

ABSTRACT
Dendritic cell (DC)-based vaccines are recognized as a promising immunotherapeutic strategy against cancer. Various adjuvants are often incorporated to enhance the modest immunogenicity of DC vaccines. More specifically, many of the commonly used adjuvants are derived from bacteria. In the current study, we evaluate the use of apoptosis inhibitor 5 (API5), a damage-associated molecular pattern expressed by many human cancer cells, as a novel DC vaccine adjuvant. We showed that API5 can prompt activation and maturation of DCs and activate NFkB by stimulating the Toll-like receptor signaling pathway. We also demonstrated that vaccination with API5-treated DCs pulsed with OVA, E7, or AH1-A5 peptides led to the generation of OVA, E7, or AH1-A5-specific CD8 + T cells and memory T cells, which is associated with long term tumor protection and antitumor effects in mice, against EG.7, TC-1, and CT26 tumors. Additionally, we determined that API5-mediated DC activation and immune stimulation are dependent on TLR4. Lastly, we showed that the API5 protein sequence fragment that is proximal to its leucine zipper motif is responsible for the adjuvant effects exerted by API5. Our data provide evidence that support the use of API5 as a promising adjuvant for DC-based therapies, which can be applied in combination with other cancer therapies. Most notably, our results further support the continued investigation of human-based adjuvants.
Keywords: API5, dendritic cells, cancer vaccines, adjuvants, TLR4
Introduction
Dendritic cell (DC)-based vaccines represent an encouraging treatment method against cancer.1,2 DCs are the most effective antigen-presenting cells, particularly in priming CD8 + T cell-mediated immune responses, because they express both major histocompatibility (MHC) class I and costimulatory molecules.3 Numerous preclinical and clinical studies on various cancers determined that DC vaccines are safe; however, only moderately immunogenic.3,4 Resultantly, adjuvants should be incorporated to increase the antitumor immune responses generated by DC vaccines.
Commonly used adjuvants include cytokines, Toll-like receptor ligands and heat shock proteins. Cytokines such as interleukin-2 (IL-2) and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been used previously as adjuvants in cancer vaccines.5,6 The majority of the current cancer vaccine adjuvants are derived from bacteria, and are mainly used to provoke innate immune responses by stimulating the Toll-like receptors (TLR).7 Moreover, extracellular and membrane heat shock proteins (HSPs) have been previously shown to enhance vaccine-induced immune responses.8 Several adjuvants have been incorporated into DC vaccines, including tumor necrosis factor-related activation-induced cytokine (TRANCE) and Picibanil (OK-432), a bacterial adjuvant.9,10 In addition, DC vaccines can be used in combination with other therapies, such as chemotherapy, which can be used to modulate the tumor microenvironment to elicit stronger antitumor immune responses.11
It has been reported that tumor cells secrete various stress proteins in response to exposure to stressing conditions such as chemotherapy. Many of these stress proteins, such as High Mobility Group Box 1 (HMGB1), function to promote tumor progression and the maintenance of malignant phenotypes.12–14 In addition to their pro-tumorgenic effects, many of these stressed proteins have also been reported to act as danger-associated molecular patterns (DAMPs) that can stimulate and activate anti-tumor immunity.15–18 For example, we have previously reported that pancreatic adenocarcinoma upregulated factor (PAUF), a DAMP that is overexpressed by many cancers and promotes cancer angiogenesis, proliferation, and metastasis, can serve as a ligand for TLR4-Myd88 signaling pathway to stimulate DCs activation, maturation, and antigen presentation.19 Thus, these DAMPs can potentially serve as a new type of novel and human-originated adjuvant for the activation of innate and adaptive immunity for cancer treatment.
Apoptosis inhibitor 5 (API5), also known as antiapoptosis clone 11 (AAC-11) or fibroblast growth factor-2-interacting factor (FIF), was first reported to support cell viability after withdrawal of growth factors,20 potentially by inhibiting caspase-3 or E2F1 induced apoptosis.21,22 It is then realized that API5 is overexpressed by many types of tumors to promote tumor progression and serve as poor prognostic marker for these cancers.23–30 However, its potential effect in modulating immune response has not been previously reported. Based on the similar functions of API5 with many other tumor stress proteins, we hypothesize that API5 may potentially act as a DAMP and stimulate the activation of immune response.
In this study, we assessed the use of API5 as an adjuvant for an antigen-specific DC vaccine in mice. We found that API5 can bind to TLR4, induce activation and maturation in DCs, and stimulate TLR signaling pathways leading to NFkB activation. DCs pulsed with OVA, E7, or AH1-A5 antigenic peptide treated with API5 can generate OVA, E7, or AH1-A5-specific CD8 + T cells and memory CD8 + T cells, which was associated with long-term tumor protection against EG.7, TC-1, and CT26 tumor challenge. Furthermore, the antigen-specific CD8 + T cell responses produced by API5-treated DC vaccines produced an antitumor effect, which led to prolonged survival. We also demonstrated that the mechanism of API5-enhanced DC vaccine potency was dependent on TLR4. Finally, we showed that the API5 protein fragment that is proximal to its leucine zipper motif is responsible for the observed DC stimulating and adjuvanting effect. Taken together, our results suggest that API5 can serve as a novel adjuvant to increase the immunogenicity of cancer vaccines.
Results
API5 is expressed by both murine and human cancers and acts as a damage associated molecular pattern that interacts and activates TLR4-NFkB signaling pathway
We first confirmed the expression of API5 by various murine and human cancer cell lines using Western Blot (Figure 1A). In addition, treating various tumor cell lines with chemotherapeutic agent doxorubicin significantly enhanced the expression of API5 by the tumor cells, suggesting that API5 functions as a DAMP in stressed tumor cells (Figure 1B). Since it has been previously reported that many DAMPs released from chemically stressed cancer cells have the ability to induce the activation of DCs and T cells through stimulating the TLR pathway,18 we sought to evaluate whether API5 can interact with TLR molecules. Recombinant human API5 proteins were generated and confirmed using Western Blot (Figure 1C). Immunoprecipitation analysis reveals that API5 has the ability to bind and interact with TLR4, but not TLR2 (Figure 1D). Furthermore, treating HEK293 cells transfected with plasmid encoding luciferase under NFkB promoter with API5 resulted in a significant increase in luciferase expression that is similar to treatment with a common TLR-4 agonist Lipopolysaccharide (LPS) (Figure 1E). Using BLItz binding affinity analysis system, the KD between API5 protein and TLR4 was assessed and found to be approximately 430nM (Fig S1). These results suggest that API5 is a DAMP molecule that has the ability to bind and trigger TLR4-NFkB signaling pathway.
Figure 1.
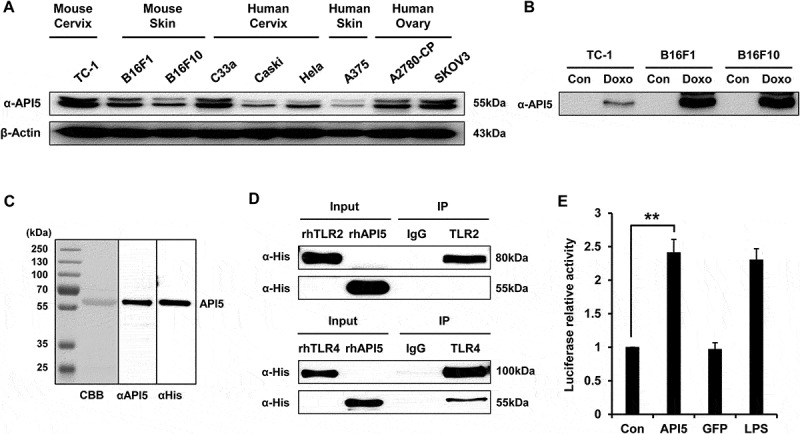
API5 as a DAMP that interacts with TLR4. (A) API5 expression level of various cancer cells were detected using western blot analysis. (B) To confirm that API5 protein is a DAMP, expression of API5 by cancer cells with or without doxorubicin treatment was measured using western blot analysis. (C) The production of recombinant human API5 protein via bacterial protein purification system was confirmed using CBB (coomassie brilliant blue) and western blot analysis. (D) Binding between API5 and TLR2 or TLR4 was assessed using immunoprecipitation. (E) TLR4 expressing HEK293 cells were transfected with plasmid encoding luciferase under NF-kB promoter, and treated with API5 (0.5μg/ml), GFP (0.5μg/ml) or LPS (100ng/ml), and assessed for luciferase signal. Data are presented as mean ± SD (** = p < 0.01).
API5 induce maturation, activation, and migration of DCs via the activation of TLR signaling pathway in vitro
Based on the identified binding interaction between API5 and TLR4, we hypothesize that API5 has the potential ability to stimulate DCs. Thus, we evaluated the effects of API5 on murine DCs by characterizing the pro-inflammatory cytokine expression and activation markers. As demonstrated in Figure 2A, incubation with API5 resulted in a dose-dependent increase for the expression of pro-inflammatory cytokines TNF-α, IL-1β, IL-6, IL-10, IL-12p70, and IFN-β in DCs compared to untreated DCs or DCs treated with irrelevant GFP protein. In addition, DCs incubated with API5 express higher levels of maturation surface markers MHC class I, CD40, CD80, and CD86 compared to untreated DCs or DCs treated with irrelevant GFP protein (Figure 2B). Further co-culturing of these API5 treated mature DCs with CD40L expressing cells re-stimulated these mature DCs to secret cytokine IL-12p70 (Fig S2), suggesting that API5 treatment does not lead to DC exhaustion as that observed in LPS-treated DCs.31 Boiling API5 protein or treating API5 protein with proteinase K before incubation with DCs abolishes its DC stimulating effects (Fig S3). Furthermore, API5 protein produced via E.coli bacteria-based protein purification method or HEK293 mammalian cell-based protein purification method exerted comparable DC stimulating ability (Fig S4), suggesting that the observed DC stimulating effects of API5 was not due to endotoxin or other bacterial contamination. Interestingly, DCs incubated with API5 also expressed higher levels of migration factor CCR7 (Figure 2D). These results indicate that API5 can activate and induce DC maturation. To demonstrate that activation of DCs by API5 is potentially induced via the interaction of TLR4 and the stimulation of TLR-signaling pathway, we cultured DCs with API5 and performed Western Blot analysis for various TLR signaling pathway proteins. Elevated levels of P-ERK, P-JNK, P-P38, P-AKT were observed in DCs after incubation with API5, while a reduction in IkB-α following API5 incubation were observed (Figure 2C). Similar to results observed in murine DCs, treating human differentiated macrophage cell line THP-1 also resulted in the upregulation of maturation markers CD80, CD83, CD86, migration marker CCR7, as well as the expression of pro-inflammatory cytokines TNF-α, IL-6, IL-10, and IFN-β (Figure 2E and F). Likewise, treating human DCs with API5 induced upregulation of HLA-DR, CD80, and CD86 expression, as well as IL-10 and IL-12p70 cytokine production by treated DCs (Fig S5). These results show that API5 can activate innate immune responses in DCs through stimulation of TLR in both murine and human setting.
Figure 2.
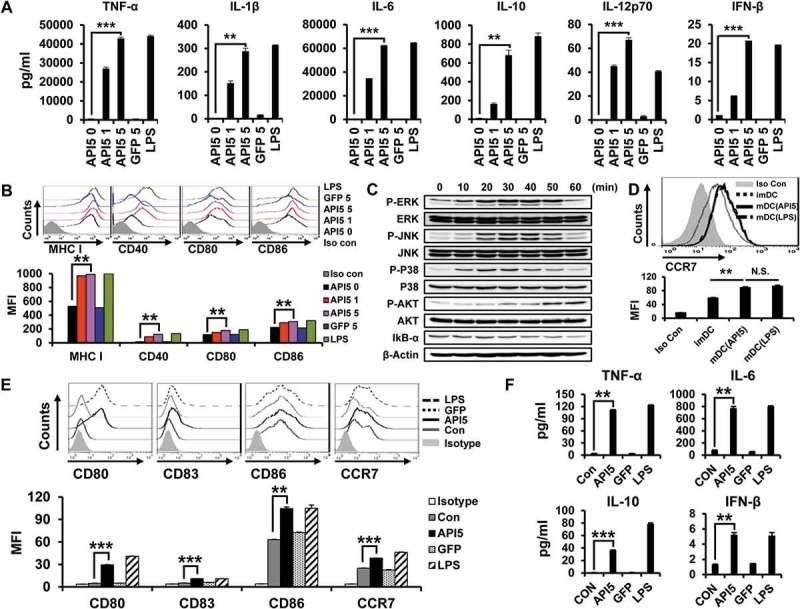
Recombinant human API5 protein matures and activates mouse and human APCs. (A) Wild type murine dendritic cells were treated with recombinant API5 (1μg/ml or 5μg/ml), GFP (5μg/ml) or LPS (100ng/ml). Supernatants were collected and levels of various cytokines were assessed using ELISA. (B) Murine DCs were treated with API5 (1μg/ml or 5μg/ml) protein, GFP (5μg/ml) or LPS (100ng/ml) and assessed for the expression of various maturation markers (CD40, CD80, CD86 and MHCI) using flow cytometry. (C) Murine DCs were treated with API5 (5μg/ml) protein for 0, 10, 20, 30, 40, 50 or 60 minutes, lysed, and assessed for the expression of various MAPk and IkB-α using western blot analysis. (D) Expression of migration marker CCR7 by murine DCs with or without API5 or LPS treatment were measured using flow cytometry. (E-F) Macrophage-like human THP-1 cells were treated with API5 (5μg/ml), GFP (5μg/ml) or LPS (100ng/ml). Cells and supernatants were collected and assessed for the expression of various macrophage maturation markers and secretion of various cytokines using flow cytometry and ELISA, respectively. Data are presented as mean ± SD (** = p < 0.01, *** = p < 0.001, N.S. = not significant).
API5-activated DC vaccine produces antigen-specific CD8 + T cells and memory T cells in vivo and provokes long-term tumor protection
We evaluated the potential of API5 in producing antigen-specific adaptive cellular immune responses using API5-activated DCs as a DC based vaccine strategy. As shown in Figure 3A, C and E, potent antigen-specific CD8 + T cell expansion were observed in mice vaccinated with API5-treated DCs pulsed with antigenic peptides. Further characterization of the immune response generated by API5 treated, OVA peptide loaded DCs demonstrated a significant activation of Th1 CD4 + T cells, generation of OVA-specific antibodies, as well as significant OVA-specific CD8 + T cell responses (Fig S6). We then sought to determine whether the induction of antigen-specific CTL proliferation by API5 treated, antigenic peptide pulsed DC vaccination translated into tumor protection. As shown in Figure 3B, D and F, all the mice vaccinated with API5 treated DCs pulsed with OVA, E7, or AH1-A5 peptide stayed tumor free for at least 50 days following tumor challenge, and 80 to 90% of mice from the group treated with LPS-treated DCs pulsed with antigenic peptide stayed tumor free for at least 50 days, while all other mice developed tumors within 20 days following tumor challenge. Depletion of various subsets of immune cells following API5 treated DC vaccination suggests that the protective anti-tumor effects elicited by the vaccination is CD8 + T cell dependent and CD4 + T cell and NK cell independent (Fig S7). These results suggest that API5-treated DCs can activate antigen-specific CD8 + T cells capable of tumor protection. Next, we assessed whether the API5-treated DC vaccines can induce long-term memory. Much to our surprise, stimulation by antigenic peptides still led to the generation of more activated antigen-specific CD8 + T cells in the splenocytes 7 weeks after last immunization (Figure 4A, C and E). Further, we observed a greater number of activated antigen-specific CD8 + T cells after tumor challenge. Additionally, the API5-treated DC vaccines maintained their tumor protection effects 7 weeks after the last vaccination, in which all the vaccinated mice stayed tumor free for at least 30 days following tumor challenge (Figure 4B, D and F). These results indicate that API5-treated DC vaccines can generate antigen-specific memory CD8 + T cells that can lead to long-term tumor prevention.
Figure 3.
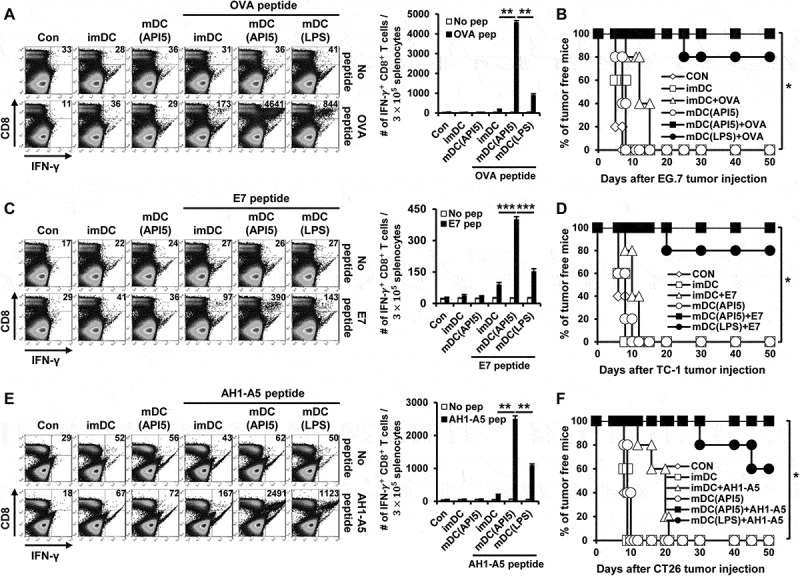
API5-mediated DC vaccination produces antigen-specific CD8 ± T cells and prevents tumor formation. Wild type C57BL/6 mice (5 mice per group) were immunized with 1) PBS, 2) immature DC, 3) immature DC loaded with antigen peptide, 4) API5 treated mature DC, 5) API5 treated mature DC loaded with antigen peptide, and 6) LPS treated mature DC loaded with antigen peptide, twice at one week intervals. One week following respective vaccination, the abundance of OVA (A), E7 (C) or AH1-A5 (E) specific CD8+ and IFN-γ+ T cells in splenocytes of mice were measured using flow cytometry. Kaplan-Meier survival analysis of mice(5 mice per group) challenged with TC-1 (B), EG.7 (D) or CT26 (F) tumor cells, at one week after the second DC vaccination. Data are presented as mean ± SD (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
Figure 4.
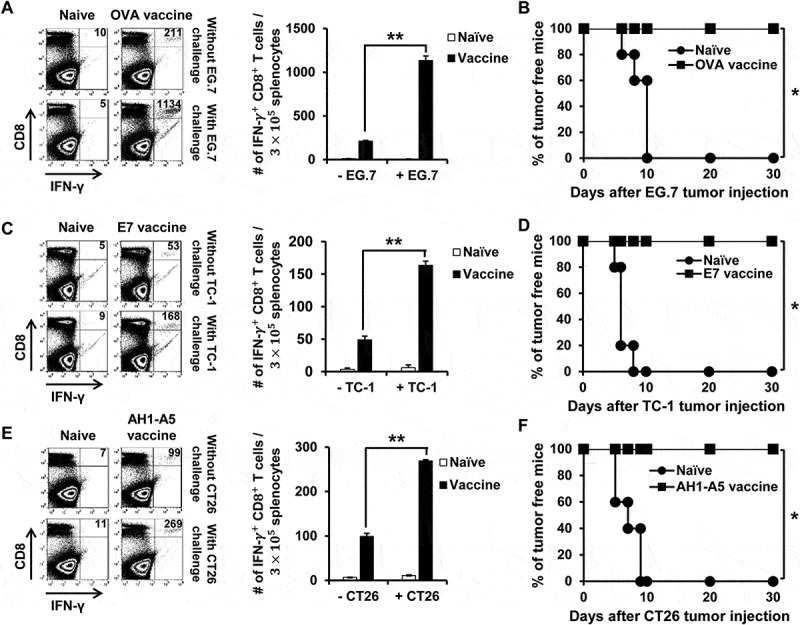
Long-term antigen-specific CD8 ± T cell memory and tumor protective effect produced by API5 treated DC vaccination. Mice (5 mice per group) were vaccinated with PBS or API5 treated mature DC loaded OVA (A), E7 (C) or AH1-A5 twice at one week intervals. (E). Seven weeks after second vaccination, mice were challenged with or without EG.7, TC-1 or CT26 tumor cells, respectively. One week after tumor challenge, splenocytes of mice were collected and assessed for the abundance of antigen specific CD8+ T cells by flow cytometry. The tumor free survival of EG.7 (B), TC-1 (D) or CT26 (F) tumor challenged mice cells were followed for up to 30 days after tumor challenge. Data are presented as mean ± SD (* = p < 0.05, ** = p < 0.01).
API5-mediated DC vaccine induces therapeutic antitumor effect and prolongs survival in tumor-bearing mice
Next, we sought to evaluate the potential of API5-treated DC vaccine in treating tumors. Interestingly, vaccination with API5-treated DCs pulsed with antigenic peptides at 3 and 10 days following tumor challenge effectively suppressed the growth of tumor for at least 20 days in mice bearing EG.7, TC-1, or CT26 tumors, while vaccination with DCs treated with API5 or antigenic peptides alone did not induce any effects in controlling tumor growth (Figure 5A, C and E). Notably, tumor-bearing mice vaccinated with API5-treated DCs pulsed with antigenic peptide demonstrated long durations of survival compared to tumor-bearing mice treated with other regimens (Figure 5B, D and F). Similar tumor control and survival results were observed when tumor bearing mice were treated with a delayed vaccination schedule, for which tumor-bearing mice received various DC vaccinations at 5 and 12 days after tumor challenge (Fig S8). These data suggest that vaccination with API5-treated DC, pulsed with antigenic peptides, can provoke potent therapeutic antitumor effect and prolonged survival.
Figure 5.
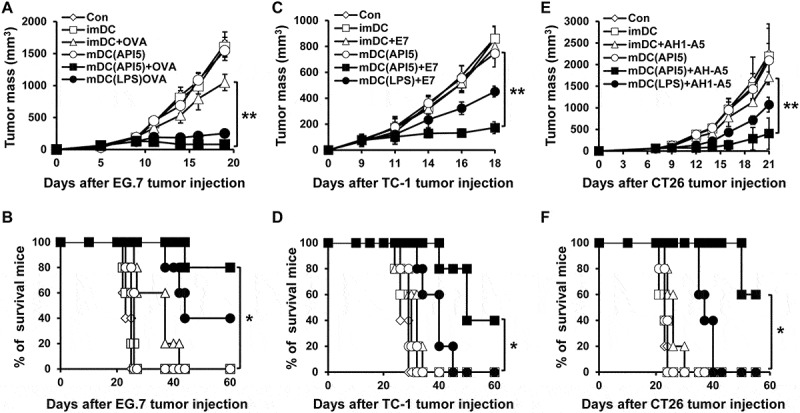
API5-treated DC vaccination reduces tumor growth and prolongs survival. Mice (5 mice per group) were challenged with EG.7 (A), TC-1 (C) or CT26 (E) tumor cells. Three and ten days after tumor challenge, mice were treated with 1) PBS, 2) immature DC, 3) immature DC loaded with antigen peptide, 4) API5 treated mature DC, 5) API5 treated mature DC loaded with antigen peptide, and 6) LPS treated mature DC loaded with antigen peptide, in the footpad twice at one week intervals. Tumor volume was measured using digital caliper. (B, D, F) Mice survival were assessed using Kaplan-Meier analysis. Data are presented as mean ± SD (* = p ≤ 0.05, ** = p ≤ 0.01).
API5-mediated activation and maturation of DCs depend on TLR4
Since we demonstrated that API5 interacts with TLR4 and activates TLR-signaling in DCs, we sought to confirm whether the DC stimulating and adjuvant effects of API5 were dependent on TLR4. To do so, we repeated in vitro experiments using either DCs from TLR2 or TLR4 knockout mice. Treating TLR4-/- DCs with API5 failed to induce the expression of pro-inflammatory cytokines TNF-α, IL-1β, IL-6, IL-10, IL-12p70, and IFN-β that was observed in wild type DCs and TLR2-/- DCs treated with API5 (Figure 6A). API5 treatment also failed to induce the expression of maturation markers MHC-I, CD40, CD80, and CD86 by TLR4 -/- DCs as compared to wild type and TLR2 -/- DCs (Figure 6B). Likewise, there was no increase in TLR signaling proteins, including P-ERK, P-JNK, P-P38, and P-AKT in TLR4 -/- DCs following API5 treatment as compared to API5-treated wild type and TLR2 -/- DCs. Also, there was no observed reduction in IkB-α in TLR4-/- DCs following API5 treatment, suggesting the lack of NFkB activation (Figure 6C). Similar to TLR4-/- DCs, knocking out the downstream molecule of TLR4 signaling pathway, MyD88 or TRIF, also abolishes the ability of API5 in stimulating the maturation of DCs and the secretion of pro-inflammatory cytokines by DCs (Fig S9). When comparing the efficacy of API5-treated, antigenic peptide loaded DCs in generating CD8 + T cell response, vaccinating mice with TLR4 -/- DCs treated with API5 and loaded with E7 peptide failed to elicit a significant E7-specific CD8 + T cells response compared to vaccinating with wild type, API5 treated, and E7-peptide loaded DCs (Figure 6D). To investigate the resultant therapeutic antitumor effects, mice were challenged with TC-1 tumor cells then vaccinated with wild type or TLR4-/- DCs that were treated with API5 and E7 peptides. Knocking out TLR4 in DCs completely abolished its therapeutic antitumor effects even after treating with API5 and E7 peptide, resulting in the inability to control tumor growth and prolong the survival of mice (Figure 6E and F). These results indicate that TLR4 is necessary for API5 to induce activation and maturation of DCs to induce antigen-specific adaptive cellular immune response and antitumor effects.
Figure 6.
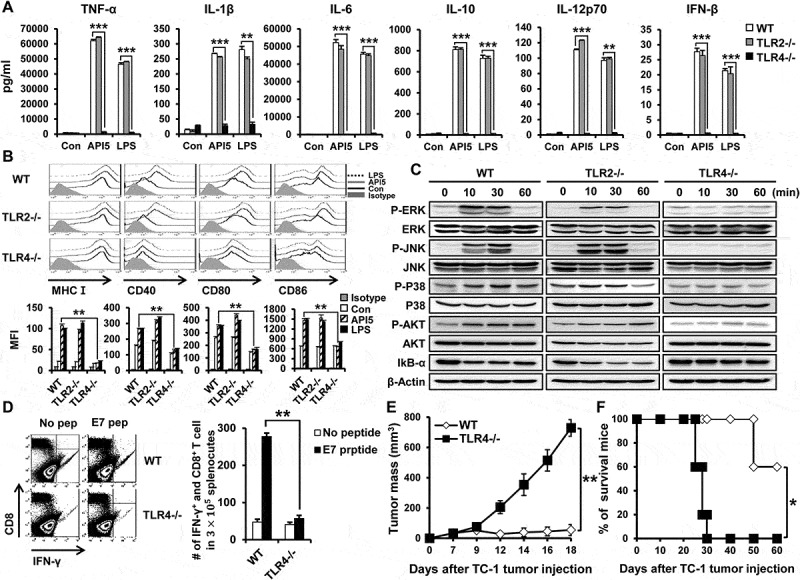
DC activating effect of API5 is TLR4 dependent. Wild type, TLR2 KO or TLR4 KO murine DCs were treated with API5 (5μg/ml) protein or LPS (100ng/ml). (A) Supernatants were collected and levels of various cytokines were assessed using ELISA. (B) Expression of various maturation markers (CD40, CD80, CD86 and MHCI) by treated cells was analyzed using flow cytometry. (C) Wild type, TLR2 KO or TLR4 KO murine DCs were treated with API5 (5μg/ml) protein for 0, 10, 30, or 60 minutes, lysed, and assessed for the expression of various MAPk and IkB-α using western blot analysis. (D) Mice (5 mice per group) were vaccinated with wild type or TLR4 KO DCs treated with API5 and pulsed with E7 peptide twice at one week intervals. One week after second vaccination, splenocytes of mice were collected and assessed for E7-specific CD8+ T cells. (E-F) Mice (5 mice per group) were challenged with TC-1 cells. Three days after tumor challenge, mice were vaccinated with wild type or TLR4 KO DCs treated with API5 and pulsed with E7 peptide twice at one week intervals. (E) Tumor growth was measured using digital caliper. (F) Mice survival were assessed using Kaplan-Meier analysis. Data are presented as mean ± SD (* = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001).
API5 protein sequence proximal to the leucine zipper motif is responsible for the observed adjuvant effects exerted by API5
After demonstrating the ability of API5 to stimulate and activate DCs, we sought to further characterize the portion of the protein that is crucial for the adjuvant functions. We cloned and generated 3 different fragments of API5 protein: a fragment (1–230) that contains a fibroblast growth factor 2 (FGF2) binding region of API5, another fragment (108–391) that contains a FGF2 binding region as well as the leucine zipper motif of API5, and a third fragment (320–504) that contains the leucine zipper motif and the Nuclear location sequence of API5, and confirmed the expression of API5 fragments by Western Blot (Figure 7A and B). When comparing the ability of the API5 protein fragments in stimulating DC activation, treating DCs with API5(1–230) fragment failed to induce the secretion of pro-inflammatory cytokines. On the other hand, treating DCs with API5(108–391) fragment as well as API5(320–504) fragment led to the secretion of noticeable amount of pro-inflammatory cytokines by DCs, although cytokine secretion was much lower compared to when treating DCs with full length API5 protein (Figure 7C). Similar to the secretion of pro-inflammatory cytokines, DCs treated with API5(1–230) fragment failed to induce the upregulation of maturation markers CD40 and CD80, while DCs treated with API5(108–391) and API5(320–504) fragments both induced a noticeable increase of expression in maturation markers compared to control, although the expressions were less compared to the full length API5 treated DCs (Figure 7D). These results demonstrated that the ability of API5 to act as an adjuvant for DC stimulation is mainly due to its protein fragment proximal to the leucine zipper motif.
Figure 7.
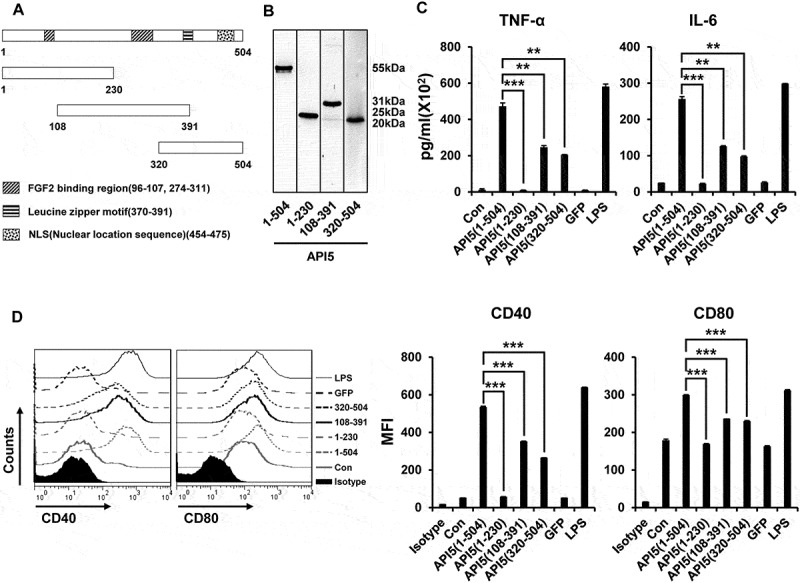
The effect of various API5 protein fragments on inducing DC activation and maturation. (A) Fragments of API5 protein were constructed (API5 full [aa1-504], API5 forward [aa1-230], API5 middle [aa108-391] and API5 end [aa320-504]). (B) Various API5 protein fragments were purified using E.coli system and confirmed using western blot analysis. (C) DCs were treated with various API5 protein fragments and assessed for secretion of TNF-α and IL-6 cytokine levels using ELISA. (D) Expression of maturation marker (CD40 and CD80) by various API5 fragment treated DCs were measured using flow cytometry. Data are presented as mean ± SD (** = p ≤ 0.01, *** = p ≤ 0.001).
Discussion
In the current study, we evaluated the effect of API5 in enhancing the potency of a DC-based vaccine in mice. We demonstrated that API5 acted as a DAMP and interacted with TLR4, and treatment with API5 induced activation and maturation of DCs, and stimulated TLR signaling pathway resulting in activation of NFkB. Additionally, API5-treated DCs pulsed with OVA, E7, and AH5-A1 peptides generated greater numbers of antigen-specific CD8 + T cells including memory T cells, which led to long term protection in mice against EG.7, TC-1, and CT26 tumor challenge. Furthermore, we demonstrated that the antigen-specific CD8 + T cell immune response elicited by the API5-treated DC vaccine leads to powerful antitumor effects and prolonged survival in tumor-bearing mice. We’ve also demonstrated that API5’s adjuvant effect in DC vaccines is TLR4 dependent, and is mediated by its protein sequence proximal to the leucine zipper motif.
Of note, we performed experiments using MyD88 and TRIF knockout mice to verify that API5 leads to stimulation of TLR signaling pathway. As shown in Fig S9, treatment with API5 does not lead to increases in the pro-inflammatory cytokines TNF-α, IL-6, IL-10, and IL-12p70 in DCs lacking MyD88 or TRIF. Furthermore, expression levels of CD40 and CD80 were not elevated following API5 treatment in DCs lacking MyD88 TRIF. These results indicate that activation of DCs by API5 is through MyD88 and TRIF-dependent TLR stimulation.
To date, API5 is more commonly known as a tumorigenic protein that has been shown to mediate immune escape and cancer metastasis by activating P-ERK signaling pathway without activating other molecules in the NFkB signaling such as P-P38 and P-AKT in tumor cells.24,26 Here we demonstrate that API5 can induce the upregulation of NFkB signaling pathway including P-ERK, P-JNK, P-P38, and P-AKT in DCs through interaction with TLR4. This is potentially due to the various binding targets of API5. The observed pro-tumorgenic effects of API5 are mainly mediated by its association with FGF-2 signaling, where as we report the association of API5 with TLR4 in this study. Further, we showed that the activation of DCs by API5 is mainly mediated by TLR4 but not TLR2 stimulation. The expression levels of TLR4 and FGF-2 can vary between DCs and tumor cells, leading to different levels of activation of various pathways, resulting in different downstream effects.
Some issues will need to be addressed before API5-treated DC vaccine can be considered translatable for human use. To keep the cost of producing this adjuvant down, E. coli can be used to produce recombinant protein. Although API5 possesses various pro-tumorigenic properties, in this study, we are only treating API5 to DC. API5 is washed off before DCs are injected into mice; no API5 is actually injected into the body. Furthermore, we found that API5 treatment leads to activation and maturation in DCs, and is unlikely to induce oncogenic properties. Thus, the API5-treated DC vaccine should not carry oncogenic effects. Nevertheless, this safety concern should be evaluated in future studies.
To date, over one hundred adjuvants have been developed and tested in vaccines.32 The majority of these adjuvants are pathogen-associated molecular patterns (PAMPs) derived from organic or inorganic compounds, or from microorganisms. In the current study, we used API5, a protein of human origin, to activate DCs for the first time. The immune stimulating properties of API5 are very similar to those of damage-associated molecular pattern molecules (DAMPs). In particular, HMGB1, a ligand for TLR4 and TLR2 with human origin, also activates DCs through a TLR4 dependent manner.33 Our results serve as a reference for further investigation of using other DAMPs such as HMGB1, to induce activation and maturation in DCs to generate antigen-specific CD8 + T cells. While PAMPs and DAMPs are danger signals that can activate DCs and other immune cells, their excessive presence may lead to toxicities and adverse effects, such as acute and chronic inflammation, which may ultimately lead to cancer.34 Future studies should investigate whether the human origin of API5 can help lessen the undesirable effects associated with danger signal derived adjuvants.
In the current study, we demonstrated the use of API5 as a novel human-derived adjuvant can improve the antigen-specific CD8 + T cell antitumor immunity of DC vaccines, by inducing activation and maturation of DCs and stimulating the innate immunity. API5 enhanced cancer vaccines may also be used in combination with other therapies, such as chemotherapy, to reduce immunosuppression in the tumor microenvironment and further increase the antitumor effects. In conclusion, API5 is a novel human-based adjuvant with encouraging translational potential.
Materials and methods
Mice
5- to 8 week old female wild-type C57BL/6 mice were purchased from OrientBio. 5- to 8 week old female C57BL/6J TLR2 knockout mice (TLR2-/-; B6.129-Tlr), C57BL/10 TLR4 knockout mice (TLR4-/-; C57BL/10ScNJ) and C57BL/6 OT-I transgenic mice (OT-I; C57BL/6-Tg-TcraTcrb- 1100Mjb/J) were obtained from The Jackson Laboratory. MyD88 knockout mice were obtained from Dr. Heung-Kyu Lee (KAIST University). TRIF knockout mice were obtained from Dr. Jong-Hwan Park (Konyang University). All mice were housed and bred under specific, pathogen-free conditions in the housing facility at Konkuk University. All procedures were performed according to approved protocols by the Institutional Animal Care and Use Committee (IACUC) at Konkuk University.
Cells
The generation of TC-1 cells has been previously described.35 EG.7 cells (EL4 lymphoma cell transduced with the gene encoding for OVA) and CT26 (mouse colon cancer cell line) were obtained from ATCC. Hela, Caski, C33a, 526mel, A375, SKOV3 and A2780 (human cancer cell lines) were obtained from ATCC and THP-1 (human monocyte cell line), B16F1, B16F10 (mouse cancer cell lines) and HEK293 cells were obtained from the Korean Cell Line Bank. Cells were maintained at 37°C under 5% CO2 atmosphere in RPMI 1640 or DMEM medium (biowest) supplemented with 10% fetal bovine serum (biowest), 50U/ml penicillin streptomycin (biowest), 2mM L-glutamine, 1mM sodium pyruvate and 2mM nonessential amino acid. THP-1 cells were used to generate macrophage-like THP-1 cells by incubating with PMA (100ng/ml, sigma) for two days. Dendritic cells were derived from mouse bone marrow precursors by culturing in RPMI 1640 medium supplemented with 10% fetal bovine serum, 50U/ml penicillin streptomycin, 2mM L-glutamine, 1mM sodium pyruvate and 2mM nonessential amino acid, 0.05mM 2-mercaptoethanol (Gibco) and 10ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Jw creagene) for 6 days. For interaction between API5 and TLR4, 293/hTLR4A-MD2-CD14 cells were obtained from invivogen and cultured following manufacturer’s protocol.
Recombinant human API5 and GFP purification
For expression and purification of recombinant human API5 (full protein: aa1-504, protein fragments aa1-230, aa108-391, and aa320-504) protein and GFP protein in E. coli, pET-28b-human API5 (encoding API5 fragment aa1-504, aa1-230, aa108-391, or aa320-504) or pET-28a-GFP, plasmids were constructed and transformed into BL21 (DE3) E. coli (NEB). After transformed E. coli cells were cultured in LB medium at 37°C for three hours, protein expression was induced by isopropyl-b-D-thiogalactopyranoside (IPTG, 1mmol/L, Bio Basic) for six hours. The cultured E. coli cells were harvested and lysed with SoluLyse buffer (Genlantis), deoxynuclease I (DNase I, 200U/ml, invitrogen), lysozyme (100μg/ml, Gibco) and DTT (5mmol/L, Bio Basic) at room temperature for two hours. For expression and purification of recombinant human API5 protein in mammalian cell system pCDNA3.1 (±)-His-API5-v5-His plasmids were constructed and transfected into HEK293 cells. Transfected HEK293 cells were cultured in DMED medium at 37°C for two days, harvested and lysed following manufacturer’s freeze and thaw method (Qiagen). Lysed cells were centrifuged at 12,000rpm for ten minutes. Supernatants were filtered at 0.45μm filter and purified Ni-NTA agarose (Qiagen) following the manufacturer’s instruction. The purified protein was concentrated with 10K centrifugal filter (Merck Millipore) and analyzed by staining Coomassie Brilliant Blue of 12% SDS-PAGE gels and by using western blotting to API5 (Novus) or His (MBL) antibody. Endotoxin contaminated protein was removed by using Triton X-114 Surfact-Amps solution (Thermo Scientific). Endotoxin levels (>0.1EU/mg) were measured by using Limulus amoebocyte lysate (LAL, Lonza). The sequence homology of human and murine API5 protein is 98.4% based on NCBI protein blast sequence alignment analysis.
NF-kB luciferase activity assay
For NF-kB activity to API5 protein, 293/hTLR4A-MD2-CD14 cells (/well) were seeded in 96-well white plate, transfected with pGL4.32[luc2P/NF-kB-RE/Hygro] vector (promega) and pRL-TK vector (promega) for 16 hours at 37°C. Incubated cells were treated with API5 (5μg/ml), GFP (0.5μg/ml) or LPS (100ng/ml) for 4 hours. Subsequently NF-kB luciferase activity of 293/hTLR4A-MD2-CD14 cells were measured following manufacturer’s protocol (promega).
Maturation and activation of dendritic cells and THP-1 macrophage
For maturation of differentiated dendritic cells from wild type, TLR2 KO, TLR4 KO, MyD88 KO, TRIF KO and THP-1 macrophage cells, cells were incubated overnight with or without recombinant API5 protein (1μg or 5μg/ml), GFP protein (5μg/ml) or LPS (100ng/ml, invivogen). Cells were collected and stained with FITC-CD11c and PE-CD40, PE-CD80, PE-CD83, PE-CD86, PE-MHC-I or PE-CCR7 (biolegend). Cells were analyzed on FACSCalibur using CELLQuest (Becton Dickinson). For activation of dendritic cells and THP-1 macrophage cells, supernatant from cultured cells was collected and cytokine levels were measured using anti-human and anti-mouse TNF-α, IL-1β, IL-6, IL-10, IL-12p70 (ebioscience) and IFN-β (pbl) ELISA kit following the manufacturer’s instructions. Dendritic cells were incubated with API5 protein (5μg/ml) for 0, 10, 20, 30, 40, 50 or 60 minutes. Cells were collected, washed and lysed with the protein extraction solution RIPA (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 1 mM phenylmethylsulphonyl fluoride [PMSF], 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40 [NP-40], and 0.5 mM EDTA) (Elpis Biotech) at 4°C for thirty minutes. Extracted protein concentration was measured using Bradford protein assay kit (pierce). Equal protein of each group were solubilized in SDS-PAGE loading buffer (250mM Tris-HCl, pH 6.8, 0.5M DTT, 10% SDS, 0.5% bromophenol blue, 50% Glycerol), boiled for 10min, separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes (Roche). The primary antibodies against phospho-AKT, total AKT, phospho-ERK, total ERK, phospho-JNK, total JNK, phospho-p38, total p38 MAPK, IkB-α (Cell signaling technology) or β-actin (Santa Cruz) were used at 1:1000 dilution in 5% BSA. Appropriate horseradish peroxidase (HRP) conjugated secondary antibodies (abbiotech and Enzo) were used at 1:10000 dilution in 5% skim milk. Immunoreactive bands were confirmed by using enhanced chemiluminescence (ECL, Merck Millipore).
Generation of monocyte-derived human immature dcs and mature DCs
Monocytes were isolated from peripheral blood MNCs obtained from healthy donors according to the protocol by the Institutional Research Board of Chonnam National University Hwasun Hospital, using a CD14± magnetic activating cell sorting (MACs) system (Miltenyi Biotec Inc., Auburn, CA, USA), and cultured at 5 × 105 cells/well in six-well plates (BD Falcon, San Jose, CA, USA) in RPMI-1640 medium (Gibco-BRL; Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone; Logan, UT, USA), 1% penicillin-streptomycin (PS), 50 ng/mL recombinant human granulocyte macrophage colony-stimulating factor (LG Biochemical, Daejon, Republic of Korea), and 20 ng/mL recombinant human IL-4 (R&D Systems; Minneapolis, MN, USA). The culture medium was replenished every 2 days. On day 6, approximately 2 × 105 imDCs were activated using LPS (200 ng/mL), GFP (5µg/mL), API5 protein (1µg/mL), or API5 protein (5 µg/mL), respectively. Maturated DCs were harvested on day 8.
Human DC phenotypes analysis
After treating immature human DCs with or without LPS (200 ng/mL), GFP (5µg/mL), API5 protein (1µg/mL), or API5 protein (5 µg/mL), the treated DCs were harvested. Monoclonal antibodies against human HLA-DR-FITC, CD80-PE, and CD86-FITC (miltenyi) were used to detect surface markers on DCs for phenotypic analysis using flow cytometry. Cell debris was eliminated by forward and side scatter gating. The samples were acquired on a FACSCalibur cell sorter (Becton Dickinson, Mountain View, CA), and analyzed using WinMDI Version 2.9 software (Biology Software Net: http://en.bio-soft.net/other/WinMDI.html).
Cytokine production by human DCs
Cytokine (IL-12p70 and IL-10) production by DCs was determined using ELISA assay (BD Bioscience). After treating immature human DCs with or without LPS (200 ng/mL), GFP (5µg/mL), API5 protein (1µg/mL), or API5 protein (5 µg/mL), the supernatant was harvested to measure IL-12p70 and IL-10 production. To assess DC re-stimulation following API5 treatment, human DCs were treated with 1μg/ml or 5μg/ml of API5 protein for 24 hours. After washing, the maturated DCs were cultured in 96-well plates at 2 × 104 cells/well, and stimulated with 5 × 104 cells/well of CD40 ligand (CD40L)-transfected J558 cells (as an analog of CD40L-expressing Th cells; kind gift from Dr. P. Lane, University of Birmingham, UK). The supernatant was harvested after 24 hours, and assessed for IL-12p70 cytokine level via ELISA. Each sample was analyzed in triplicate, and the mean absorbance values of each set of standards and samples were calculated.
In vitro endotoxin-free API5 experiment
Recombinant API5 protein (5μg/ml) or LPS (100ng/ml) was incubated with proteinase K (100μg/ml) (promega) at 45°C for three hours or polymyxin B (10μg/ml) (sigma) at room temperature for ten minutes and boiled to eliminate proteinase K effect. Dendritic cells were treated with prepared API5 protein and LPS. Cells and supernatants were collected and measured with flow cytometry (CD40 and MHC-I) and ELISA (TNF-α, IL-1β, IL-6, IL-10, IL-12p70 and IFN-β), respectively.
In vivo experiment
For antigen specific CD8 + T cell production in response to DC vaccine, mice were injected with PBS, immature DC, immature DC loaded with antigen peptides, mature DC (treated API5), mature DC (treated API5) loaded with antigen peptides, or mature DC (treated LPS) loaded with antigen peptides (five mice per group) in footpad twice at one week intervals. Antigen peptides (E7 [RAHYNIVTE], OVA [SIINFEKL], AH1-A5 [SPSYAYHQF]) (Anygen) against three tumor model were used. Seven days after second DC vaccination, splenocytes were obtained from immunized mice, re-stimulated with antigen peptide overnight and assessed using intracellular cytokine staining protocol following manufacturer’s instructions (BD). For production and boosting of antigen-specific CD8± memory T cells, mice were immunized with PBS or mature DC (treated API5) loaded with antigen peptides twice at one week intervals. Seven weeks after last DC inoculation, mice were subcutaneously injected with or without tumor cells (2x105 TC-1, 1 × 106 EG.7 or 2 × 105 CT26). One week after tumor injection splenocytes from mice were collected and measured for CD8± IFN-γ± memory T cells using flow cytometry analysis. For demonstration of anti-tumor effect of long-term memory CD8 ± T cell, mice were vaccinated twice, at one week intervals with PBS or mature DC (API5) loaded with antigen peptides. Seven weeks after second DC vaccination, mice were inoculated with tumor cells. Tumor growth was monitored for 30 days via digital caliper.
Tumor prevention and immune cell depletion
For tumor prevention experiment, mice were immunized with PBS, 2 × 106 immature DC, immature DC loaded with antigen peptides, mature DC (treated API5), mature DC (treated API5) loaded with antigen peptides, or mature DC (treated LPS) loaded with antigen peptides (five mice per group) in footpad twice at one week intervals. One week after the second vaccination, mice were challenged with 2.5x105 TC-1, 5 × 106 EG.7 or 2.5x105 CT26 cells. The mice were defined as tumor bearing when a 3mm3 tumor mass was detected. For tumor prevention under immune cell depleted setting, mice were immunized using conditions stated above. Starting four days after the second vaccination, mice were injected intraperitoneally with CD4 (MAb GK1.5), CD8 (MAb 2.43) or NK1.1 (MAb PK136) depletion antibodies,35 at two day intervals. One week after the second vaccination mice were injected with 2.5x105 TC-1, 5 × 106 EG.7 or 2.5x105 CT26 cells. Mice tumor growth was monitored every 2–3 days via digital caliper.
Tumor treatment
For tumor treatment experiments, 5- to 8-week-old C57BL/6 were subcutaneously inoculated with 2 × 105 TC-1, 1 × 106 EG.7 or 2 × 105 CT26. On day three and day ten after tumor challenge, mice were injected with PBS, 2 × 106 immature DC, immature DC loaded with antigen peptides, mature DC (treated API5), mature DC (treated API5) loaded with antigen peptides, or mature DC (treated LPS) loaded with antigen peptides (five mice per group) in footpad twice at one week intervals. Tumor growth was measured every 2–3 days using digital calipers. For the treatment effect of late-treatment of API5-mediated DC-based vaccine, mice (five mice per group) were inoculated with EG.7, TC-1 or CT26 tumor cells, as mentioned above. After five days and twelve days, mice were vaccinated with PBS, 2 × 106 immature DC, immature DC loaded with antigen peptides, mature DC (treated API5), mature DC (treated API5) loaded with antigen peptides, or mature DC (treated LPS) loaded with antigen peptides. Tumor mass was monitored using the same method mentioned above.
Immunoprecipitation
For binding between API5 and TLR2 or TLR4, recombinant human API5 protein (1μg) and recombinant human TLR2 or TLR4 (0.5μg) (R&D) were incubated at 4°C overnight in 1ml NP-40 buffer (Elpis Biotech), added with TLR2 or TLR4 detection antibodies (1μg) (Novus) or rabbit-IgG antibodies (Santa Cruz) at 4°C overnight and added with protein G-agarose (20μl) (Roche) at 4°C overnight. Protein G-agarose was centrifuged at 13,000rpm for 1 minute. Supernatants were removed and washed with 1ml NP-40 three times at five minute intervals. Protein bound protein G-agarose was eluted with 1xSDS loading buffer (25μl) (Bio Basic).
Th type and antibody induction by API5 protein
For CD4 T cells Th type induction by API5 protein experiment, mice were subcutaneously inoculated with PBS, OVA protein (5μg), API5 protein (200μg) or OVA+API5 twice at one week intervals. One week after the second injection, mouse splenocytes were isolated, re-stimulated with OVA (ISQAVHAAHAEINEAGR)(Anygen) peptide (5μg/ml) and stained with APC-CD3, FITC-CD4 (ebioscience), PE-IFN-γ, PE-IL4, and IL17 (MACS). OVA-specific CD4 + T cells were measured using flow cytometry.
To detect the production of antigen specific antibodies following API5 injection, mice were injected as stated above. One week after the last injection, serum was isolated and the abundance of OVA-specific antibodies were measured via ELISA.
API5 and TLR4 binding affinity assay
The binding affinity between API5 and TLR4 was confirmed using BLItz system (ForteBio) according to vendor’s protocol. Briefly, protein A biosensors (ForteBio) were hydrated in PBS for 10 minutes. Anti-human TLR4 antibody (Santa Cruz), recombinant TLR4 protein 0.5μg/μl (Sino Biological), recombinant API5 protein (0.075μg/μl or 0.0375μg/μl), or BSA (0.075μg/μl) were gradually incubated with protein A biosensor. Program setting was as follows: initial base line for 30sec, loading for 150sec, base line for 30sec, loading for 150sec, base line for 30sec, association for 150sec, dissociation for 150sec. The KD between API5 and TLR4 was calculated using BLItz pro software analysis.
Statistical analysis
All data was presented as mean ± SD of three independent experiments with at least five samples per group. Individual data points were compared by Student’s t-test. Comparison of mice survival between two groups was analyzed by the Kaplan-Meier method followed by the log rank test (SPSS software Version22.0). For all tests, a p value < 0.05 was considered significant.
Funding Statement
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2016R1A5A2012284, NRF-2015R1A2A1A13001713) and supported by Basic Research Laboratory Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2013R1A4A1069575). This study was also supported by a grant of Korea Health Technology R&D project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI13C0826, HI15C2524). This study was also supported by the United States National Institutes of Health (NIH) Cervical Cancer Specialized Program of Research Excellence (SPORE) (P50 CA098252) and R01 (CA114425).
Disclosure of Interest
The authors report no conflict of interest.
Authors’ Contributions
Conception and design: S.H. Lee, T.H. Kang, Y.M. Park
Development of methodology: Y.S. Kim, T.H. Kang
Acquisition of data (Animal experiments, in vitro experiments etc.): Y.S. Kim, H.J. Park, J.H. Park, E.J. Hong, G.Y. Jang, M.C. Vo, J.J. Lee
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): I.D. Jung, H.D. Han, S.H. Lee, T.H. Kang, Y.M. Park
Writing, review, and/or revision of the manuscript: Y.S. Kim, A. Yang, E. Farmer, T.-C. Wu, T.H. Kang, Y.M. Park
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): H.J. Park, J.H. Park, E.J. Hong
Study supervision: T.H. Kang, Y.M. Park
Supplemental material
Supplemental data for this article can be accessed here.
References
- 1.Davis ID, Jefford M, Parente P, Cebon J.. Rational approaches to human cancer immunotherapy. J Leukoc Biol. 2003;73:3–29. doi: 10.1189/jlb.0502261. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Reviews Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 3.Figdor CG, De Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 4.Morisaki T, Matsumoto K, Onishi H, et al. Dendritic cell-based combined immunotherapy with autologous tumor-pulsed dendritic cell vaccine and activated T cells for cancer patients: rationale, current progress, and perspectives. Hum Cell. 2003;16:175–182. doi: 10.1111/j.1749-0774.2003.tb00151.x. [DOI] [PubMed] [Google Scholar]
- 5.Baek S, Kim YM, Kim SB, Kim CS, Kwon SW, Kim Y, Lee H. Therapeutic DC vaccination with IL-2 as a consolidation therapy for ovarian cancer patients: a phase I/II trial. Cell Mol Immunol. 2014.;12:87–95. doi: 10.1038/cmi.2014.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D, et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immun. 2014;63:175–183. doi: 10.1007/s00262-013-1502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbanski HF, Sorwell KG. Age-related changes in neuroendocrine rhythmic function in the rhesus macaque. Age (Dordr). 2012;34:1111–1121. doi: 10.1007/s11357-011-9352-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Yu C, Zhao J, Fu L, Yi S, Liu S, Yu T, Chen W. Vaccination with a DNA vaccine based on human PSCA and HSP70 adjuvant enhances the antigen-specific CD8+ T-cell response and inhibits the PSCA+ tumors growth in mice. J Gene Med. 2007;9:715–726. doi: 10.1002/jgm.1067. [DOI] [PubMed] [Google Scholar]
- 9.Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, RM Steinman, Choi Y. TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J Exp Med. 2000;191:495–502. doi: 10.1084/jem.191.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Endo H, Saito T, Kenjo A, Hoshino M, Terashima M, Sato T, Anazawa T, Kimura T, Tsuchiya T, Irisawa A, et al. Phase I trial of preoperative intratumoral injection of immature dendritic cells and OK-432 for resectable pancreatic cancer patients. J Hepatobiliary Pancreat Sci. 2012;19:465–475. doi: 10.1007/s00534-011-0457-7. [DOI] [PubMed] [Google Scholar]
- 11.Liu JY, Wu Y, Zhang XS, Yang JL, Li HL, Mao YQ, Wang Y, Cheng X, Li Y-Q, Xia J-C, et al. Single administration of low dose cyclophosphamide augments the antitumor effect of dendritic cell vaccine. Cancer Immunol Immun. 2007;56:1597–1604. doi: 10.1007/s00262-007-0305-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4:e631. doi: 10.1038/cddis.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013;73:629–639. doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 15.Nace G, Evankovich J, Eid R, Tsung A. Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J Innate Immun. 2012;4:6–15. doi: 10.1159/000334245. [DOI] [PubMed] [Google Scholar]
- 16.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 17.Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang KY, Ferrone S, Gameiro SR. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. 2013;133:624–636. doi: 10.1002/ijc.28070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang H, Ang B, Xu X, Huang X, Wu Y, Sun Y, Wang W, Li N, Cao X, Wan T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol Immunol. 2014;11:150–159. doi: 10.1038/cmi.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang TH, Kim YS, Kim S, Yang B, Lee JJ, Lee HJ, Lee J, Jung ID, Han HD, Lee S-H, et al. Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget. 2015;6:27751–27762. doi: 10.18632/oncotarget.v6i29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tewari M, Yu M, Ross B, Dean C, Giordano A, Rubin R. AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res. 1997;57:4063–4069. [PubMed] [Google Scholar]
- 21.Rigou P, Piddubnyak V, Faye A, Rain JC, Michel L, Calvo F, Poyet J-L. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. EMBO J. 2009;28:1576–1588. doi: 10.1038/emboj.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris EJ, Michaud WA, Ji JY, Moon NS, Rocco JW, Dyson NJ. Functional identification of Api5 as a suppressor of E2F-dependent apoptosis in vivo. PLoS Genet. 2006;2:e196. doi: 10.1371/journal.pgen.0020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basset C, Bonnet-Magnaval F, Navarro MG-J, Touriol C, Courtade M, Prats H, Garmy-Susini B, Lacazette E. Api5 a new cofactor of estrogen receptor alpha involved in breast cancer outcome. Oncotarget. 2017;8. doi: 10.18632/oncotarget.v8i32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song KH, Cho H, Kim S, Lee HJ, Oh SJ, Woo SR, Hong S-O, Jang HS, Noh KH, Choi CH, et al. API5 confers cancer stem cell-like properties through the FGF2-NANOG axis. Oncogenesis. 2017;6:e285. doi: 10.1038/oncsis.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho H, Chung JY, Song KH, Noh KH, Kim BW, Chung EJ, Ylaya K, Kim JH, Kim TW, Hewitt SM, et al. Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer. 2014;14:545. doi: 10.1186/1471-2407-14-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noh KH, Kim SH, Kim JH, Song KH, Lee YH, Kang TH, Han HD, Sood AK, Ng J, Kim K, et al. API5 confers tumoral immune escape through FGF2-dependent cell survival pathway. Cancer Res. 2014;74:3556–3566. doi: 10.1158/0008-5472.CAN-13-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Liu H, Liu B, Ma W, Xue X, Chen J, Gene Zhou Q.. Gene expression levels of CSNK1A1 and AAC-11, but not NME1, in tumor tissues as prognostic factors in NSCLC patients. Med Sci Monit. 2010;16:CR357–64. [PubMed] [Google Scholar]
- 28.Krejci P, Pejchalova K, Rosenbloom BE, Rosenfelt FP, Tran EL, Laurell H, Wilcox WR. The antiapoptotic protein Api5 and its partner, high molecular weight FGF2, are up-regulated in B cell chronic lymphoid leukemia. J Leukoc Biol. 2007;82:1363–1364. doi: 10.1189/jlb.0607425. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki H, Moriyama S, Yukiue H, Kobayashi Y, Nakashima Y, Kaji M, Fukai I, Kiriyama M, Yamakawa Y, Fujii Y. Expression of the antiapoptosis gene, AAC-11, as a prognosis marker in non-small cell lung cancer. Lung Cancer. 2001;34:53–57. doi: 10.1016/S0169-5002(01)00213-6. [DOI] [PubMed] [Google Scholar]
- 30.Kim JW, Cho HS, Kim JH, Hur SY, Kim TE, Lee JM, Kim I-K, Namkoong SE. AAC-11 overexpression induces invasion and protects cervical cancer cells from apoptosis. Lab Invest. 2000;80:587–594. doi: 10.1038/labinvest.3780063. [DOI] [PubMed] [Google Scholar]
- 31.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 32.Sayers S, Ulysse G, Xiang Z, He Y. Vaxjo: a web-based vaccine adjuvant database and its application for analysis of vaccine adjuvants and their uses in vaccine development. J Biomed Biotechnol. 2012;2012:1–13. doi: 10.1155/2012/831486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, Sundqvist S, Larsson M, Hayashi T, Minev B, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211. doi: 10.1186/1479-5876-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, TC Wu. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.