of Crigler-Najjar syndrome type 1. Am J Hum Genet. 1998;62(3):585–592. doi: 10.1086/301756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Available upon request (raw data of the sequencing results are available).