

 This work is licensed under a
This work is licensed under a Summary
Mitochondrial diseases are rare, heterogeneous conditions affecting organs dependent on high aerobic metabolism. Presenting symptoms and signs vary depending on the mutation and mutant protein load. Diabetes mellitus is the most common endocrinopathy, and recognition of these patients is important due to its impact on management and screening of family members. In particular, glycemic management differs in these patients: the use of metformin is avoided because of the risk of lactic acidosis. We describe a patient who presented with gradual weight loss and an acute presentation of hyperglycemia complicated by the superior mesenteric artery syndrome. His maternal history of diabetes and deafness and a personal history of hearing impairment led to the diagnosis of a mitochondrial disorder.
Learning points:
The constellation of diabetes, multi-organ involvement and maternal inheritance should prompt consideration of a mitochondrial disorder.
Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) and maternally inherited diabetes and deafness (MIDD) are the most common mitochondrial diabetes disorders caused by a mutation in m.3243A>G in 80% of cases.
Metformin should be avoided due to the risk of lactic acidosis.
There is more rapid progression to insulin therapy and higher prevalence of diabetic complications compared to type 2 diabetes.
Diagnosis of a mitochondrial disorder leads to family screening, education and surveillance for future complications.
Superior mesenteric artery syndrome, an uncommon but important cause of intestinal pseudo-obstruction in cases of significant weight loss, has been reported in MELAS patients.
Background
Mitochondrial disease occurs when there are mutations in either the mitochondrial or nuclear DNA. Mitochondrial DNA mutations are more common, occurring in 20 per 100 000 adults (1) and are transmitted through maternal inheritance. Nuclear DNA mutations are less common (2.9:100 000) and are inherited in an autosomal dominant or recessive pattern. Clinical symptoms correlate poorly with mutation load in the blood (2). Classification of mitochondrial diseases is dependent on the mutation and organ involved. This heterogeneity makes it challenging to diagnose mitochondrial diseases.
Diabetes mellitus is the most common endocrinopathy reported in mitochondrial diseases, with the highest prevalence detected in those with the m.3243A>G mutation (3). Metformin, usually considered first-line oral hypoglycemic therapy, is avoided due to the risk of lactic acidosis.
Case presentation
A 31-year-old white male information technology worker presented to our emergency department with a 2-day history of vomiting and abdominal pain, on the background of a 4-month history of polydipsia, polyuria and 10 kg loss of body weight.
On presentation, his BMI was 15 kg/m2 (43 kg, height 175 cm), blood pressure 117/85 mmHg without a postural drop. He had a sinus tachycardia with a pulse rate of 136 beats per minute, marfanoid facies and pectus excavatum. Abdomen was soft to palpation. There was bilateral horizontal nystagmus without diplopia, and reduced eye abduction, left more than right.
Past history was significant for birth prematurity at 28 weeks, childhood hearing impairment, migraine headaches, retinopathy of prematurity, right retinal detachment, cataracts and bilateral rectus muscle weakness. There was no history of stroke-like episodes or epilepsy. He had normal developmental milestones.
Family history was notable in that his mother had insulin-requiring diabetes, deafness and chronic kidney disease (Fig. 1). She died at age 54 years from a stroke. His father passed away from mesothelioma. His only sibling was a brother who was stillborn. He recalled two maternal aunts had dementia and his maternal grandmother had diabetes. He was on no medications prior to admission.
Figure 1.
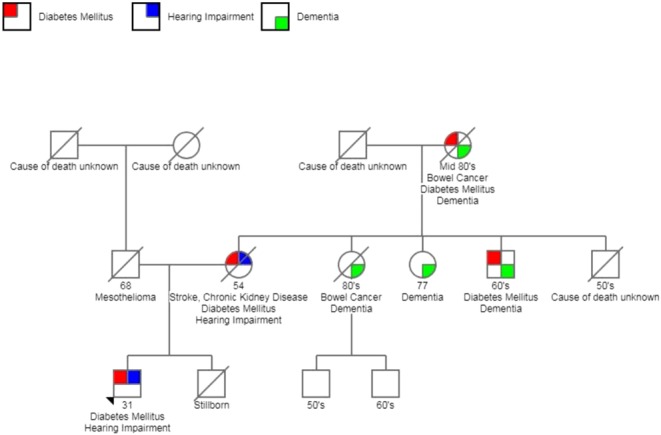
Family pedigree.
Investigations
His pathology is summarized in Table 1.
Table 1.
Investigation results.
| Investigation (units) | Admission | 1-year follow up | Reference values |
| Blood glucose level (mmol/L) | 32 | 3.9–7 | |
| Venous blood gas | |||
| pH | 7.34 | 7.35–7.45 | |
| PCo2 (mmHg) | 54 | 41–51 | |
| Bicarbonate (mmol/L) | 29 | 21–28 | |
| Base excess (mmol/L) | 1.7 | −3.0 to 3.0 | |
| Lactate (mmol/L) | 8.1 | 1.4–2.5 | 0.5–2.2 |
| Biochemistry | |||
| Sodium (mmol/L) | 134 | 142 | 135–145 |
| Potassium (mmol/L) | 4.3 | 5.4 | 3.5–5.2 |
| Chloride (mmol/L) | 75 | 101 | 95–110 |
| Urea (mmol/L) | 16.4 | 9.7 | 2.8–8.1 |
| Creatinine (µmol/L) | 99 | 86 | 60–110 |
| Estimated glomerular filtration rate | 87 | >90 | >90 |
| Urine albumin creatinine ratio (mg/mmol) | 1.5 | 1.2 | <2.5 |
| Liver function test | |||
| Bilirubin (µmol/L) | 15 | <22 | |
| Alanine aminotransferase (IU/L) | 22 | 5–40 | |
| Gamma glutamyl transferase (IU/L) | 24 | 10–71 | |
| Alkaline phosphatase (IU/L) | 51 | 30–110 | |
| Albumin (g/L) | 53 | 34–47 | |
| Lipase (IU/L) | 22 | 13–60 | |
| Hemoglobin A1c (%) | 13.9 | 7.1 | 4.0–6.0 |
| Islet-cell antibody (units/mL) | <0.3 | <15 | |
| Glutamic acid decarboxylase antibody (units/mL) | <0.6 | <5.0 |
Brain MRI showed global cerebral atrophy and small vessel ischemic changes.
Treatment
An insulin infusion was commenced resulting in normalization of glucose, beta hydroxy-butyrate, lactate levels and improved renal function. His vomiting however persisted. CT of the abdomen (Figs 2 and 3) showed gross gastric dilatation secondary to pseudo-obstruction caused by superior mesenteric artery syndrome. A nasogastric tube was inserted, and supplemental parenteral nutrition was commenced.
Figure 2.
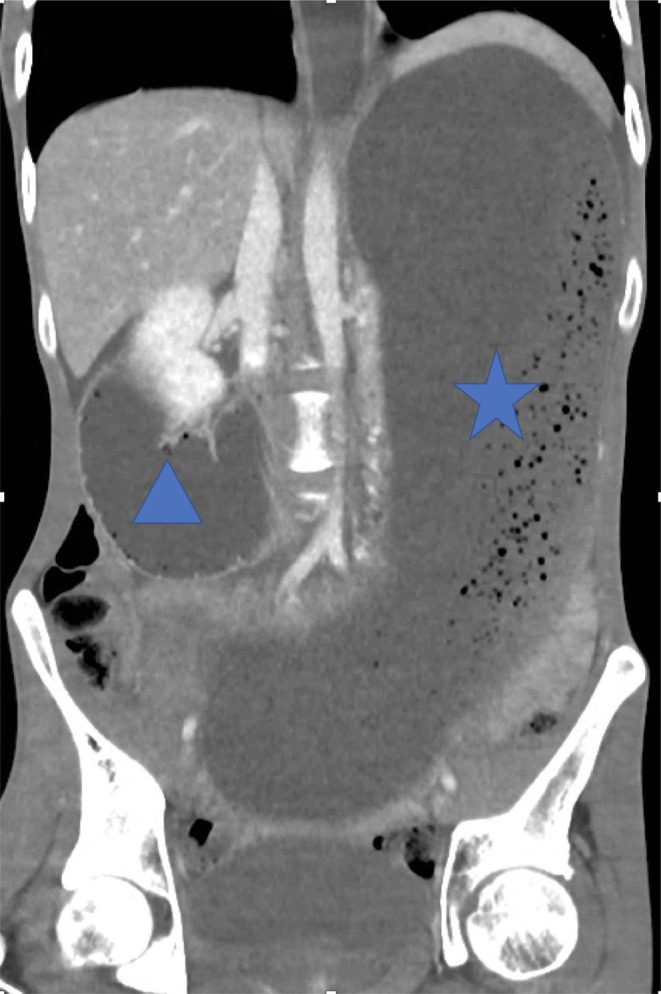
Coronal image of CT abdomen/pelvis demonstrates gross dilatation of stomach (star) and proximal duodenum (triangle).
Figure 3.
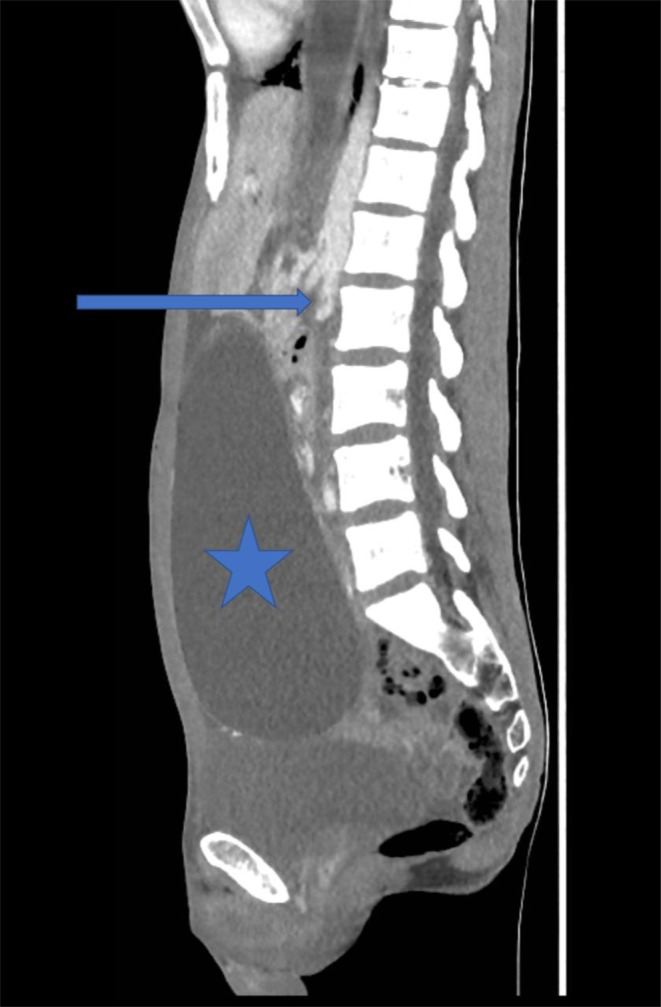
Sagittal image of CT abdomen/pelvis demonstrates narrowing of the aorto-mesenteric angle (arrow) and massive gastric distention due to duodenal compression (star).
Outcome and follow-up
Our patient improved with conservative management, and he was transferred to a rehabilitation facility Day 14 of hospital admission. He returned home 2 weeks later. In view of his maternal history, elevated serum lactate, negative glutamic acid decarboxylase and islet-cell antibodies, a mitochondrial disorder was considered.
Peripheral blood analysis through PCR amplification identified a mutation in the mitochondrial DNA, characterized by replacement of adenosine by guanine at location 3243 (m.3243A>G) of the mitochondrial encoded gene affecting transfer RNA leucine 1 (MTTL1). Mutant load was 30%, and a diagnosis of an overlap between MELAS and MIDD was made.
Currently our patient remains insulinopenic – C-peptide 0.1 nmol/L (0.4–1.5 nmol/L). His insulin regimen comprises glargine 16 units daily and aspart four units with dinner. His repeat serum lactate levels fluctuate between normal range to mild elevation, 1.4–2.5 mmol/L (0.5–2 mmol/L). A trial of citrulline and folinic acid resulted in improvement in energy levels and appetite, and he has gained 10 kg in weight over the past 2 years. During follow-up, he also reported symptoms suggestive of peripheral neuropathy. This has since improved with commencement of pregabalin.
Discussion
Mitochondrial diseases are rare, heterogeneous conditions caused by mutations in either the mitochondrial or nuclear DNA and can present with a variety of clinical symptoms. Mitochondrial mutation is more common and is transmitted through maternal inheritance. Mitochondrial diabetes most frequently results from the m.3243A>G mutation, responsible for both MELAS and MIDD.
Clinical features that raise suspicion for mitochondrial diabetes include multi-organ involvement, elevated serum lactate levels, a more rapid progression to insulin therapy and the earlier onset of diabetes-related complications compared to individuals with type 2 diabetes. The inability of dysfunctional mitochondria to produce sufficient ATP results in multi-organ defects and affects predominantly organs with high energy requirements such as the central nervous system, muscle, retina, kidney and pancreas.
The main clinical characteristics of MELAS are stroke-like episodes below the age of 40 years, encephalopathy defined by seizures and/or dementia, myopathy with/without red ragged fibers, recurrent headaches and vomiting. Lactic acidosis occurs in up to 94% of MELAS patients due to impaired glucose oxidation with subsequent accumulation of pyruvate and shunting to lactate.
MIDD is characterized by diabetes and sensorineural deafness in the individual and maternal relatives. Other features include proximal myopathy, macular dystrophy and renal focal segmental glomerular sclerosis (4). The classic ophthalmic finding in these individuals is pigmentary retinal dystrophy rather than the diabetic retinopathy of type 2 diabetes (4, 5). These features can also be found in MELAS (6).
Diabetes in mitochondrial disease can present insidiously and is usually diagnosed in the late fourth decade (7). This is thought to be primarily due to insulin deficiency secondary to beta-cell dysfunction, although insulin resistance may also occur. Twenty percent present acutely with hyperglycemia as in our patient and 8% present with diabetic ketoacidosis. Within 2–4 years, 45.2% require insulin (7). Other endocrinopathies reported in patients with mitochondrial disease include hypogonadism, adrenal dysfunction, hypoparathyroidism and growth hormone deficiency (8, 9).
Our patient had elevated lactate levels at presentation and intermittently when he was well. This feature combined with ischemic changes on brain MRI and confirmed genetic mutation supports the diagnosis of MELAS (6). His retinopathy was thought to be related to prematurity and he did not have the pigmentary retinopathy typical of MIDD (5, 10). However, his maternal history of diabetes and deafness strongly suggested MIDD. Therefore, our patient was considered to have an overlap of MELAS and MIDD.
The mutant mitochondrial load in our patient’s peripheral blood analysis was 30%. In general, there is weak correlation between mutant load measured in the blood and the severity of clinical features (2). Urine epithelial cell sediment analysis for mutated mitochondrial DNA genome is useful and may be a better alternative. Where the diagnosis cannot be confirmed by DNA testing, tissue biopsy typically from the vastus lateralis muscle is considered the gold standard (11). A muscle biopsy was not performed in our patient as he remained clinically stable. Furthermore, his phenotypic features and DNA result were considered sufficient to support the diagnosis of a mitochondrial disorder (12).
The other prominent clinical feature in our patient was vomiting due to the superior mesentery artery syndrome (SMAS). Gastrointestinal complaints are common in mitochondrial disorders, estimated to affect 50% of patients. Symptoms include anorexia, constipation, dysphagia, gastroparesis and vomiting (13). The etiology remains unclear but may involve loss of the intestinal cells of Cajal and marked atrophy of the muscularis propia. SMAS is an uncommon cause of small bowel obstruction that has been reported in MELAS patients (14). The 3rd part of the duodenum is usually located between the abdominal aorta and superior mesenteric artery at its origin. The presence of the retroperitoneal fat pad in this region protects the duodenum from being compressed, with the usual aorto-mesenteric angle at 25–60°. However, factors such as significant weight loss, correction of scoliosis or esophagectomy can lead to loss of the retroperitoneal fat pad, resulting in narrowing of the aorto-mesenteric angle and compression of the duodenum. Case reports of SMAS and weight loss averaged between 10 and 12 kg, similar to our patient. Management of SMAS is largely conservative with gastric decompression, although duodeno-jejunostomy may be required if there is no improvement.
Management of patients with MELAS and MIDD is predominantly supportive. Low levels of nitric oxide have been detected in MELAS patients, and nitric oxide precursors such as arginine and citrulline have been observed to reduce lactate levels. The use of intravenous arginine was associated with a reduction in severity and frequency of stroke-like episodes (15). The use of citrulline in mitochondrial disease has not been studied and is the subject of a current trial (16). Folinic acid can correct cerebral folate deficiency and has been shown to reverse leukoencephalopathy and improve cerebral hypomyelination, ataxia and hypotonia; however, results are mixed and further research is required. Females with mitochondrial disease may avoid transmitting the defect by utilizing ‘three parent IVF’ with the father’s sperm combined with mitochondria and cytoplasm from a donated healthy ovum and nuclear DNA from the mother.
In summary, mitochondrial diseases are rare but important conditions to recognize and diagnose due to their impact on glycemic management, screening of family members and surveillance of complications.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The primary author is supported by the Eastern Health Foundation Grant; funds however were not utilized in the preparation of this report.
Patient consent
Written informed consent for publication of their clinical details and clinical images was obtained from the patient.
Author contribution statement
M L Yee: Literature review, report draft, registrar involved in patient’s care. R Wong: Senior staff who provided supervision, assistance and amendments. M Datta: Radiology Head of Department who assisted with reporting of radiology films. T N Fazio: Metabolic Diseases Unit Physician involved in patient’s care, review of manuscript. M M Ebrahim: Assistance with literature review, registrar involved in patient’s care. E C McNamara: Attending registrar during the patient’s acute hospital presentation. G De Jong: Metabolic Diseases Unit Head of Department involved in patient’s care. C Gilfillan: Head of Department and attending senior medical staff involved in the patient’s care.
References
- 1.Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of Neurology 2015. 77 753–759. ( 10.1002/ana.24362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehrazin M, Shanske S, Kaufmann P, Wei Y, Coku J, Engelstad K, Naini A, De Vivo DC, DiMauro S. Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS. American Journal of Medical Genetics: Part A 2009. 149A 584–587. ( 10.1002/ajmg.a.32703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whittaker RG, Schaefer AM, McFarland R, Taylor RW, Walker M, Turnbull DM. Prevalence and progression of diabetes in mitochondrial disease. Diabetologia 2007. 50 2085–2089. ( 10.1007/s00125-007-0779-9) [DOI] [PubMed] [Google Scholar]
- 4.Murphy R, Turnbull DM, Walker M, Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabetic Medicine 2008. 25 383–399. ( 10.1111/j.1464-5491.2008.02359.x) [DOI] [PubMed] [Google Scholar]
- 5.Naing A, Kenchaiah M, Krishnan B, Mir F, Charnley A, Egan C, Bano G. Maternally inherited diabetes and deafness (MIDD): diagnosis and management. Journal of Diabetes and its Complications 2014. 28 542–546. ( 10.1016/j.jdiacomp.2014.03.006) [DOI] [PubMed] [Google Scholar]
- 6.El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Molecular Genetics and Metabolism 2015. 116 4–12. ( 10.1016/j.ymgme.2015.06.004) [DOI] [PubMed] [Google Scholar]
- 7.Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatric Diabetes 2015. 16 1–9. ( 10.1111/pedi.12223) [DOI] [PubMed] [Google Scholar]
- 8.Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Molecular and Cellular Endocrinology 2013. 379 2–11. ( 10.1016/j.mce.2013.06.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction. Nature Reviews Endocrinology 2017. 13 92–104. ( 10.1038/nrendo.2016.151) [DOI] [PubMed] [Google Scholar]
- 10.Chinnery PF. Mitochondrial disorders overview In Gene Reviews (R). Eds Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K. & Amemiya A. University of Washington: Seattle, WA, 1993. [PubMed] [Google Scholar]
- 11.Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genetics in Medicine 2015. 17 689–701. ( 10.1038/gim.2014.177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wortmann SB, Mayr JA, Nuoffer JM, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics 2017. 48 309–314. ( 10.1055/s-0037-1603776) [DOI] [PubMed] [Google Scholar]
- 13.Finsterer J, Frank M. Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therapeutic Advances in Gastroenterology 2017. 10 142–154. ( 10.1177/1756283X16666806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon OY, Lim SG, Park SH. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode leading to recurrent superior mesenteric artery syndrome. American Journal of Emergency Medicine 2014. 32 951.e951–951.e952. ( 10.1016/j.ajem.2014.01.059) [DOI] [PubMed] [Google Scholar]
- 15.Koenig MK, Emrick L, Karaa A, Korson M, Scaglia F, Parikh S, Goldstein A. Recommendations for the management of strokelike episodes in patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes. JAMA Neurology 2016. 73 591–594. ( 10.1001/jamaneurol.2015.5072) [DOI] [PubMed] [Google Scholar]
- 16.El-Hattab AW, Zarante AM, Almannai M, Scaglia F. Therapies for mitochondrial diseases and current clinical trials. Molecular Genetics and Metabolism 2017. 122 1–9. ( 10.1016/j.ymgme.2017.09.009) [DOI] [PMC free article] [PubMed] [Google Scholar]