

ABSTRACT
The dismal prognosis of glioblastoma is attributed in part to the existence of stem-like brain tumor-initiating cells (BTICs) that are highly radio- and chemo-resistant. New approaches such as therapies that reprogram compromised immune cells against BTICs are needed. Effective immunotherapies in glioblastoma, however, remain elusive unless the mechanisms of immunosuppression by the tumor are better understood. Here, we describe that while the conditioned media of activated T lymphocytes reduce the growth capacity of BTICs, this growth suppression was abrogated in live co-culture of BTICs with T cells. We present evidence that BTICs produce the extracellular matrix protein tenascin-C (TNC) to inhibit T cell activity in live co-culture. In human glioblastoma brain specimens, TNC was widely deposited in the vicinity of T cells. Mechanistically, TNC inhibited T cell proliferation through interaction with α5β1 and αvβ6 integrins on T lymphocytes associated with reduced mTOR signaling. Strikingly, TNC was exported out of BTICs associated with exosomes, and TNC-depleted exosomes suppressed T cell responses to a significantly lesser extent than control. Finally, we found that circulating exosomes from glioblastoma patients contained more TNC and T cell-suppressive activity than those from control individuals. Taken together, our study establishes a novel immunosuppressive role for TNC associated with BTIC-secreted exosomes to affect local and distal T lymphocyte immunity.
Keywords: cancer stem cells, glioma, adaptive immunity, extracellular matrix, exosome
Introduction
Glioblastomas are devastating brain tumors that arise in the adult central nervous system (CNS). Despite aggressive surgery, chemotherapy and radiotherapy, glioblastomas have one of the lowest median survival rates of all human cancers, with a median survival of 14.6 months.1 This dismal prognosis is contributed by stem-like cells termed brain tumor-initiating cells (BTICs), a rare population in the glioblastoma microenvironment that is crucial in maintaining tumor growth and recurrence.2 BTICs are more resistant to chemotherapy and radiotherapy in contrast to their more differentiated progenies.3,4 This resistance to current treatment modalities emphasizes the need for new approaches, particularly immunotherapy that reprograms the compromised immune cells of patients against BTICs.
Despite major advances in cancer immunotherapy, clinical efficacy is significantly attenuated by immunosuppressive mechanisms exerted by the tumor. The major immune suppressive cell types in the glioblastoma milieu that have been co-opted to promote tumor health are regulatory T cells, tumor-associated macrophages, and myeloid-derived suppressor cells.5,6 Soluble molecules made by these cells, and also by tumor cells, to suppress tumor killing responses include interleukin (IL)-10, transforming growth factor-β, prostaglandin E2, and indolamine 2,3-dioxygenase 1-generated kynurenine.7 The challenges in overcoming immunosuppression, however, are not exclusive to the CNS tumor microenvironment, as several inhibitory factors exist outside of the brain. As a manifestation, circulating T cells from glioblastoma patients are defective in their effector activity,8 and they exhibit lower levels of tyrosine phosphorylation of signaling molecules compared to controls.9 These distal effects of systemic immunosuppression are likely mediated by inhibitory factors secreted into the circulation by glioblastoma cells. Overcoming these factors is of significant interest in order to foster an effective anti-tumor T cell response.
Extracellular matrix (ECM) proteins are a key component of a variety of factors secreted by glioblastoma cells. There are several reports of the elevation of ECM proteins in the serum of glioblastoma patients, with an association with poorer prognosis.10 While the roles of ECM proteins in modulating glioma cell phenotype and cancer progression have been widely investigated,10 their potential roles in immune suppressive characteristics in glioblastomas remain largely undefined. The composition of ECM in glioblastomas is unique and includes hyaluronan, osteopontin, collagen-I and -IV, vitronectin, and tenascin-C (TNC).11 Prior evidence suggests that one of the most important ECM proteins in glioblastoma is (TNC), as it is abundantly expressed in the tumor microenvironment.12,13 TNC has been found to be advantageous for cancer cells, and its level in brain specimens is associated with increased glioma grade and poorer survival.14,15 TNC also plays a critical role in maintaining the stemness of cancer stem cells.16,17 We reported that TNC is produced by BTICs to promote their growth.12 In addition, there are reports of the capacity of TNC to inhibit T cell activation,18,19 and TNC overexpression in prostate cancer is a major mechanism by which prostate cancer stem cells inhibit T cell responses.20 A single study has shown an inhibitory role of TNC on T cell migration in glioblastoma.21 However, the effects of TNC on other critical aspects of T cell activation, in addition to its potential role in BTIC-mediated immunosuppression, remain unknown.
TNC is elevated in the serum of cancer patients 22,23 and likely has the potential to inhibit systemic immunity. Proteins with a molecular weight under 400–500 Daltons cross the blood-brain barrier (BBB),24 so whether TNC, a hexameric macromolecule consisting of subunits of ~180 to ~300kDa,25 crosses the BBB from the glioblastoma tumor to alter systemic immunity remains unclear. One plausible mechanism by which large extracellular matrix molecules may exit the CNS parenchyma to affect system immunity is by transport in extracellular vesicles. Extracellular vesicles including exosomes mediate cell-cell communication through transportation of proteins, mRNAs, and microRNAs.26 Moreover, cancer-derived exosomes are known to trigger the destruction of the BBB.27,28It has also been documented that glioblastoma cells release exosomes into their local microenvironment to promote their proliferation and to suppress immune responses.29,30 Based on these observations, we tested the hypothesis that BTIC-derived exosomes carry TNC into the circulation, providing the means by which TNC can suppress systemic T cell responses.
Results
Failure of syngeneic murine T cells to inhibit sphere forming capacity of BTICs in live co-cultures
To determine whether T cells can inhibit the sphere forming capacity of BTICs in live co-culture, we used two mouse BTIC lines from the C57BL/6 background, previously generated and propagated as stem-like cells.31 Purified T cells from C57BL/6 mouse spleen were activated with anti-CD3 and anti-CD28 antibodies for 2 days, or were left unactivated in NeuroCult™ Basal Medium. In order to confirm that T cells are properly activated in this medium, we determined T cell proliferation and effector functions in NeuroCult compared to RPMI-1640 medium. We found similar T cell proliferation in NeuroCult and RPMI-1640 medium (Supplementary Figure 1a). Moreover, there was not significant difference of IFN-γ and IL-2 production between T cells activated in NeuroCult and RPMI-1640 medium (Supplementary Figure 1b-g). T cells were then collected and co-cultured with BTICs for 3 days (Figure 1a). We found that increasing numbers of activated or non-activated T cells in co-culture with BTICs were unable to reduce the sphere forming capacity of BTICs (Figure 1b, c). In contrast, conditioned medium (CM) from syngeneic T cells activated in isolation inhibited BTIC growth (Figure 1d, e). To identify putative mediators responsible for the inhibition of BTIC growth, we measured TNF-α and IFN-γ concentrations in CM of activated T cells co-cultured with BTICs; we and others have previously shown that TNF-α and IFN-γ reduced tumor cell growth in glioblastomas.32,33 We found that the levels of these cytokines were downregulated in T cells co-cultured with BTICs compared to T cells activated in isolation (Figure 1f). These results suggest that BTICs in co-culture with T cells were producing inhibitory molecules that suppressed T cell effector functions.
Figure 1.
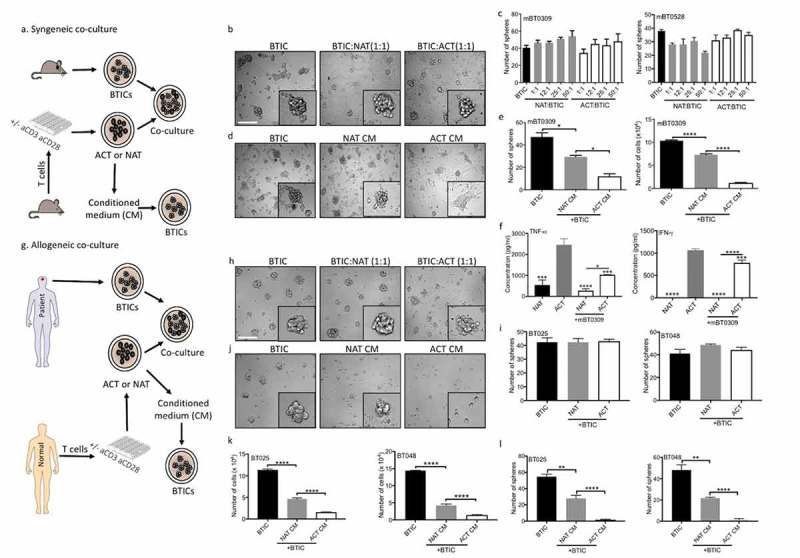
Live T cells are inefficient at reducing sphere forming capacity of BTICs in contrast to their conditioned media. (a) T cells were purified from mouse spleen and activated (ACT) with anti-CD3 and anti-CD28 antibodies, or were left non-activated (NAT) for 2 days. The conditioned medium (CM) and live cells were collected and exposed to syngeneic mouse BTICs. The resultant number of spheres above the 60 µm diameter cutoff was monitored after 3 days by photographing multiple fields per well with subsequent analyses. (b) Outcome images of spheres from mBT0309 co-cultured with T cells at ratio 1:1. (c) Increasing numbers of T cells in co-culture with two mouse BTIC lines were unable to reduce the sphere forming capacity of BTICs. (d) In contrast to live T cells, their CM reduced the sphere forming capacity of mBT0309. (e) T cell CM reduced the number of spheres and the total number of cells. (f) CM were taken after 48 h from ACT and NAT in the presence or absence of mouse BT0309 at ratio 10:1 (T cell:BTIC) to determine the concentration of TNF-α and IFN-γ. (g) T cells were isolated from normal individuals and activated (ACT) with anti-CD3 and anti-CD28 antibodies or were left non-activated (NAT) for 2 days. Cells and CM were collected and co-incubated with human BTIC lines for 3 days. (h) Images of 72h outcomes of spheres from BT025 co-cultured with T cells at ratio 1:1. (i) Activated T cells were inefficient to reduce sphere forming capacity of BTICs. (j) Images of spheres from BT025 co-incubated with T cell CM. (k, l) Conditioned medium from activated T cells significantly reduced the number of spheres and the total number of cells compared to non-activated T cell CM. All bars are mean ± SEM of triplicate cultures. Data in all panels are analyzed by 1-way ANOVA with Tukey’s multiple comparisons post-hoc test. *P < 0.05, **P < 10–2, ****P < 10–4 compared to ACT group unless otherwise displayed. BTIC, Brain tumor initiating cell; ACT, activated T cell; NAT, non-activated T cell; CM, Conditioned medium. Scale bar, 60 μm.
Alloreactive human T cells are inefficient in reducing BTIC growth compared to their conditioned medium
BTICs in culture express MHC-I (Supplementary Figure 2). Since alloreactive T cells would recognize MHC molecules expressed on BTICs, here, we used T cells isolated from healthy individuals to address their capacity to reduce allogeneic human BTIC sphere formation (Figure 1g). Activated or non-activated T cells in co-culture with allogeneic BTICs did not affect the sphere-forming capacity of BTICs (Figure 1h, i). In contrast, a sphere-and cell number-reducing effect was observed for CM of T cells activated in isolation (Figure 1j-l). Taken together, and corroborating the mouse data, human BTICs in co-culture produce factors that attenuate the tumor suppressive activity of T cells.
Inhibitory effects of BTIC conditioned medium on T cells
The inhibitory effects of differentiated glioma cells on T cells have been studied,34,35 but the suppressive mechanisms of BTICs remain to be addressed. We determined that inhibitory component(s) were secreted by BTICs since human T cells that were activated in the presence of BTIC CM were significantly reduced in their proliferative response (Figure 2a) and their expression level of CD69 (Figure 2b, c), one of the earliest T cell activation markers. In correspondence, the percentage of T cells expressing IFN-γ and IL-2 was significantly lowered by BTIC CM (Figure 2d, e). Moreover, flow cytometry analyses showed a reduction in the MFI (mean fluorescence intensity) of T cells positive for IFN-γ (Figure 2f), although the MFI of IL-2 positive T cells was not altered (Figure 2g). BTICs are, therefore, able to attenuate T cell activation without the requirement of cell-to-cell contact.
Figure 2.
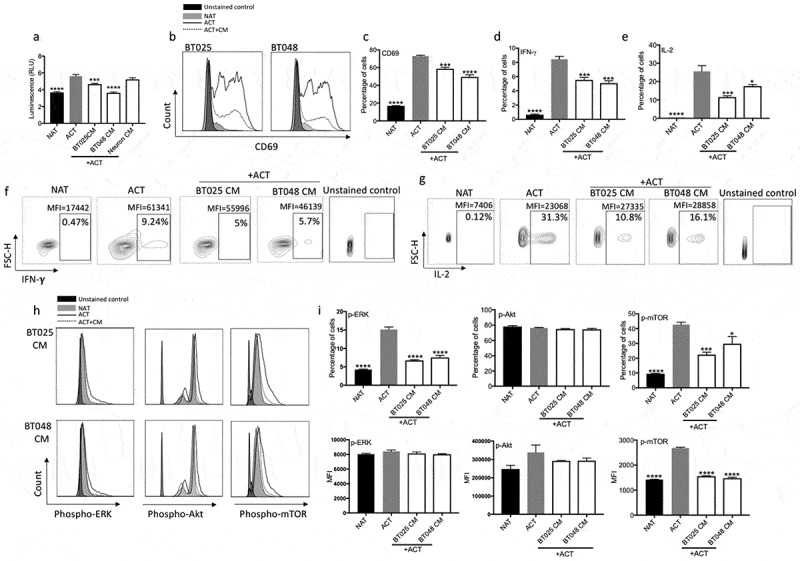
BTIC conditioned medium inhibits proliferation and effector functions of T cells.
(a) Conditioned medium (CM) from two human BTIC lines reduced human T cell proliferation while CM from human neurons, as non-transformed cells, did not inhibit T cell proliferation as determined by ATP Luminescence assay. (b, c) The expression of CD69, an early T cell activation marker, was significantly reduced following exposure to BTIC CM (b: Representative flow plots of CD69 expression). (d-g) Percentage of cells expressing both IFN-γ and IL-2 was significantly lowered after exposing T cells to BTIC CM (f, g: Representative flow plots). (h, i) T cells incubated with BTIC CM decreased their level of signaling molecules compared to activated T cells as determined by flow cytometry (h: Representative flow plots). All bars are mean ± SEM of triplicate cultures. Data in all panels are analyzed by 1-way ANOVA with Tukey’s multiple comparisons post-hoc test. *P < 0.05, ***P < 10–3, ****P < 10–4 compared to ACT group. BTIC, Brain tumor initiating cell; ACT, activated T cell; NAT, non-activated T cell; CM, Conditioned medium; MFI, Mean fluorescence Intensity.
We sought to address the mechanisms by which BTIC CM inhibits T cell activation. Since TCR signaling is a crucial step towards T cell activation and effector functions, we examined the expression levels of phospho-proteins downstream of TCR by flow cytometry analyses. Indeed, BTIC CM attenuated TCR signaling as documented by the significant reduction in p-ERK and p-mTOR, but not p-Akt, expression in T cells (Figure 2h, i).
Tenascin-c in BTIC conditioned medium is a major immunosuppressive protein
The CM from BTICs contains several products that may be responsible for T cell suppression. We focused on the secreted ECM factor, TNC, as we have previously demonstrated that it is produced prominently by BTICs.12 Western blot confirmed the expression of TNC in human and mouse BTIC lysates and their corresponding conditioned media (Figure 3a, b). Furthermore, by flow cytometry analyses we found that BTICs express TNC on their surfaces (Figure 3c).
Figure 3.
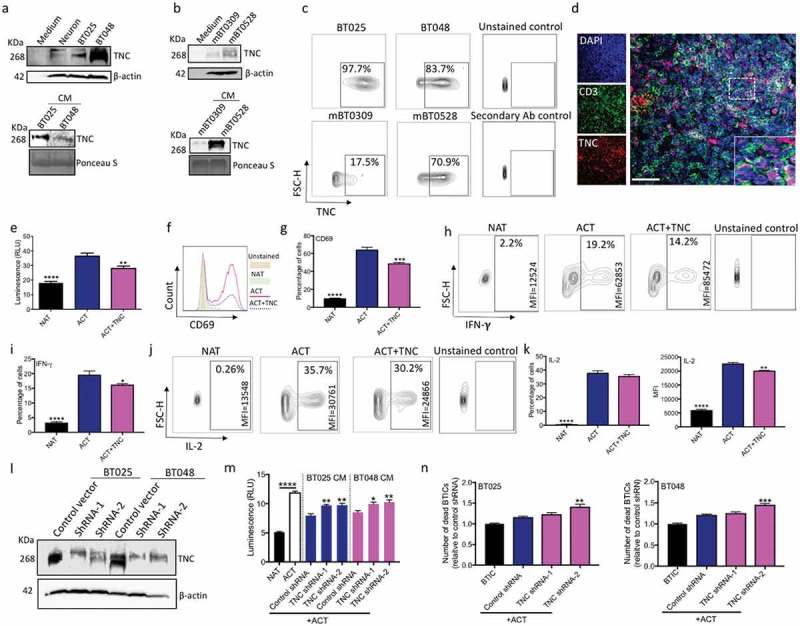
TNC produced by BTICs is in proximity to T cells in the brain tumor microenvironment; in culture TNC inhibits T cell responses. (a, b) Immunoblots of lysates prepared from human and mouse BTIC lines show TNC expression; neuronal lysate is included as a non-transformed control comparison. Moreover, TNC is present in the conditioned medium (CM) of human and mouse BTIC lines. (c) Flow cytometry analysis show surface expression of TNC on human and mouse BTIC lines. (d) Representative image from immunofluorescence staining shows the high level of expression of TNC in the glioblastoma microenvironment, in proximity to CD3-positive T cells in a tumor area with high T cell infiltration. (e) Purified TNC significantly reduced T cell proliferation as determined by ATP Luminescence assay. (f, g) Activated T cells show a decreased level of CD69 expression following exposure to TNC. (h-k) T cells exposed to TNC significantly lowered their expression of IFN-γ and IL-2 compared to activated T cells. (l) Following shRNA transduction, Western blot analysis confirmed the reduction of TNC in two human BTIC lines. (m) CM from TNC knock-down BTICs inhibited T cell proliferation to a significantly lesser extent than control shRNA. (n) Silencing of TNC in BTICs results in the enhanced cytotoxic ability of T cells as shown by increased number of dead BTICs compared to control shRNA. All bars are mean ± SEM of triplicate cultures. Data in all panels are analyzed by 1-way ANOVA with Tukey’s multiple comparisons post-hoc test. *P < 0.05, **P < 10–2, ***P < 10–3, ****P < 10–4 compared to ACT group (p values in panel m and n are relative to control shRNA unless otherwise displayed). BTIC, Brain tumor initiating cell; ACT, activated T cell; NAT, non-activated T cell; CM, Conditioned medium; MFI, Mean flourescence Intensity. Scale bar, 50 μm.
Next, we examined the spatial localization of TNC in relation to T cells in human glioblastoma specimens. Figure 3d is a representative immunofluorescence image demonstrating a high level TNC expression in the glioblastoma microenvironment, in proximity to CD3-positive T cells within the tumor. This suggests the possibility of interactions between TNC and T cells in the glioblastoma microenvironment. Therefore, we evaluated the effects of TNC on human T cells activated by anti-CD3 and anti-CD28 antibodies. Tenascin-C was added at a concentration of 20 µg/ml as previous studies had shown that application of 15 µg/ml, 10 µg/ml and 5 µg/ml elicited biological responses in embryonic hippocampal neurons,36 embryonic cortical 37 and embryonic spinal cord derived stem cells,38 respectively. We found that TNC reduced T cell proliferation (Figure 3e) and cell surface level of CD69 (Figure 3f, g). Moreover, T cells exposed to TNC lowered their expression of IFN-γ and IL-2 (Figure 3h-k). Altogether, TNC mirrored the effects of BTIC CM on T cell proliferation, CD69 expression, and cytokine production.
To determine whether TNC underlies the suppressive properties of BTIC CM, two human BTIC lines were transduced with lentiviral vectors encoding either two different short hairpin RNAs (shRNAs) specific for TNC or scrambled shRNA (control). Western blot analysis confirmed the silencing of TNC (Figure 3l). When exposed to activated T cells, the CM from TNC-knock down BTICs suppressed T cell proliferation to a significantly lesser extent than control shRNA CM (Figure 3m). We also investigated whether TNC knock-down in BTICs leads to their vulnerability to T cell-mediated cytotoxicity. After 6h of co-culture, PI staining of dying/dead BTICs showed that TNC-silenced BTICs became susceptible to killing by T cells (Figure 3n). Since TNC was incompletely eliminated in CM from TNC knock-down BTICs, we reduced TNC further in CM by ultrafiltration columns. Western blot analysis confirmed TNC depletion in CMs (Supplementary Figure 3a). TNC-depleted CM still showed inhibitory effects on T cell proliferation (Supplementary Figure 3b). Moreover, T cells exposed to TNC-depleted CM still lowered their expression of CD69, IFN-γ and IL-2 (Supplementary Figure 3c-h). These findings show that in addition to TNC, other immunomodulatory mediators are produced by BTICs.
Finally, we treated T cells with TNC in the presence of a series of monoclonal antibodies previously shown to affect TNC-altered neuronal biology; none of these, however, neutralized the inhibitory effect of TNC on T cells (Supplementary Figure 4). Thus, epitopes of TNC that affect neuronal physiology appear to be irrelevant to modulating T cell suppression.
TNC produced by BTICs attenuates TCR signaling pathways through interaction with the α5β1 and αvβ6 integrins on T cells
We first examined whether TNC displays any toxicity to T cells through live/dead cell flow cytometry discrimination. We found no effect for TNC on T cell viability (Figure 4a). TNC binds to different receptors, in which integrins such as α2β1, α5β1, α9β1 and αvβ6 comprise the major class.39 Thus, we activated T cells in the presence of monoclonal antibodies to these integrins and exogenous TNC. After 2 days, T cell proliferation was quantified. Blocking antibodies to α5β1and αvβ6 inhibited the suppressive activity of TNC on T cell proliferation, whereas α2β1- and α9β1-neutralizing antibodies had no effect on TNC suppressive activity (Figure 4b). Moreover, T cells treated with both α5β1- and αvβ6-neutralizing antibodies had elevated proliferation level than cells incubated with the single antibody (Figure 4c).
Figure 4.
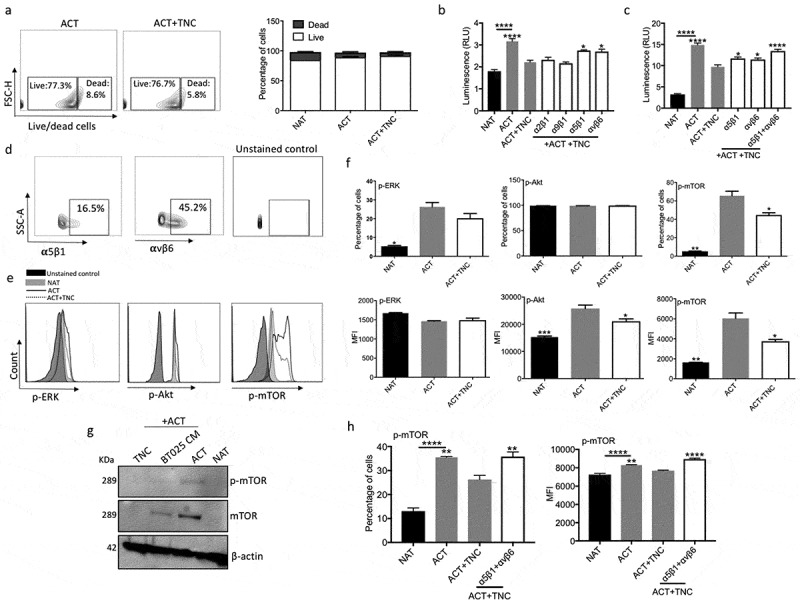
TNC inhibits T cell functions through integrin α5β1 and αvβ6. (a) Live/dead cell FACS analysis shows that TNC is not toxic to activated T cells after 2 days incubation. (b) Huaman T cells were activated with anti-CD3 and anti-CD28 antibodies in the presence of exogenous TNC and blocking antibodies to α2β1, α5β1, α9β1 and αvβ6 integrin receptor. Blocking antibodies to α5β1and αvβ6 abrogated the suppressive activity of TNC on T cell proliferation. (c) Combination of blocking antibodies to α5β1 and αvβ6 had a greater effect on the reversion of T cell proliferation that either antibody alone. (d) Activated T cells express α5β1and αvβ6 receptor as documented by flow cytometry analysis. (e, f) Purified TNC reduced the expression of p-Akt (MFI) and p-mTOR (% cells and MFI). (g) Down-regulation of p-mTOR and total mTOR in T cells after treatment with BT025 CM and TNC. (h) Blocking both α5β1and αvβ6 receptors following treatment with TNC elevated p-mTOR expression in T cells.
All bars are mean ± SEM of triplicate cultures. Data in all panels are analyzed by 1-way ANOVA with Tukey’s multiple comparisons post-hoc test. *P < 0.05, **P < 10–2, ***P < 10–3, ****P < 10–4 compared to ACT group (p values in panel b, c and h are relative to ACT+TNC group unless otherwise displayed). ACT, activated T cell; NAT, non-activated T cell.
Next, we determined the expression levels of these integrin receptors on activated T cells and found that 16.5% and 45.2% of cells express α5β1and αvβ6, respectively (Figure 4d). These results suggest that TNC inhibits T cell proliferation through α5β1and αvβ6 integrin receptors.
As noted earlier, CM from BTICs downregulated signaling molecules downstream of TCR (Figure 2h, i); we therefore determined whether TNC has the same activity. Flow cytometry analysis showed down-regulation of p-mTOR (% cells and mean fluorescence intensity, MFI) and p-Akt (MFI) in T cells after exposure to TNC (Figure 4e, f). We confirmed the downregulation of the mTOR pathway in activated T cells (Figures 2i and 4f) by Western blot (Figure 4g).
To corroborate the hypothesis that TNC inhibits TCR signaling through interaction with α5β1and αvβ6 integrins, we examined the expression of p-mTOR in T cells in the presence of TNC and the blocking integrin antibodies. Indeed, the data showed that the down-regulation of p-mTOR level in TNC treated T cells was prevented by the integrin blocking antibodies (Figure 4h).
TNC is a major immunosuppressive gene expressed in the glioblastoma microenvironment
To further establish the relevance of TNC in glioblastoma biology, we assessed the level of TNC mRNA expression across 20 major tumor types including glioblastoma in The Cancer Genome Atlas (TCGA). As shown in Figure 5a, TNC expression in glioblastoma was second to head and neck cancers. Also, TNC was expressed at the 99th percentile in TCGA glioblastoma specimens (data not shown), further establishing the importance of TNC expression in the glioblastoma microenvironment.
Figure 5.
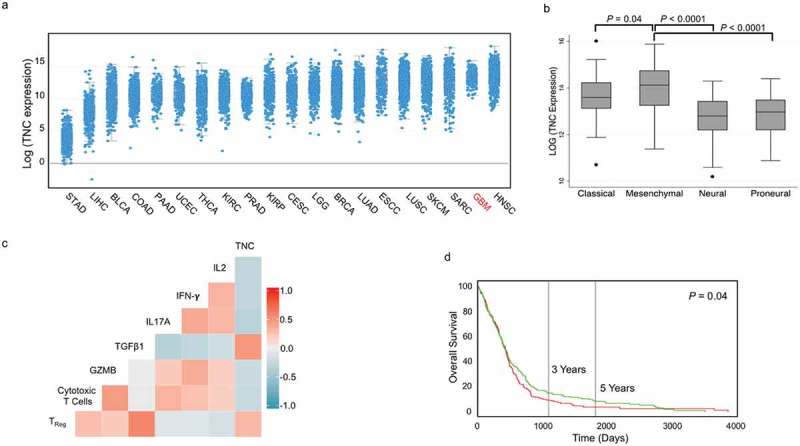
TNC expression in glioblastoma is associated with an aggressive and immuno-suppressive tumor phenotype. (a) TNC mRNA expression plotted based on increasing median expression across 20 major solid tumor types in TCGA. (b) TNC mRNA expression in TCGA patients belonging to four glioblastoma subtypes. (c) Spearman correlation matrix of TNC expression with interleukin 2 (IL2), interferon-γ (IFNG), interleukin 17A (IL17A), tumor growth factor β (TGFB1), intra-tumoral cytolytic activity (GZMB expression), tumour infiltrating CD8+ T cells (expression of CD3E and CD8A) and regulatory T cells (expression of CD3E, FOXP3 and IL2RA). The colors denote positive (increasing intensity of red) and negative (increasing intensity of blue) correlation. (d) Kaplan-Meier curves showing the association between TNC mRNA expression (green: above median; red: below median) and overall survival in TCGA glioblastoma patients.
STAD, stomach adenocarcinoma; LIHC, Liver hepatocellular carcinoma; BLCA, bladder cancer; COAD, colorectal adenocarcinoma; PAAD, pancreatic adenocarcinoma; UCEC, uterine corpus endometrial carcinoma; THCA, thyroid cancer; KIRC, clear cell renal cell carcinoma; PRAD, prostate adenocarcinoma; KIRP, Kidney Renal Papillary Cell Carcinoma; CESC, cervical squamous cell carcinoma; LGG, low-grade glioma; BRCA, breast cancer; LUAD, lung adenocarcinoma; ESCC, esophageal squamous cell carcinoma; LUSC, lung squamous cell carcinoma; SKCM, skin cutaneous melanoma; SARC, sarcoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma.
We evaluated the expression of TNC mRNA in TCGA samples classified into classical, mesenchymal, proneural and neural subtypes of glioblastoma. We found that TNC expression was significantly higher in the aggressive mesenchymal subtype compared to all other subtypes (Figure 5b). To identify an association between TNC and selected immune genes, we performed correlation analysis on gene expression data from TCGA glioblastoma patients (Figure 5c). TNC expression was negatively correlated with the expression of the pro-inflammatory cytokines (IL-2, rho = – 0.34, P = 5.8 x 10−12; IFN-γ, rho = – 0.35, P = 2.5 x 10−12 and IL-17A, rho = – 0.40, P = 2.1 x 10−15), CD8+ T cell infiltration (rho = – 0.32, P = 4.0 x 10−9) and Granzyme B (rho = – 0.30, P = 5.1 x 10−8). Conversely, TNC expression was positively correlated with regulatory T cell infiltration in the tumour microenvironment (rho = 0.37, P = 4.5 x 10−13) and the immunosuppressive cytokine TGF-β (rho = 0.56, P = 4.8 x 10−33). Above median TNC expression was also associated with worse overall survival in glioblastoma patients (log rank P = 0.04; Figure 5d).
BTICs associate TNC with exosomes for secretion
Since cancer-derived exosomes can disrupt the BBB,27,28 we examined whether TNC is packaged within BTIC exosomes to enable its migration from the CNS to the periphery through the BBB. Exosomes were isolated from supernatants of two human BTIC lines and verified by transmission electron microscopy (Figure 6a) and the Western blot expression of exosome markers, TSG101 and Flotillin 1 40,(Figure 6b). Moreover, CD63, an exosome surface antigen, was detected by flow cytometry of isolated vesicles (Figure 6c). Through Western blot, TNC was detected in exosomes from two mouse and two human BTIC lines (Figure 6d, e). Upon centrifugation at 100,000 xg to deplete exosomes (Exosome-dep.), the CM of BTICs no longer exhibited TNC expression (Figure 6d, e). We also used Size Exclusion Chromatography (SEC) followed by centrifugation at 100,000 xg for further purification of exosomes. Western blot analyses showed TNC and exosome marker, Flotillin 1, expression in isolated exosomes (Figure 6f). To confirm the purity of the exosome preparations, we found no expression of cis-Golgi marker GM130 and endoplasmic reticulum marker Calnexin in exosomes (Figure 6f). These observations confirm that BTICs release TNC in association with exosomes.
Figure 6.
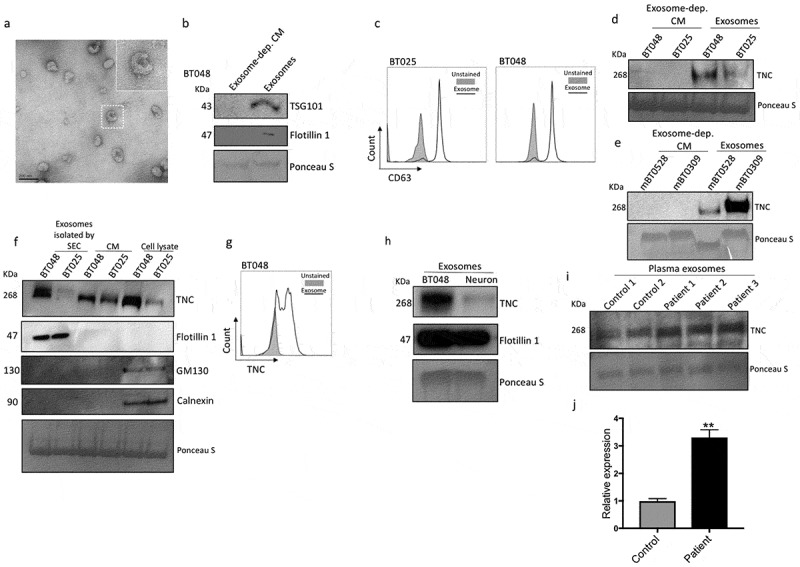
BTICs export TNC through exosomes. (a) Representative TEM image of exosomes derived from BT012. (b) The expression of the exosome markers TSG101 and Flotillin 1 was verified by Western blot. (c) Moreover, CD63, an exosome surface antigen, was detected by flow cytometry of isolated vesicles. (d, e) TNC was detected in exosomes from two human and two mouse BTIC lines. Upon centrifugation to deplete (dep.) exosomes, the CM of BTICs no longer exhibited TNC expression. (f) Exosomes were isolated from BTIC conditioned media by Size Exclusion Chromatography (SEC) followed by ultracentrifugation. The expression of TNC, exosome marker Flotillin 1, cis-Golgi marker GM130 and endoplasmic reticulum marker Calnexin was determined by Western blot. (g) Exosome particles expressed TNC on their surface as documented by flow cytometry analysis. (h) Exosomes isolated from BTICs contain a higher amount of TNC than exosomes derived from neurons. (i) Western blot analysis of TNC expression in isolated exosomes from the blood of three glioblastoma patients and two control individuals. (j) Densitometric analysis of blot normalized to Ponceau S staining shows that circulating exosomes in the blood of three glioblastoma patients contained significantly more TNC than those from control individuals. Data in panel j are analysed by unpaired t-test (2-tailed). **P < 10–2.
We investigated TNC expression on exosome particles by flow cytometry analysis and found that exosomes expressed high levels of TNC on their surface (Figure 6g). Furthermore, exosomes produced by BTICs contained higher levels of TNC compared to exosomes derived from normal brain cells as exemplified by neurons (Figure 6h). Finally, we found that circulating exosomes in the blood of glioblastoma patients contained significantly more TNC than those from control individuals (Figure 6i, j). Collectively, these data show that TNC is associated with, and perhaps packaged within, BTIC-secreted exosomes and circulates in the blood stream of glioblastoma patients.
BTICs-derived exosomes inhibit T cells activation in part by the expression of TNC
We labeled exosomes from BTICs with BODIPY TR Ceramide and used fluorescent live cell imaging to visualize their interactions with DiO-tagged T cells after 16h of co-culture. Figure 7a shows that exosomes isolated from two human BTIC lines were closely associated with T cells particularly on their cell surface. Next, we investigated the inhibitory effects of exosomes on T cell proliferation and found that, in contrast to exosomes isolated from neurons, BTIC-derived exosomes suppressed T cell proliferation (Figure 7b). Moreover, exosomes from TNC knock-down BTICs lost their inhibitory effects on T cell proliferation compared to exosomes from BTICs treated with control shRNAs (Figure 7c).
Figure 7.
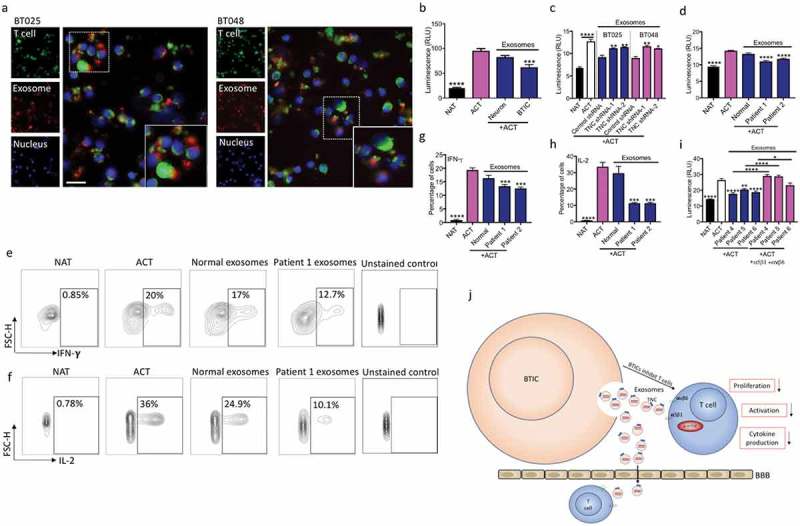
Exosomes from BTICs exert their inhibitory effects on T cells through TNC expression. (a) Exosomes isolated from human BTIC lines were labeled red with BODIPY TR Ceramide. T cells were purified from healthy individual and stained green with DiO. Representative images show exosome association with T cells. (b) In contrast to exosomes from neurons, BTIC exosomes suppressed T cell proliferation. (c) TNC-depleted exosomes had lower inhibitory effects on T cell proliferation compared to control shRNA exosomes. (d) Circulating exosomes from two glioblastoma patients induced a higher reduction in T cell proliferation than healthy control exosomes. (e-h) Exosomes from glioblastoma patients caused greater reduction of IFN-γ and IL-2 levels in activated T cells than normal exosomes (e, f: Representative flow plots). (i) Blocking both α5β1and αvβ6 receptors following treatment with plasma exosomes from three glioblastoma patients elevated T cell proliferation. (j) The postulated mechanism of T cell suppression by TNC expressed on exosomes. The cartoon depicts that BTICs release exosomes that carry TNC. TNC through interaction with integrin receptors α5β1 and αvβ6 attenuates the expression of p-mTOR in T cells. This results in the reduction of T cell proliferation, activation and cytokine production. Moreover, exosomes pass through the BBB in glioblastoma patients to alter systemic immunity. All bars are mean ± SEM of triplicate cultures. Data in all panels are analyzed by 1-way ANOVA with Tukey’s multiple comparisons post-hoc test. *P < 0.05, **P < 10–2, ***P < 10–3, ****P < 10–4 compared to ACT group unless otherwise displayed (p values in panel c are relative to control shRNA unless otherwise displayed). BTIC, Brain tumor initiating cell; ACT, activated T cell; NAT, non-activated T cell. Scale bar, 20 μm.
We also compared exosomes isolated from the blood of glioblastoma patients to that from healthy controls. The glioblastoma exosomes produced a greater reduction in T cell proliferation compared to healthy control exosomes (Figure 7d). Moreover, in contrast to normal exosomes, exosomes from glioblastoma patients suppressed IFN-γ and IL-2 production by T cells (Figure 7e-h). To corroborate the hypothesis that TNC associated with exosomes inhibits T cells through interaction with α5β1and αvβ6 integrins, we examined T cell proliferation in the presence of glioblastoma exosomes and the blocking integrin antibodies. The data showed that reduced proliferation of exosome treated T cells was rescued by the integrin blocking antibodies (Figure 7i).
We conclude that BTICs secrete high amounts of TNC into the circulation through exosomes to suppress systemic immune responses in glioblastoma patients (Figure 7j).
Discussion
Within glioblastomas, BTICs have the principal properties of self-renewal, clonal tumor initiation capacity, and clonal long-term repopulation potential.41 Sphere-forming capacity is a particular trait of stem-like cells which enable them to recapitulate cell growth even at low numbers. It has been shown that one cancer stem cell has the potential to proliferate and form a sphere in vitro.42 It is this capacity of BTICs that makes them very refractory to many commonly employed cancer therapies. Similar to normal stem cells, BTICs are resident in niches which provide cues to support and initiate stem-like programs in cancer cells.43 In addition, niches manipulate immune responses to protect BTICs from immune cells. Therefore, there has been significant interest in studying BTIC niches to identify and target inhibitory factors as a possible tool to enhance the vulnerability of BTICs to immunotherapy. A principal component of these niches are the proteins comprising the ECM, including TNC as a prominent constituent44; arguably, the most prominent ECM protein in BTIC niches is TNC which is involved in glioma growth, angiogenesis and invasion.12 We stained human glioblastoma brain specimens and found TNC to be widely deposited within the tumor, including in areas of T cell infiltration; therefore, there is the potential for interactions between BTIC-derived TNC and T cells. Moreover, we found a high level of TNC protein in the supernatant of BTIC lines supporting the notion that BTICs secrete TNC protein into the microenvironment. However, little information exists regarding the influence of BTIC-derived TNC on immune cell activity.
In the current study, we observed that while T cells activated in isolation produce factors that inhibit BTIC growth, this is lost in co-culture of activated T cells and BTICs, suggesting a dynamic mechanism whereby BTICs prevent T cell activation. Several experimental results led us to conclude that the release of TNC is an important mechanism by which BTICs inhibit T cell responses. Besides similarity in BTIC CM and purified TNC protein in reducing T cell activity, the depletion of TNC in BTICs attenuated the CM-mediated T cell suppression. TNC depletion by filtration, however, did not completely abrogate the immunosuppressive activity of BTIC CM, suggesting that other mechanisms are involved in T cell inhibition.
It is known that TNC binds to several receptors, of which integrins are the major class.45 We found that TNC inhibits T cells through two integrin receptors α5β1 and αvβ6. Focal adhesion kinase (FAK) is a key component of the signaling pathways triggered by integrins.46 FAK in the upstream of PI3K/Akt signaling pathway46 induces tumor cell growth12 and enhances cell migration and survival.47 TNC/integrin interaction promotes survival and growth through FAk activation and by inducing the PI3K/Akt pathway in cancer cells.48 Conversely, we found that TNC downregulates the Akt/mTOR pathway in T cells, a signaling cascade that appears to be crucial for their activation. Consistent with our results, TNC produced by prostate cancer stem-like cells suppresses T cell activation.20 TNC interaction with α5β1 inhibits GTPases such as Rho in T cells.20 Interestingly, Rho regulates the activaty of mTOR.49 TNC as a large protein with several domains inhibits T cells by different mechanisms. For instance, the FnIII 1–5 repeats of TNC suppresses T cells through inhibition of α5β1-dependent T cell adhesion to fibronectin,50 while domain TnFnIII A1A2 inhibits T cells through an unknown mechanism.19 Fibronectin engages the integrin α/β of T cells to activate Akt/ERK signaling pathway and T cell proliferation.21 The signaling molecules affected by BTIC CM and TNC, however, are not exactly the same. Moreover, following complete depletion of TNC, BTIC CM still exhibited suppressive activity. This suggests that there are other factors in the CM besides TNC that suppress T cell responses as reported by other groups.7,29
An important finding herein is that TNC is exported by BTICs associated with exosomes, and that these TNC-containing exosomes can be found in the circulation of patients with glioblastoma. The inhibitory effects of exosomes on immune cells, and T cells in particular, is a growing area of investigation. It was shown that exosomes from non-BTIC glioblastoma cells at concentrations more than 100 μg/ml suppress cytokine production by T cells, whereas low concentrations have no effect.29,51,52 In our study, we used exosome concentrations of 200 μg/ml which approximately represents the concentration of exosomes within the blood of cancer patients.51 Of note, in the tumor microenvironment, the concentration of exosomes may be around 2000 μg/ml.51 During the course of this work, it was reported that exosomes from glioblastoma cancer stem cells also mediate T cell suppression 29 although the precise inhibitory mechanisms of glioblastoma-derived exosomes from that study were not investigated. Herein, through shRNA knockdown experiments, we found that BTIC-derived exosomes contain high levels of TNC and that this is an important protein in exosomes that is involved in T cell suppression. Other BTIC exosome-derived molecules are likely also important for immunosuppressive actions. Previously, it was reported that the interaction of Fas ligand on exosomes with Fas receptor on T cells is involved in the suppression of the T cell response through the promotion of apoptosis.52 We found that TNC is expressed on the surface of BTICs and their derived exosomes. Because TNC does not comprise a transmembrane domain it most probably interacts with membrane receptors. For example, it has been shown that TNC interacts with annexin II and integrins on cell surface.39,53 Moreover, interaction with lipid rafts is another mechanism for cell surface expression.54
Systemic immunosuppression is a major immunological feature of glioblastoma patients. Glioma cells secrete humoral factors to attenuate the systemic immune response; however, it has been unclear how soluble molecules migrate through the BBB to reach the circulation. Cancer cells may produce factors that perturb the BBB and it was recently shown that cancer cell-derived exosomes trigger BBB breakdown through the expression of different microRNAs.27,28 Therefore, considering the capacity of exosomes to harbor high amounts of TNC which has an inhibitory effect on T cells, we conclude that circulating exosomes in glioblastoma patients are a major inhibitory mechanism that cancer cells employ to manipulate immunity. With this in mind, exosomes carry a wide variety of molecules and it seems difficult to conclude that a single exosome-protein can explain the entire mechanism of immunosuppression in glioblastomas. Nevertheless, we have detected an increased level of TNC in exosomes from glioblastoma patients as well as more inhibitory activity in exosomes from glioblastoma patients, suggesting an important role of TNC in systemic immunosuppression. We acknowledge, however, that our focus on TNC is a biased approach and future studies will need to be performed to contrast the relative activity of TNC to other immunosuppressive factors to be uncovered by an unbiased screen. In conclusion, we propose that TNC in circulating exosomes is an important target for therapeutic approaches to improve the prognosis of glioblastomas.
Materials and methods
T cell isolation and activation
Mice
Splenocytes from C57BL/6 mice (Charles River Laboratories) of 6–8 weeks of age were collected and purified using Ficoll-Plaque PLUS (GE Healthcare) centrifugation. Cells were resuspended in MACS Separation Buffer (Miltenyi Biotec). T cells were purified by EasySep™ Mouse T Cell Isolation Kit (STEMCELL Technologies). Purified T cells were resuspended in NeuroCult™ Basal Medium supplemented with NeuroCult™ Proliferation Supplement, EGF, FGF, and heparin sulfate (Stem Cell Technologies), and plated in a 96-well round bottom plate that was coated with 1 μg/ml anti-CD3 antibodies (BD Biosciences) for 2–3 h at 37°C. T cells were plated at 100,000 cells per well in a total volume of 100 μl. Also, 1 μg/ml of soluble anti-mouse CD28 antibodies (BD Pharmingen) were added to the coated wells. Non-activated T cells were plated on uncoated wells. Conditioned media and cells from each condition were harvested after 48 h of activation. Ethics approval from the University Animal Care Committee was received for the use of mice.
Human
Blood was drawn from healthy adult human volunteers and PBMCs were purified by Ficoll-Plaque PLUS centrifugation. Briefly, heparinized blood was diluted 1:1 with PBS, carefully layered over the Ficoll, and centrifuged; the cell interface layer was collected, washed twice in PBS and resuspended in MACS Separation Buffer. T cells were purified by EasySep™ Human T Cell Isolation Kit (STEMCELL Technologies). Isolated T cells were resuspended in NeuroCult™ Basal Medium and seeded in a 96-well round bottom plate. Wells of a 96-well round bottom plate were coated with 1 μg/ml anti-human CD3 (BD Biosciences) for 2–3 h at 37°C. Wells were left uncoated to harvest non-activated T cells. Isolated T cells were plated at 100,000 cells per well in a final volume of 100 μl. Also, 1 μg/ml of soluble anti-human CD28 antibodies (BD Pharmingen) were added to the coated wells. Conditioned media and cells from each condition were harvested after 48 h of activation. In some experiments, T cells were incubated in the presence of 20 μg/ml TNC purified protein (Millipore, CC065). For integrin inhibition experiments, T cells were incubated with 4 μg/ml blocking antibodies to α2β1 (Clone P1E6, Abcam), α5β1 (Clone M200, Novus Biologicals), α9β1 (Clone Y9A2, Millipore), and αvβ6 (Clone 10D5, Millipore) integrins followed by TNC after 1 hour. For TNC neutralization, antibodies specific to different epitopes of TNC, and found to be function blocking in bioassays affecting neuronal growth,36 were used. In particular, the rat mAbs 576, 19H12TNC and 20A1TNC have been mapped to the alternatively spliced FNIII domain A2 while mAb 578 recognizes the FNIII domain D of the alternatively spliced region of TNC.13,55 All human samples were collected with informed consent and approved by a local institutional ethics committee.
Derivation of mouse and human BTICs
Human BTICs were generated from resected specimens of glioblastoma patients as described before.33 In the present study, we used two human BTIC lines designated BT025 and BT048 for all tissue culture experiments. The lines have been previously referred as 25EF and 48EF, respectively.33 These lines were cultured chronologically, maintained, and authenticated within the University of Calgary BTIC Core. The stemness features of these two human cell lines have been previously described in our publications.33,56 Cell lines were initiated in culture from patient specimens in the years 2005 (BT025) and 2007 (BT048). We also used two mouse BTIC lines including mBT0309 and mBT0528 in the C57BL/6 background, previously generated and propagated as stem-like cells.31 These lines have been previously shown to recapitulate key features of a human glioblastoma.
BTIC growth evaluation
For the sphere forming growth assay, BTIC cells were plated at 10,000 cells/well in 100µl of NeuroCult™ Basal Medium supplemented with NeuroCult™ Proliferation Supplement, EGF, FGF, and heparin sulfate (Stem Cell Technologies) into a 96-well plate. The resultant number of spheres above the 60 µm diameter cutoff, a convenient parameter to describe growth characteristics, was monitored after 3 days by bright-filed photographing multiple fields per well with subsequent analyzes as previously described.33 Where total cell number was shown, BTICs were collected, centrifuged, resuspended in 25 µL of Accumax™, mixed with Trypan Blue (1:1) and counted using a TC20™ automated cell counter (Bio-Rad). For conditioned medium (CM) experiment, BTICs were plated at 10,000 cells per well in a 96-well plate for 3 days, after which CM was collected.
Flow cytometry
Human T cells after 2 days in culture were resuspended in PBS and incubated with the following antibodies (BD Biosciences except where indicated): PE-conjugated anti-CD69 (Clone FN50), APC-conjugated anti-IFN-γ (Clone B27), PE-conjugated anti-IL-2 (Clone MQ1-17H12), PE-conjugated anti-HLA-I (Clone G46-2.6), PE-conjugated anti-p-ERK (Clone MILAN8R, Thermo Fisher Scientific), APC-conjugated anti-p-Akt (Clone SDRNR, Thermo Fisher Scientific), and PE-Cy7-conjugated anti-p-mTOR (Clone MRRBY, Thermo Fisher Scientific). For intracellular cytokine staining, cell were treated with Leukocyte Activation Cocktail, with GolgiPlug (BD Biosciences) for 4–6 h. Cells were analyzed on an Attune® Acoustic Focusing Flow Cytometer (Applied Biosystems), gated and quantified using FlowJo (version 10.0.7) software. The percentage of live/dead cells was detected using the LIVE/DEAD™ Fixable Green Dead Cell Stain Kit as described by the manufacturer (Thermo Fisher Scientific). The percentage of lymphocytes, calculated via light scatter gating, was used as an indirect measure of cell viability. The presence of CD63 exosomal marker and surface expression of TNC on exosomes was analyzed by flow cytometry. Exosomes isolated by ultracentrifugation were resuspended in PBS and bound to magnetic CD63-coated Dynabeads (Thermo Fisher Scientific) during an overnight incubation at 4°C. The following day, the Dynabeads-bound exosomes were stained with the PE-conjugated anti-CD63 (Clone H5C6) and polyclonal anti-human TNC (Novus Biologicals, 41660002) antibodies along with isotype controls.
Exosome isolation and co-incubation with T cells
Exosomes were isolated from the plasma and CM by ultracentrifugation.40 Briefly, blood plasma (after dilution 1:1 in PBS) and conditioned media were sequentially centrifuged at 300 × g for 10 minutes at 4°C and 2000 × g for 10 minutes at 4°C. After filtration through a 0.22-μm (Millipore) filter, collected supernatants were ultracentrifuged at 100,000 × g for 90 minutes at 4°C. Exosome pellets were washed with PBS and centrifuged at 100,000 × g for 90 minutes at 4°C and resuspended in 20 μL of PBS. Izon’s qEV10 (iZON Science) column was used for exosome isolation based on Size Exclusion Chromatography (SEC). Transmission electron microscopy (TEM) analysis was carried out as previously described.40 Exosomes preparations were conserved at −80°C for later use. For fluorescence labeling of exosomes, 2ul of BODIPY TR ceramide solution (Thermo Fisher Scientific) was added to 100 μg exosomes to obtain 20 μM concentration of final dye. After incubation at 37°C for 20 min, the exosome solution was washed with Exosome Spin Columns (MW 3000) as described by the manufacturer (Thermo Fisher Scientific). For the co-incubation experiments, T cells were stained with DiO (Thermo Fisher Scientific). After seeding in a 96-well plate coated with 1μg/ml anti-CD3 and anti-CD28 antibodies, 20 μg of labeled exosomes was added to T cells and incubated for 16 hours at 37°C with 5% CO2. The uptake of exosome was monitored by ImageXpress (Molecular Devices), a high-throughput cellular imaging system, using a 20× objective. NucBlue (Thermo Fisher Scientific) was used to stain cell nuclei.
Silencing of TNC expression
TNC expression was stably downregulated in BT025 and BT048 using lentivirus-mediated transduction. We used TNC shRNA constructs or Scrambled control from Sigma Mission shRNA Library inserted in pLKO.1-puro Vector. Transduction for these two cell lines was done as described previously.12
TNC depletion by ultrafiltration
Conditioned media collected from human BTICs were subjected to ultrafiltration columns (Sterlitech) with 200kDa cutoff to eliminate TNC.
Immunofluorescence staining of human glioblastoma specimens
We evaluated paraffin sections from 2 autopsied glioblastoma patients (obtained from the University of Calgary Neuropathology archive) for the expression of TNC and CD3. Paraffin-embedded tissues were sectioned (6 μm) using a Leica RM2135 Microtome. After deparaffinization and antigen retrieval with 10 mM sodium citrate buffer, sections were blocked with 3% BSA in PBS and then subjected to antibody staining with anti-CD3 (Abcam, Clone CD3-12, 1:200) and anti-human TNC antibody (Novus Biologicals, 41660002, 1:200) overnight at 4°C. After three washes of PBS at room temperature, Alexa 488-conjugated anti-rat or Alexa 546-conjugated secondary antibodies (1:500; Jackson ImmunoResearch Laboratories) were added for 90 min. Sections were then mounted in Gelvatol medium and cover-slipped. Fluorescence images were taken on a confocal microscope (Fluoview FV10i; Olympus). Images were captured using a 60× objective.
Western blot analysis
Cells were collected, washed in PBS, and resuspended in RIPA Buffer (Thermo Fisher Scientific) containing Protease Inhibitor Cocktail and phosphoSTOP (Roche). Lysates (25 μg per lane) were loaded on a NuPAGE™ 3–8% Tris-Acetate Protein Gel, transferred to PVDF membranes, blocked with 5% w/v milk in PBS for 1 h, and then immuno-blotted overnight with the following antibodies: anti-TSG101 (Abcam, Clone EPR7131, dilution 1:500), anti-Flotillin 1 (Abcam, Clone EPR6041, 1:2000), anti-GM130 (Abcam, Clone EP892Y, 1:1000), anti-Calnexin (Santa Cruz Biotechnology, sc-11397, 1:100), anti-p-mTOR (Ser2448) (Cell Signaling, 1:500), anti-mTOR (Cell Signaling, 1:500), anti-human TNC (Novus Biologicals, 41660002, 1:1000), anti-mouse TNC (Novus Biologicals, Clone 4C8MS, dilution 1:250) antibodies. For beta-actin probing, membranes were incubated for 90 min with HRP-conjugated anti-beta-actin antibody (Abcam, Clone mAbcam 8226, dilution 1:5000). All antibodies were diluted in Tris Buffered Saline with Tween 20 and 0.05% BSA. For the unconjugated antibodies, membranes were incubated for 90 min with the corresponding HRP-linked secondary antibody (Cell Signaling, dilution 1:5000) for chemiluminescence imaging by ECL Select Western Blotting Detection Reagent (GE Healthcare). We quantified the resulting blots by densitometry analysis using Image Lab software (Bio-Rad).
T cell cytotoxicity assay
T cells were seeded at different densities in a 96-well plate that was coated with 1 μg/ml anti-CD3 and anti-CD28 antibodies. Non-activated T cells were plated in uncoated wells. After 1 hour incubation at 37°C, BTICs labeled with DiO solution (Thermo Fisher Scientific) were added to wells to yield the desired T cell:BTIC ratios. After incubation for 4 h at 37°C, the percentage of dead BTICs was determined by propidium iodide (PI) staining. NucBlue (Thermo Fisher Scientific) was used to stain cell nuclei. We quantified the percentage of dead cells by ImageXpress (Molecular Devices) system.
ELISA
Isolated mouse T cells were plated at 100,000 cells per well in a final volume of 100 μl in the presence on anti-CD3 and anti-CD28 antibodies. In some wells, T cells were activated in the presence of 10,000 mouse BT0309. After 48h, medium was taken for TNF-α and IFN-γ measurement according to the manufacturer’s instructions (Invitrogen).
Human neurons
Neurons were obtained from brain tissues from human abortuses of 10–18 week fetal age based on protocol reported previously.57 The use of the specimens was approved by local institutional ethics committee.
Bioinformatic analyses
Visualization of TNC expression in The Cancer Genome Atlas (TCGA) cancer types was performed on cBioPortal (http://www.cbioportal.org/) using RNAseq data. RNAseq (level 3) data were downloaded from the Broad Institute Firehose GDAC (https://gdac.broadinstitute.org/). We quantified the TNC expression in glioblastoma subtypes by the STATA 15 statistical software package (College Station, TX). For the association study between TNC expression and survival in glioblastoma patients, we used Kaplan Meier curves in the PROGgene V2 Prognostic Database (http://watson.compbio.iupui.edu/chirayu/proggene/database/index.php) and data derived from TCGA HT-HG-U133A microarray (Affymetrix, San Diego, CA). TNC expression was dichotomized at the median and significance was assessed using the log rank test. Spearman correlation was used to assess the relationship between TNC and intratumoral cytokines (IL2, IFNG, IL17A and TGFB1), Granzyme B (GZMB), cytotoxic T cells (geometric mean of CD3E and CD8A) and regulatory T cells (geometric mean of CD3E, FOXP3 and IL2RA). Correlation matrices were constructed using ggcorr plot and visualization was performed using ggplot2 R plugins.
Statistics
All results are expressed as mean ± SEM. Statistical analysis was performed with Prism version 7.0 (GraphPad Software). For multiple group comparisons to controls or other subgroups, a 1-way ANOVA with a Tukey’s multiple comparisons post-hoc test was used. For analysis of data between 2 groups, an unpaired t-test (2-tailed unless otherwise stated) was used. The p values <0.05 were considered significant. All experiments were reproduced at least twice.
Funding Statement
This work was supported by operating grants from Alberta Innovates – Health Solutions in collaboration with the Alberta Cancer Foundation, and from the Canadian Institutes of Health ResearchGovernment of Canada | Canadian Institutes of Health Research (CIHR).
Acknowledgments
We thank the University of Calgary BTIC Core headed by Drs. Sam Weiss and Greg Cairncross for isolating BTIC lines from patient-resected specimens. RM is supported by a University of Calgary Eyes High postdoctoral scholarship. LD is supported by Alberta Innovates – Health Solutions (AIHS) Summer Studentship. KSR is supported by a Vanier Canada Graduate Scholarship. AF gratefully acknowledges grant support by the German Research Foundation (DFG, SPP 1757/2, Fa 159/20-2). VWY is a Canada Research Chair (Tier 1) in Neuroimmunology for which salary support is gratefully acknowledged. This work was supported by operating grants from AIHS in collaboration with the Alberta Cancer Foundation, and from the Canadian Institutes of Health Research.
Supplemental meterial
Supplemental data for this article can be accessed here.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF.. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–1241. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, Grotenhuis JA, Hoogerbrugge PM, De Vries IJM, Adema GJ. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. 2009;11:394–402. doi: 10.1215/15228517-2008-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9:67–78. doi: 10.1158/1535-7163.MCT-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100:216–232. doi: 10.1016/S0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 9.Ashkenazi E, Deutsch M, Tirosh R, Weinreb A, Tsukerman A, Brodie C. A selective impairment of the IL-2 system in lymphocytes of patients with glioblastomas: increased level of soluble IL-2R and reduced protein tyrosine phosphorylation. Neuroimmunomodulation. 1997;4:49–56. doi: 10.1159/000097315. [DOI] [PubMed] [Google Scholar]
- 10.Reinhard J, Brosicke N, Theocharidis U, Faissner A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int J Biochem Cell Biol. 2016;81:174–183. doi: 10.1016/j.biocel.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol. 2004;36:1046–1069. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar S, Mirzaei R, Zemp FJ, Wei W, Senger DL, Robbins SM, Yong VW. Activation of NOTCH signaling by tenascin-C promotes growth of human brain tumor-initiating cells. Cancer Res. 2017;77:3231–3243. doi: 10.1158/0008-5472.CAN-16-2171. [DOI] [PubMed] [Google Scholar]
- 13.Brosicke N, Van Landeghem FK, Scheffler B, Faissner A. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013;354:409–430. doi: 10.1007/s00441-013-1704-9. [DOI] [PubMed] [Google Scholar]
- 14.Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH. Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer. 2002;98:362–369. doi: 10.1002/ijc.10233. [DOI] [PubMed] [Google Scholar]
- 15.Leins A, Riva P, Lindstedt R, Davidoff MS, Mehraein P, Weis S. Expression of tenascin-C in various human brain tumors and its relevance for survival in patients with astrocytoma. Cancer. 2003;98:2430–2439. doi: 10.1002/(ISSN)1097-0142. [DOI] [PubMed] [Google Scholar]
- 16.Garikapati KR, Patel N, Makani VK, Cilamkoti P, Bhadra U, Bhadra MP. Down-regulation of BORIS/CTCFL efficiently regulates cancer stemness and metastasis in MYCN amplified neuroblastoma cell line by modulating Wnt/beta-catenin signaling pathway. Biochem Biophys Res Commun. 2017;484:93–99. doi: 10.1016/j.bbrc.2017.01.066. [DOI] [PubMed] [Google Scholar]
- 17.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hibino S, Kato K, Kudoh S, Yagita H, Okumura K. Tenascin suppresses CD3-mediated T cell activation. Biochem Biophys Res Commun. 1998;250:119–124. doi: 10.1006/bbrc.1998.9258. [DOI] [PubMed] [Google Scholar]
- 19.Puente Navazo MD, Valmori D, Ruegg C. The alternatively spliced domain TnFnIII A1A2 of the extracellular matrix protein tenascin-C suppresses activation-induced T lymphocyte proliferation and cytokine production. J Immunol (Baltimore, Md: 1950). 2001;167:6431–6440. doi: 10.4049/jimmunol.167.11.6431. [DOI] [PubMed] [Google Scholar]
- 20.Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, Piras IS, Restuccia U, Calcinotto A, Freschi M, et al. Tenascin-C protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res. 2015;75:2095–2108. doi: 10.1158/0008-5472.CAN-14-2346. [DOI] [PubMed] [Google Scholar]
- 21.Huang JY, Cheng YJ, Lin YP, Lin HC, Su CC, Juliano R, Yang B-C. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J Immunol (Baltimore, Md: 1950). 2010;185:1450–1459. doi: 10.4049/jimmunol.0901352. [DOI] [PubMed] [Google Scholar]
- 22.Didem T, Faruk T, Senem K, Derya D, Murat S, Murat G, Oznur K. Clinical significance of serum tenascin-c levels in epithelial ovarian cancer. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2014;35:6777–6782. doi: 10.1007/s13277-014-1923-z. [DOI] [PubMed] [Google Scholar]
- 23.Riedl S, Bodenmuller H, Hinz U, Holle R, Moller P, Schlag P, Herfarth C, Faissner A. Significance of tenascin serum level as tumor marker in primary colorectal carcinoma. Int J Cancer. 1995;64:65–69. doi: 10.1002/(ISSN)1097-0215. [DOI] [PubMed] [Google Scholar]
- 24.Pardridge WM. Blood-brain barrier drug targeting enables neuroprotection in brain ischemia following delayed intravenous administration of neurotrophins. Adv Exp Med Biol. 2002;513:397–430. [DOI] [PubMed] [Google Scholar]
- 25.Erickson HP, Bourdon MA. Tenascin: an extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu Rev Cell Biol. 1989;5:71–92. doi: 10.1146/annurev.cb.05.110189.000443. [DOI] [PubMed] [Google Scholar]
- 26.Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30:836–848. doi: 10.1016/j.ccell.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O’Connor S, Chin A, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, Lötvall J, Nakagama H, Ochiya T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun. 2015;6:6716. doi: 10.1038/ncomms7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, Manini I, Beltrami AP, Ius T, Skrap M, Di Loreto C, et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS One. 2017;12:e0169932. doi: 10.1371/journal.pone.0169932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skog J, Wurdinger T, Van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pisklakova A, McKenzie B, Zemp F, Lun X, Kenchappa RS, Etame AB, Rahman MM, Reilly K, Pilon-Thomas S, McFadden G, et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro Oncol. 2016. doi: 10.1093/neuonc/now006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kominsky S, Johnson HM, Bryan G, Tanabe T, Hobeika AC, Subramaniam PS, Torres B. IFNgamma inhibition of cell growth in glioblastomas correlates with increased levels of the cyclin dependent kinase inhibitor p21WAF1/CIP1. Oncogene. 1998;17:2973–2979. doi: 10.1038/sj.onc.1202217. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci. 2014;17:46–55. doi: 10.1038/nn.3597. [DOI] [PubMed] [Google Scholar]
- 34.Zou JP, Morford LA, Chougnet C, Dix AR, Brooks AG, Torres N, Shuman JD, Coligan JE, Brooks WH, Roszman TL, et al Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J Immunol (Baltimore, Md: 1950). 1999;162:4882–4892. [PubMed] [Google Scholar]
- 35.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17(Suppl 7):vii9–vii14. doi: 10.1093/neuonc/nov151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lochter A, Vaughan L, Kaplony A, Prochiantz A, Schachner M, Faissner A. J1/tenascin in substrate-bound and soluble form displays contrary effects on neurite outgrowth. J Cell Biol. 1991;113:1159–1171. doi: 10.1083/jcb.113.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcion E, Halilagic A, Faissner A, ffrench-Constant C. Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development. 2004;131:3423–3432. doi: 10.1242/dev.01202. [DOI] [PubMed] [Google Scholar]
- 38.Karus M, Denecke B, ffrench-Constant C, Wiese S, Faissner A. The extracellular matrix molecule tenascin C modulates expression levels and territories of key patterning genes during spinal cord astrocyte specification. Development. 2011;138:5321–5331. doi: 10.1242/dev.067413. [DOI] [PubMed] [Google Scholar]
- 39.Tucker RP, Chiquet-Ehrismann R. Tenascin-C: its functions as an integrin ligand. Int J Biochem Cell Biol. 2015;65:165–168. doi: 10.1016/j.biocel.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit3.22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 41.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 42.Cao L, Zhou Y, Zhai B, Liao J, Xu W, Zhang R, Li J, Zhang Y, Chen L, Qian H, et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011;11:71. doi: 10.1186/1471-230X-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 44.Faissner A, Roll L, Theocharidis U. Tenascin-C in the matrisome of neural stem and progenitor cells. Mol Cell Neurosci. 2017;81:22–31. doi: 10.1016/j.mcn.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 46.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 47.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paron I, Berchtold S, Voros J, Shamarla M, Erkan M, Hofler H, Esposito I, Gottardi C. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS One. 2011;6:e21684. doi: 10.1371/journal.pone.0021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell. 2011;42:50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hauzenberger D, Olivier P, Gundersen D, Ruegg C. Tenascin-C inhibits beta1 integrin-dependent T lymphocyte adhesion to fibronectin through the binding of its fnIII 1-5 repeats to fibronectin. Eur J Immunol. 1999;29:1435–1447. doi:. [DOI] [PubMed] [Google Scholar]
- 51.Hellwinkel JE, Redzic JS, Harland TA, Gunaydin D, Anchordoquy TJ, Graner MW. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 2016;18:497–506. doi: 10.1093/neuonc/nov170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ren Y, Yang J, Xie R, Gao L, Yang Y, Fan H, Qian K. Exosomal-like vesicles with immune-modulatory features are present in human plasma and can induce CD4+ T-cell apoptosis in vitro. Transfusion. 2011;51:1002–1011. doi: 10.1111/trf.2011.51.issue-5. [DOI] [PubMed] [Google Scholar]
- 53.Esposito I, Penzel R, Chaib-Harrireche M, Barcena U, Bergmann F, Riedl S, Kayed H, Giese N, Kleeff J, Friess H, et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J Pathol. 2006;208:673–685. doi: 10.1002/(ISSN)1096-9896. [DOI] [PubMed] [Google Scholar]
- 54.Czopka T, Von Holst A, ffrench-Constant C, Faissner A. Regulatory mechanisms that mediate tenascin C-dependent inhibition of oligodendrocyte precursor differentiation. J Neuroscience: Official Journal Soc Neurosci. 2010;30:12310–12322. doi: 10.1523/JNEUROSCI.4957-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gotz B, Scholze A, Clement A, Joester A, Schutte K, Wigger F, Frank R, Spiess E, Ekblom P, Faissner A Tenascin-C contains distinct adhesive, anti-adhesive, and neurite outgrowth promoting sites for neurons. J Cell Biol. 1996;132:681–699. doi: 10.1083/jcb.132.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lun X, Wells JC, Grinshtein N, King JC, Hao X, Dang NH, Wang X, Aman A, Uehling D, Datti A, et al. Disulfiram when combined with copper enhances the therapeutic effects of temozolomide for the treatment of glioblastoma. Clin Cancer Res. 2016;22:3860–3875. doi: 10.1158/1078-0432.CCR-15-1798. [DOI] [PubMed] [Google Scholar]
- 57.Vecil GG, Larsen PH, Corley SM, Herx LM, Besson A, Goodyer CG, Yong VW. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J Neurosci Res. 2000;61:212–224. doi: 10.1002/(ISSN)1097-4547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.