

ABSTRACT
The tumor suppressor p53 responds to genotoxic and oncogenic stresses by inducing cell cycle arrest and apoptosis. Recent studies suggest that p53 also participates in the regulation of cellular immune responses. Here, we have investigated the potential of p53 gene therapy to augment immune checkpoint inhibition by combining an anti-programmed cell death protein 1 (PD1) antibody with SGT-53, our investigational nanomedicine carrying a plasmid encoding human wild-type p53. In three syngeneic mouse tumor models examined including a breast cancer, a non-small cell lung carcinoma, and a glioblastoma, SGT-53 sensitized otherwise refractory tumors to anti-PD1 antibody. The involvement of p53 in enhancing anti-PD1 immunotherapy appears to be multifaceted, since SGT-53 treatment increased tumor immunogenicity, enhanced both innate and adaptive immune responses, and reduced tumor-induced immunosuppression in a 4T1 breast tumor model. In addition, SGT-53 alleviates a fatal xenogeneic hypersensitivity associated with the anti-PD1 antibody in this model. Our data suggest that restoring p53 function by SGT-53 is able to boost anti-tumor immunity to augment anti-PD1 therapy by sensitizing tumors otherwise insensitive to anti-PD1 immunotherapy while reducing immune-related adverse events.
Keywords: checkpoint inhibitor, gene therapy, nanomedicine, p53, tumor-targeted delivery
Introduction
In recent years, manipulation of immune checkpoints or pathways has emerged as an important and effective form of immunotherapy of human cancer.1 In particular, monoclonal antibodies that target the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) or the programmed cell death protein 1 pathway (i.e., PD1/PD-L1) are the most widely studied and have demonstrated clinical effectiveness in various types of cancer.2,3 However, despite the clinical potential of these immunomodulatory antibodies, a large number of patients either do not respond or develop resistance.4,5 Moreover, some treatment-related toxicities have been observed which limit the use of these drugs, especially when both anti-PD1 and anti-CTLA-4 were combined.6 To overcome the current limitations of immune checkpoint immunotherapy, methods to enhance the efficacy and safety would be advantageous.7 In this regard, a large number of trials are now underway that combine checkpoint blockade with a wide range of therapeutic agents.8–10
Our approach to improve immunotherapy focuses on the use of tumor-targeting nanoparticles delivering the tumor suppressor gene p53 to tumor cells and to restore p53 function. The tumor suppressor p53 is a transcription factor that regulates cell cycle arrest and apoptosis in response to genotoxic and oncogenic stresses. Given that over 50% of all of human cancers have a mutational inactivation of p53 and alterations in the p53 pathway11,12 and p53-null mice are highly predisposed to cancer development,13 restoring p53 function has long been recognized as an attractive cancer therapeutic strategy.14,15 Our understanding of the cellular and molecular processes that link p53 activity to host immune regulation remains incomplete. Nonetheless, recent experimental and clinical results suggest that p53 participates in immune regulation, and that the p53 pathway can be exploited to alter the immunological landscape of tumors for improved cancer therapies.16,17 Thus, we hypothesized that the restoring p53 function would boost anti-tumor immunity to augment anti-PD1 therapy. In the current study, we have investigated the potential of p53 gene therapy to augment checkpoint blockade utilizing the investigational agent SGT-53 in combination with a checkpoint inhibitor (an anti-PD1 antibody) in several syngeneic mouse tumor models. SGT-53 is a novel tumor-targeted nanomedicine based on a nanocomplex carrying a plasmid encoding human wild-type p53 (wtp53) for gene therapy. The proprietary nanodelivery system employed in SGT-53 displays exquisite specificity in delivery of payloads to tumors based on the ability of ~100 nm nanocomplex to extravasate into the tumor interstitial space and then enter tumor cells via endocytosis mediated by transferrin receptors (TfRs) that are highly elevated on tumor cells including cancer stem cells.18
The combination of SGT-53 and the anti-PD1 antibody resulted in a significantly enhanced inhibition of tumor growth compared to either agent individually in all three of the syngeneic mouse tumor models examined in this study including a breast cancer, a non-small cell lung carcinoma, and a glioblastoma. SGT-53 treatment increased immunogenic cell death (ICD) in tumors and enhanced both innate and adaptive immune responses in combination with anti-PD1, while alleviating tumor-induced immunosuppression. In addition, we have evidence that SGT-53 can alleviate the fatal xenogeneic hypersensitivity reaction to an anti-PD1 antibody seen in at least one of the syngeneic tumor models (4T1, a model for metastatic breast cancer in BALB/c mice). Collectively, our data suggests that combining SGT-53 with an anti-PD1 antibody might not only improve outcomes for cancer patients but also reduce immune-related adverse events that are sometimes seen with immunotherapies.
Results
SGT-53 increases immunogenicity of 4T1 cells
Following exposure of 4T1 mouse breast cancer cells in culture to a tumor-targeting nanocomplex loaded with a plasmid encoding human wtp53 (SGT-53) or with an empty vector control plasmid (scL-vec), quantitative RT-PCR was performed to assess expression of human p53 (Figure 1A) as well as mouse genes associated with immune responses (Figure 1B). A high level of human p53 mRNA (> 3 logs above the background signal of untreated cells when normalized to mouse GAPDH) was detected at 24, 48, and 72 h only in the cells treated with SGT-53 (Figure 1A). Following SGT-53 treatment, increased expression of type I interferon (IFNα1) and several cytokines related to innate immunity (CCL2, CXCL1 and IL15) were evident at 48 and 72 h after treatment (Figure 1B). Increased expression of DEC1, indicative of cellular senescence was also observed (Figure 1B). Notably, we observed a significant increase in the level of programmed death-ligand 1 (PD-L1) mRNA in cultured 4T1 cells after SGT-53 treatment (Figure 1B). We have also observed increased release of high mobility group box 1 (HMGB1) and ATP in the culture media following SGT-53 treatment (Figure 1C), which supports induction of ICD. To assess whether introduction of human wtp53 altered 4T1 cell survival, we examined apoptotic activity using an Annexin V assay (Figure 1D). Both Annex V+/7-AAD− (apoptotic) and Annex V+/7-AAD+ (dead) cells were significantly increased after SGT-53 treatment compared to either untreated cells or those exposed to the control nanocomplex loaded with a plasmid encoding GFP (scL-GFP). FACS analysis of 4T1 cells revealed significantly increased surface expression of calreticulin (CRT), Fas cell surface death receptor (FAS), and PD-L1 following SGT-53 treatment while scL-GFP did not increase the surface expression of any these markers (Figure 1E). Surface expression of the endoplasmic reticulum (ER) protein CRT is an indicative of ICD as are release of innate immune receptor ligands (HMGB1 and ATP). Together, these data indicate that expression of functional p53 resulting from treatment with SGT-53 is responsible for both induction of ICD and alterations in the immunogenicity of 4T1 cells in vitro and that these effects are not merely due to the introduction of a generic plasmid DNA.
Figure 1.
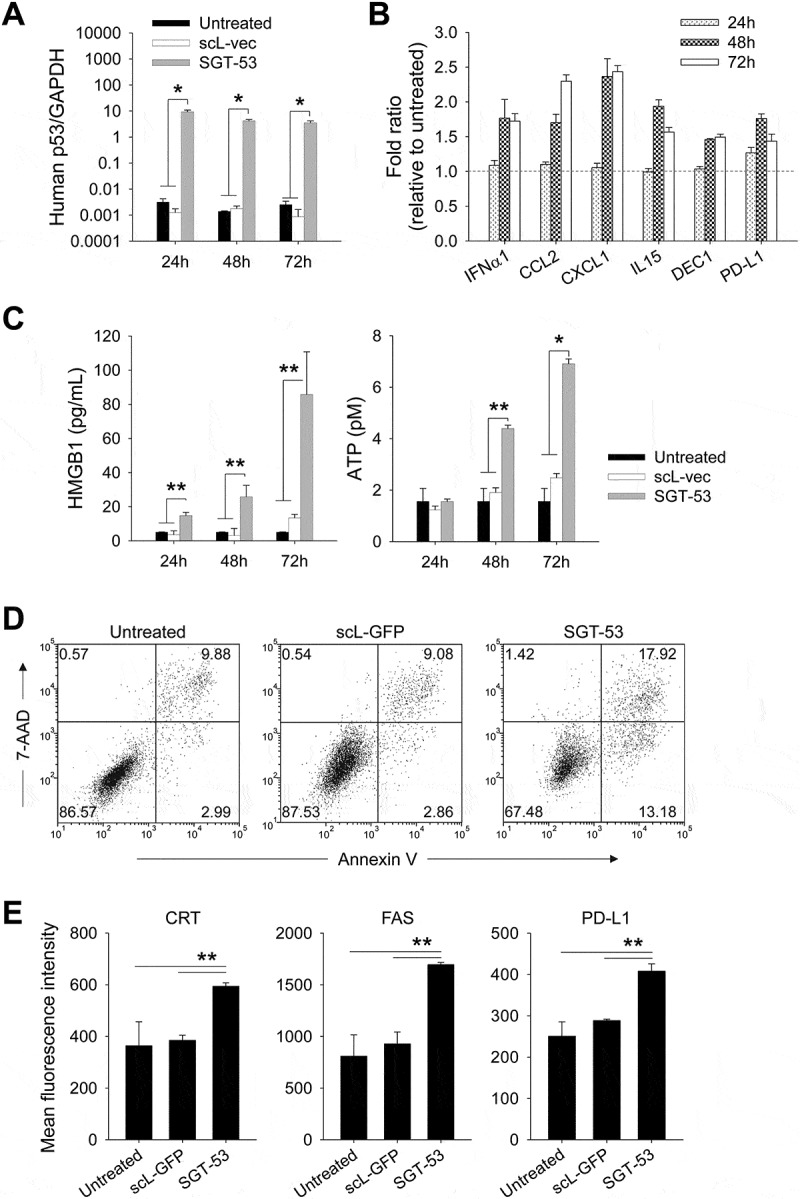
SGT-53 increases immunogenicity and induces ICD. (A) 4T1 cells were treated with either SGT-53 or scL-vec nanocomplex. Expression of human p53 was assessed by quantitative RT-PCR. The fold-change relative to mouse GAPDH mRNA is shown on a log scale (n = 6). (B) Expression of mouse genes associated with immune responses was assessed by RT-PCR in the cells treated with SGT-53 (n = 6). (C) Release of HMGB1 and ATP was assessed in the culture media (n = 6). (D) Induction of apoptosis was assessed via Annexin V/7-AAD staining at 48 h after transfection. Numbers in the quadrants indicate the percentage of cells in that quadrant. (E) Expression of cell surface components of immunogenicity was assayed at 48 h after transfection via FACS (n = 4). Data are shown as mean ± SEM. *p < 0.001, **p < 0.05, 1-way ANOVA with Bonferroni t-test.
Following SGT-53 treatment, the altered immunogenicity of 4T1 tumor cells was further evaluated in vivo (Figure 2). Mice bearing subcutaneous (s.c.) syngeneic 4T1 tumors were treated with SGT-53 via tail vein injection, and the impact of SGT-53 treatment on a number of immune-relevant markers examined. Similar to our findings in vitro, FACS analysis of harvested tumors revealed significantly increased surface expression of immune cell recognition molecules including CRT, FAS, PD-L1, CD80, CD86, ICAM1 and MHC class I (H2-Kd/H2-Dd) after SGT-53 treatment (Figure 2). Gene expression analysis of tumors using NanoString showed the increased mRNA levels of transporter associated with antigen processing 1 (TAP1) and TAP2 after SGT-53 treatment, which are implicated as important TP53 dependent components of antigen presenting machinery and mediators of ICD (Supplementary Fig. S1).19,20 Together with the in vitro results, these in vivo data strongly suggest that p53 alters the expression of immunogenic markers on the surface of tumor cells and induces ICD of tumor cells. In short, the tumors treated with SGT-53 appear to be more immunologically “hot”, and this change would be expected to result in an increased immune response to tumor cells expressing the p53 encoded by the DNA payload of SGT-53.
Figure 2.

SGT-53 increases immunogenicity of tumor in vivo. Mice bearing established 4T1 tumor were i.v. treated with SGT-53 (30 µg DNA/mouse). Expression of cell surface components of immunogenicity was assayed 48 h later via FACS and compared with those in tumor from untreated mice (n = 4). Tumor cells were dissociated by enzymatic digestion and identified by gating CD45−CD31− live cells. Data are shown as mean ± SEM. *p < 0.001, **p < 0.05, Student’s t test.
The combination of an anti-PD1 antibody and SGT-53 is more effective than either agent individually in inhibiting tumor growth and metastasis
Given the above data demonstrating the increased immunogenicity triggered by SGT-53, mice bearing s.c. established 4T1 tumors were treated with an anti-PD1 antibody alone or in combination with SGT-53 using the treatment schedule shown in Figure 3A. No significant inhibition of tumor growth was seen with anti-PD1 monotherapy indicating that the 4T1 tumors display inherent therapeutic resistance to anti-PD1 checkpoint blockade (Figure 3B). SGT-53 treatment alone resulted in only a modest inhibition of 4T1 tumor growth. However, the combination of anti-PD1 plus SGT-53 treatment resulted in a significantly diminished tumor growth leading to markedly smaller tumors compared to those in mice given either agent individually (Figure 3B&C). No significant decrease in body weight, which would be suggestive of toxicity, accompanied the tumor growth inhibition seen with the combination treatment (Figure 3D).
Figure 3.

Enhanced tumor growth inhibition by the combination of anti-PD1 and SGT-53. BALB/c mice with s.c. 4T1 tumor were randomized to therapy with anti-PD1, either alone or in combination with SGT-53 (n = 10). (A) Treatment schedule. Mice received total 5 injections of SGT-53 and/or 5 injections of anti-PD1. (B) Changes in tumor sizes were plotted versus the number of days after initiation of the treatment. (C) Quantification of tumor weight at harvest on day 17. For group treated with anti-PD1, tumors were harvested on day 16 or 17 when the mice were moribund. *p < 0.05, **p < 0.001, 1-way ANOVA with Bonferroni t-test. (D) Changes in body weight are shown. Data are shown as mean ± SEM.
The observation of enhanced anti-tumor effect of anti-PD1 plus SGT-53 combination treatment was not limited to the 4T1 breast cancer model. Using the same treatment regimen, we have also seen improved anti-tumor efficacy in other syngeneic tumor models including LL2 non-small cell lung carcinoma (Figure 4A) and GL261 glioblastoma (Figure 4B). In both tumor models, neither anti-PD1 antibody nor SGT-53 treatment individually altered tumor growth dramatically. Similar to the effect seen with 4T1 tumors, the combination of anti-PD1 and SGT-53 significantly delayed tumor growth of both LL2 and GL261. Moreover, the combination of anti-PD1 and SGT-53 clearly showed a strong, statistically significant survival benefit in both tumor models compared to both of the single agent treatments (Supplementary Fig. S2). Thus, all three tumor models examined here were relatively unresponsive to anti-PD1 treatment, but SGT-53 sensitized each to an anti-PD1 antibody. We have also previously observed a similar enhancement of anti-tumor activity in mouse syngeneic model of head and neck cancer.21 Collectively, our results suggest that the SGT-53 treatment has the potential to convert tumors resistant to anti-PD1 therapy to more responsive tumors. Extrapolating to human cancers, this conversion to sensitivity has the potential to bring patients that do not respond to anti-PD1 therapy into the category of responders and thereby enable checkpoint blockade agents to benefit a larger percentage of the total patient population.
Figure 4.
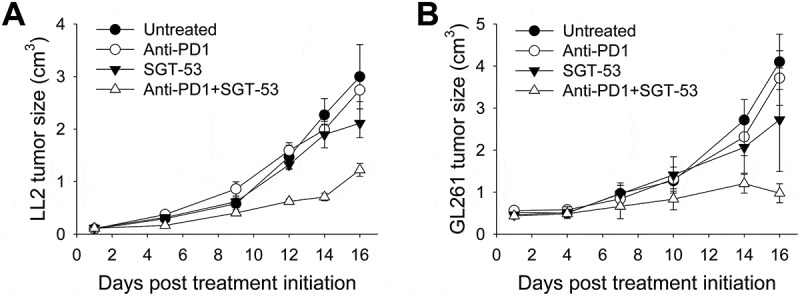
Enhanced tumor growth inhibition by anti-PD1 plus SGT-53 combination in mouse syngeneic tumor models. C57BL/6 mice with s.c. LL2 tumors (A) or GL261 tumors (B) were randomized to therapy with anti-PD1 (200 µg antibody/mouse/injection, i.p.), either alone or in combination with SGT-53 (30 µg DNA/mouse/injection, i.v.) (n = 6–10). Tumor sizes were plotted versus the number of days after initiation of the treatment. Data are shown as mean ± SEM.
Our observation of enhanced inhibition of 4T1 tumor growth by the combination of SGT-53 and anti-PD1 treatment was supported by IHC staining showing increased active caspase-3 (Casp3) and TUNEL indicative of tumor cell apoptosis as well as by decreased Ki-67 expression reflecting reduced tumor cell proliferation (Figure 5A). Tumors in mice treated with SGT-53 alone or in combination with the anti-PD1 antibody treatment exhibited significantly higher levels of Casp3-positive and TUNEL-positive cells when compared to tumors in untreated mice or those treated with anti-PD1 alone (Figure 5B). Combining anti-PD1 and SGT-53 showed only a modest increase of apoptotic cells compared to SGT-53 treatment alone consistent with the known role of p53 as a driver of apoptosis. Tumors in mice treated with SGT-53 alone or in combination with the anti-PD1 antibody exhibited a significantly lower level of Ki-67-positive cells compared to tumors in mice either untreated or treated with anti-PD1 monotherapy. Our data indicate that Ki-67 levels were not affected by anti-PD1 as a single agent, nor did anti-PD1 accentuate the inhibition of Ki-67 expression seen in mice treated with SGT-53 alone.
Figure 5.

SGT-53 increases apoptosis and inhibits proliferation of 4T1 tumor in vivo. IHC staining of tumors treated as shown in Figure 3A. Representative IHC staining (A) and quantification (B) of Casp3, TUNEL, and Ki-67 are shown. Scale bar, 50 µm. At least five fields of view from three tumor sections were counted using the ImmunoRatio, an automated cell counting software (http://153.1.200.58:8080/immunoratio/). Data are shown as mean ± SEM. *p < 0.05, 1-way ANOVA with Bonferroni t-test.
SGT-53 dramatically reduces metastases of 4T1 tumor in the lungs
Mice bearing the 4T1 breast tumors experience metastases to the lungs resembling some breast cancer patients in this regard. Indeed, 4T1 is considered to be a mouse model for metastatic breast cancer. It is possible to quantify microscopically 4T1 metastatic nodules in sections of the lungs of the 4T1-bearing mice (Figure 6A). Treatment of the mice with SGT-53 alone was able to reduce substantially metastases of 4T1 tumor in the lungs whereas treatment with anti-PD1 alone was essentially ineffective (Figure 6A&B). In mice treated with the combination of SGT-53 plus anti-PD1 antibody, virtually no 4T1 lung nodules were detected. These results are consistent with our earlier findings showing improved anti-tumor activity with the combination treatment and suggests that the combination treatment may be able to reduce the lung metastases from breast cancers that contributes to breast cancer mortality.
Figure 6.

SGT-53 substantially reduces lung metastases of 4T1 tumors. Lung tissues were harvested from tumor-bearing BALB/c mice treated as shown in Figure 3A. (A) Representative images of H&E stain of lung. T, tumor. Scale bar, 100 µm. (B) Metastatic lung nodules were microscopically counted per 4× field of view and plotted. At least five fields of view from three tissue sections were counted and averaged. Data are shown as mean ± SEM. *p < 0.001, 1-way ANOVA with Bonferroni t-test.
SGT-53 enhances immune responses in 4T1 tumor in vivo
Our results thus far are consistent with enhancement of both innate and adaptive immunity. To extend these observations, FACS analyses of tumor infiltrating myeloid cells were conducted revealing an increase of CD80+ activated DCs and tumor infiltrating macrophages following SGT-53 treatment either as a single agent or in combination with an anti-PD1 (Figure 7A). Treatment with anti-PD1 alone did not affect the number of tumor infiltrating macrophages or DCs. When SGT-53 was combined with anti-PD1 antibody, the number of activated tumor-infiltrating lymphocytes (TILs) was substantially increased. However, neither anti-PD1 antibody nor SGT-53 alone led to significant increase of activated TILs. We also observed a significant increase in granzyme B (GzmB)-positive or IFN gamma (IFNγ)-positive cytotoxic T cells (CTLs) in tumors following treatment with anti-PD1 plus SGT-53, whereas no significant increase of CTLs were observed with anti-PD1 or SGT-53 individually (Figure 7B). Tumor infiltrating immunosuppressive cells including myeloid derived suppressor cells (MDSCs) and Tregs were significantly altered by SGT-53 treatment (Figure 7C). Tumors treated with SGT-53 alone or in combination with anti-PD1 showed significantly less MDSCs compared with tumors in untreated mice or those treated with anti-PD1 alone. A significant reduction of Tregs was observed when mice were treated with anti-PD1 plus SGT-53. Gene expression analysis of tumors using NanoString revealed a significant down-modulation of genes associated with immunosuppression including indoleamine 2,3-dioxygenase 1 (IDO1) and FoxP3 following SGT-53 treatment (Figure 7D). Quantitative RT-PCR analysis further confirmed the down-modulation of immunosuppressive enzymes both IDO1 and tryptophan 2,3-dioxygenase 2 (TDO2) by SGT-53 treatment via modulating bridging integrator 1 (BIN1, negative regulator) and cytochrome c oxidase 2 (COX2, positive regulator) (Figure 7E). These data all suggest that p53 expression from SGT-53 alters the tumor microenvironment (TME) in such a way to reduce tumor evasion and to enhance anti-tumor immunity and adaptive immune response in combination with anti-PD1 treatment. Tumors appear to be more immunologically “hot” in mice treated with the combination of the two agents, i.e., these tumors are more prone to trigger responses by both the innate and adaptive immune systems. The primary mechanism of the anti-PD1 checkpoint inhibitors is thought to be release of the PD1-mediated immunosuppression of T cells, it is perhaps not surprising that the anti-PD1 as a single agent did not have an impact on the level of tumor infiltration by cells of the innate immune system i.e., DCs and macrophages (Figure 7A, left two panels). The effect of the anti-PD1 antibody was apparent in combination with SGT-53 in the T-cell-related results (Figure 7A, right two panels). Collectively, these results indicate that the role of SGT-53 is to prime the TME and that the resultant “hot” tumors are more sensitive to the release from PD1-mediated immunosuppression of T cells by the anti-PD1 component of the combination treatment.
Figure 7.
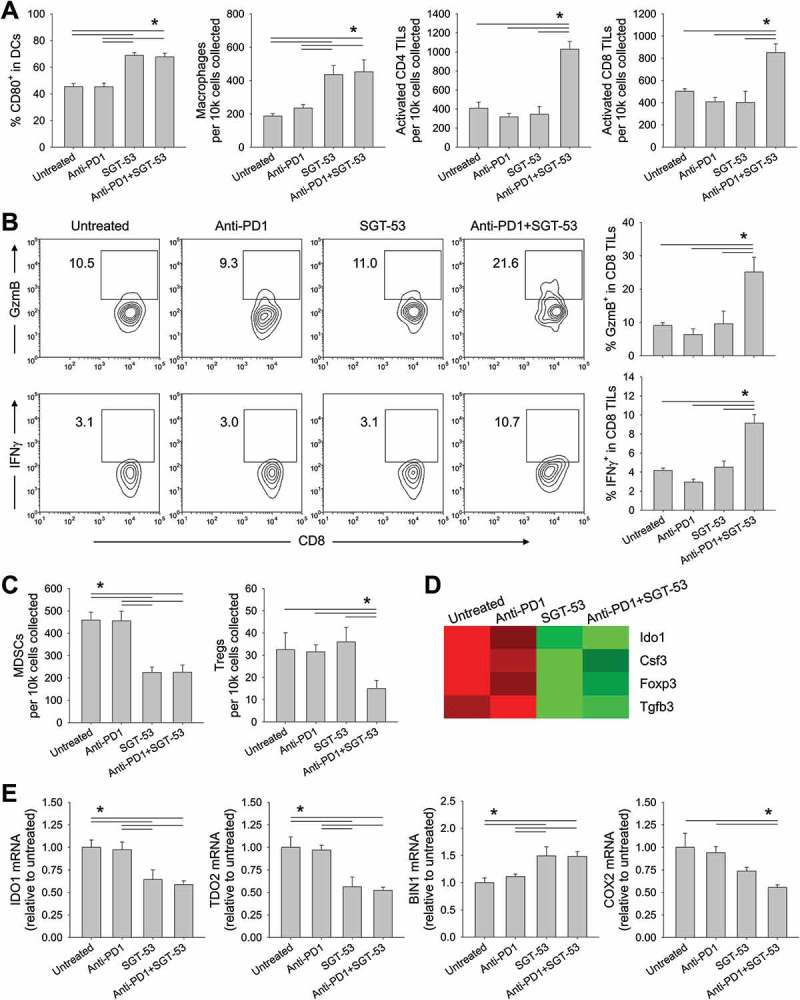
SGT-53 enhances immune responses in 4T1 tumor in vivo. Tumor cells were dissociated and immune cell infiltration was assessed via FACS. (A) Infiltrating immune cells were identified by gating CD45+ live cells including DCs (CD11c+I-A/I-E+), macrophages (CD11b+F4/80+), activated CD4 TILs (CD3+CD4+CD107a+), and activated CD8 TILs (CD3+CD8+CD107a+). Infiltrating cells are shown in terms of absolute number of cells per 1 × 104 live cells collected (n = 6–8). (B) Representative FACS plots (left panels) and graphs (right panels) of CTLs (CD3+CD8+GzmB+ or CD3+CD8+IFNγ+) are shown. Numbers in the plots indicate the percentage of cells (n = 6–8). (C) Immunosuppressive MDSCs (CD11b+Gr1+) and Tregs (CD3+CD4+FoxP3+) were identified (n = 6–8). (D) Altered expression of genes associated with immunosuppression in the tumor tissue were assessed via NanoString. (E) Expression of genes associated with immunosuppressive enzymes was assessed by RT-PCR in the tumors (n = 6–8). The fold-change relative to untreated tumors is shown. Data are shown as mean ± SEM. *p < 0.05, 1-way ANOVA with Bonferroni t-test.
SGT-53 prevents fatal xenogeneic hypersensitivity following repeated anti-PD1 administration in a syngeneic 4T1 breast cancer model
We observed that BALB/c mice bearing 4T1 tumors died after multiple injections of anti-PD1 antibody (clone RMP1-14; rat IgG). This death of the mice occurred within one hour after either the fourth or fifth injection, prior to their tumors having grown large enough to kill the animals (Figure 8A). However, non-tumor bearing BALB/c mice survived the same treatment regimen and C57BL/6 mice bearing LL2 or GL261 tumors were not killed by the same anti-PD1 antibody (data not shown). Others have also observed this fatal hypersensitivity after repeated injections of anti-PD1 (clone J43; hamster IgG) or anti-PD-L1 (clone 10F.9G2; rat IgG) in the syngeneic 4T1 tumor model and suggested that mortality was associated with neutrophil-mediated anaphylaxis involving accumulation of neutrophils in the lungs.22 Surprisingly, we observed that this fatal hypersensitivity seen with anti-PD1 antibody was not seen when the PD1 blockade was in combination with SGT-53 (Figure 8A). Autopsies of our mice that had received five injections with anti-PD1 revealed massive infiltration of neutrophils and macrophages in the lung and liver (Figure 8B), and FACS analysis confirmed abnormalities reflecting neutrophil and macrophage infiltration in the lung (Figure 8C). The lung infiltration by neutrophils seen after treatment with anti-PD1 antibody alone was eliminated if the SGT-53 was given in conjunction with anti-PD1. Similar to the report by others, multiple injections of anti-PD1 antibody triggered neutrophil-mediated anaphylaxis that kills the 4T1-bearing mice. Most interestingly, concurrent treatment with SGT-53 prevented both the neutrophil invasion of the lungs and livers and the death of the mice that were otherwise killed by anti-PD1 antibody treatment. Although rare and generally of low severity, hepatitis due to immunotherapy with anti-PD1 has been reported.23
Figure 8.
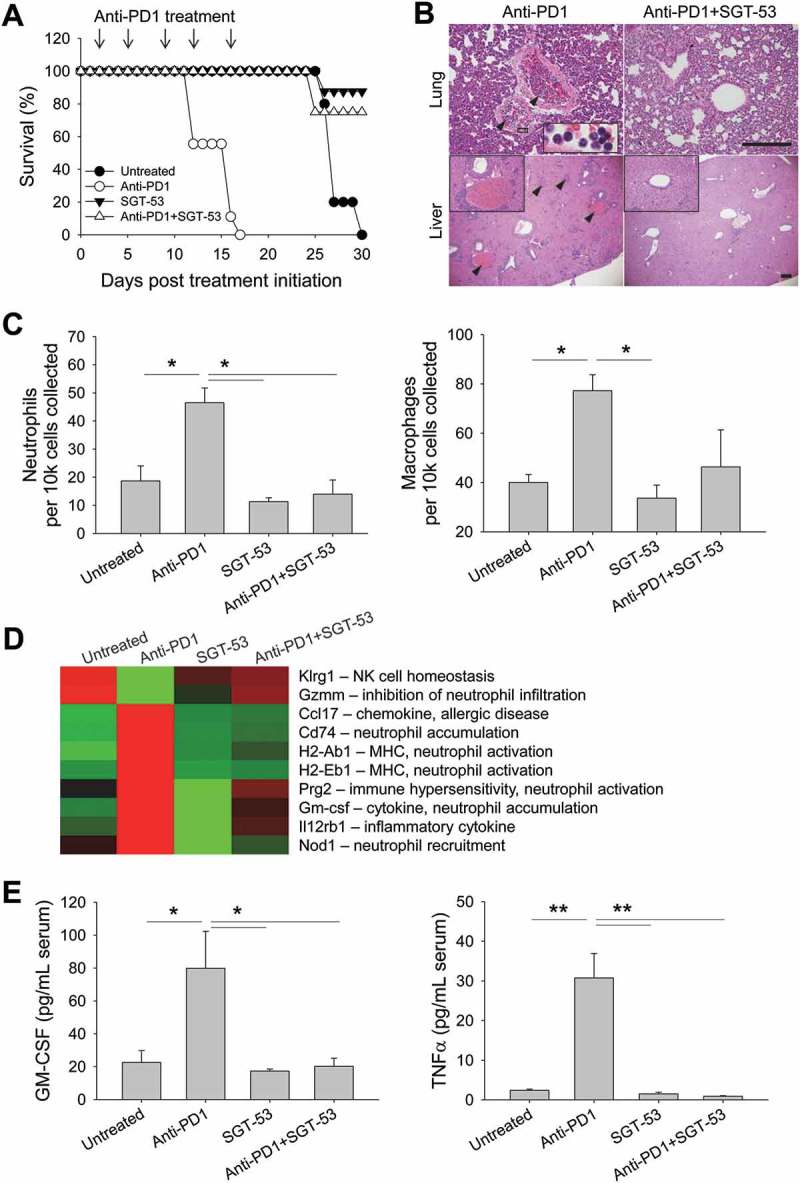
SGT-53 prevents fatal xenogeneic hypersensitivity following repeated anti-PD1 administration in a syngeneic 4T1 breast cancer model. (A) Survival of BALB/c mice bearing 4T1 tumor after repeated dosing with anti-PD1 antibody alone or in combination with SGT-53. Mice received total five injections of SGT-53 and/or five injections of anti-PD1 following the treatment schedule shown in Figure 3A. Arrows indicate the anti-PD1 injection. (B) Representative H&E stains of lungs and livers of mice treated with either anti-PD1 alone or in combination with SGT-53. Arrowheads indicate neutrophilic accumulation. Scale bars, 200 µm. (C) Infiltrating neutrophils (CD11b+F4/80−Gr1hi) and macrophages (CD11b+F4/80+) were assayed in the lung via FACS (n = 4). Infiltrating cells are shown in absolute number of cells per 1 × 104 live cells collected. (D) Heatmap of significantly altered gene expression in tumor tissue from mice treated with anti-PD1 antibody that were reversed in tumor tissue from mice treated with anti-PD1 plus SGT-53. (E) Sera from tumor bearing mice receiving the indicated treatment were collected and analyzed for levels of mouse GM-CSF or TNFα using ELISA (n = 8). Data are shown as mean ± SEM. *p < 0.05, **p < 0.001, 1-way ANOVA with Bonferroni t-test.
To understand the molecular basis of these observations, we performed gene expression profiling of tumor tissues from the mice. For these analyses, we employed the NanoString technique and specifically looked for candidate genes that were modulated by anti-PD1 treatment alone but not when SGT-53 was added to anti-PD1 (Figure 8D). The logic here was that some change(s) in gene expression would be linked to the anaphylactic death of 4T1-bearing mice, and that if SGT-53 prevents this death, then SGT-53 might prevent the underlying alteration in gene expression. Multiple genes involved in immune hypersensitivity and neutrophil recruitment, priming, and activation (Ccl17, Cd74, Prg2, Nod1, and GM-CSF) were identified as being upregulated by anti-PD1 monotherapy, while genes involved in inhibiting excessive neutrophil infiltration (e.g., Gzmm) were downregulated. The changes in expression of this set of genes were not observed with the combination treatment with anti-PD1 plus SGT-53. Of note is that GM-CSF has been directly linked to lung-damaging neutrophil accumulation.24 GM-CSF mRNA was up-regulated with anti-PD1 treatment, but when SGT-53 was added to the checkpoint blockade, GM-CSF expression in tumors was similar to that seen in the tumors of untreated animals. A similar result was observed with serum concentration of GM-CSF as assessed by ELISA (Figure 8E). Serum GM-CSF was significantly increased (~4-fold) in 4T1 tumor-bearing mice receiving anti-PD1 treatment alone, but when SGT-53 treatment was added, GM-CSF levels in the sera were similar to that seen in untreated animals. Anti-PD1 treatment also increased the serum level of the pro-inflammatory cytokine TNFα, in this case by ~12.8-fold (Figure 8E). TNFα is known to be related to the cytokine-release syndrome seen after infusion of certain monoclonal antibodies.25 Once again, SGT-53 treatment added to anti-PD1 treatment resulted in TNFα levels equivalent to those seen in untreated animals. We have thus identified a set of genes that are candidates in the fatal xenogeneic hypersensitivity reaction that we see in 4T1-bearing mice. SGT-53 treatment prevents the modulation of the expression of these genes by anti-PD1 monotherapy and “rescues” the mice from the otherwise fatal treatment with this checkpoint inhibitor. Taken together with above efficacy data, these results suggest that SGT-53 may render checkpoint blockade not only more effective but also safer.
Discussion
It has long been appreciated that the restoration of p53 function via gene therapy has potential to combat cancer.14 While the killing of cancer cells by p53 does not require a fully functional immune system, p53 can push cancer cells toward apoptotic death and tends to sensitize tumors to conventional therapeutic modalities (chemotherapy and radiotherapy) that result in DNA damage.26 Indeed, we have shown that the restoration of p53 function via SGT-53, a tumor-targeted nanomedicine, could inhibit tumor growth and sensitize xenografts of human tumors in nude mice lacking T cells to both chemotherapy27,28 and radiotherapy.29 Nonetheless, it has become clear that p53 also participates in various aspects of immune modulation in cancer.30 In the present study, we are reporting that introducing functional wtp53 gene via SGT-53 can induce immunogenic changes of cancer cells, effectively promote anti-tumor immunity, and reduce tumor-induced immunosuppression in the TME. By augmenting anti-PD1 immune checkpoint blockade, the SGT-53 was able to convert mouse syngeneic tumors that were relatively insensitive to anti-PD1 into tumors that were more responsive to anti-PD1 therapy. This priming or sensitization of tumors to anti-PD1 resulted in a significant inhibition of tumor growth. In support of our observation is a recent report suggested that the tumoricidal effects of p53 following nutlin-3a treatment (which inhibits the interaction between p53 and mdm2 that blunts p53 activity) promoted systemic anti-tumor immunity.31 The induction of immunogenic conditions in the TME with an immunogenic chemotherapy or radiation therapy is thought to convert immunologically “cold” tumors into “hot” tumors via release of tumor neoantigens.32,33 Similarly, the pushing of tumor cells toward immunogenic death by SGT-53 may be quite effective in promoting in tumor elimination by the immune system.
The involvement of p53 in immune regulation appears to be multifaceted in that SGT-53 treatment modified immunogenicity of tumor cells, increased both innate and adaptive immunity, and reduced tumor-induced immunosuppression in the TME. In cultured 4T1 mouse breast cancer cells, we observed changes indicative of ICD after SGT-53 treatment including increased surface expression of the ER protein CRT and release of cell death associated molecular patterns (HMGB1 and ATP).34 Several additional alterations in immunogenicity were also observed with SGT-53 treatment including type I IFN responses and increased tumor expression of CD80, CD86, FAS, TAP1/2, and MHC I molecules in vivo. We have previously shown that SGT-53 treatment of MOC1 head and neck cancer cells could upregulate the expression of genes involved in antigen processing and presentation including CRT, calnexin, endoplasmic reticulum amino peptidase 1, and TAP1/2 involving stimulator of interferon genes (STING) pathway.21 The increased expression of MHC I molecules could also be linked to the increased type I IFN responses.35 Importantly, resistance to anti-PD1 therapy could be mediated by suppression of type I IFN signaling in a preclinical model of Kras-mutated p53-deficient lung cancer.35 In this model, induction of IFNβ by radiotherapy was able to elevate MHC I expression and restore the responsiveness of resistant tumors to anti-PD1 therapy. Increased FAS expression with SGT-53 treatment could also increase CTL-mediated apoptosis of tumor cells since CTLs use FAS/FAS ligand binding to induce apoptosis of target cells during the CTL-tumor cell interaction.36,37 These observations recapitulate previous findings,20,21,38–40 and it is reasonable to believe that elevated expression of these component of immunogenicity would be instrumental in achieving efficient anti-tumor immune responses.
Moreover, we have observed increased production of cytokines (e.g., CCL2, CXCL1, and IL15) related to recruitment of innate immune cells after SGT-53 treatment of 4T1 cells. In line with this data, SGT-53 resulted in a significant increase of activated DCs and macrophage infiltration in TME in vivo. In addition, the increases in the number of activated TILs and GzmB-positive/IFNγ-positive CTLs in the tumor were associated with the increased anti-tumor efficacy in 4T1 tumor-bearing mice treated with the combination of SGT-53 plus anti-PD1. Supporting our data, there is increasing evidence indicating that p53 can regulate the cell-mediated adaptive immune response to tumors and ultimately promote CTL-induced cancer cell death.20,37–39 Moreover, introduction of p53 into tumor cells was shown to enhance induction of apoptosis following exposure to CTL-mediated cytotoxic insults39 and p53 accumulation in tumor cells is an indispensable component in the GzmB-induced apoptotic signaling pathway.41,42
The cytotoxicity of CD8 TILs against the tumor can be influenced by multiple immunosuppressive factors in the local TME, such as suppressive cytokines, suppressor cells (e.g., Tregs and MDSCs), and signaling through inhibitory immune ligands.43–45 Tregs and MDSCs are crucial populations in enforcing immunosuppression in the TME.31 Although further investigations on underlying mechanisms are needed, our study showed that elevated p53 expression resulting from SGT-53 treatment can be exploited as a new means of eliminating these immunosuppressive cells, accounting for the increase in activated TILs and anti-tumor immunity seen in vivo when SGT-53 is used in combination with checkpoint blockade. Importantly, gene expression profiling revealed the significant down-modulation of IDO1, an enzyme known for its key immunosuppressive role in many human cancers, following SGT-53 treatment. It has been previously shown that tryptophan depletion by cancer cell-expressed IDO1 could lead to the T cell anergy and activation of immunosuppressive Tregs and MDSCs.46,47 Currently, drugs targeting IDO1 pathway are in clinical trials to reverse the tumor-induced immunosuppression.47 In accordance with our data, a recent report demonstrated that restoration of p53 activity via nutlin-3a was able to induce ICD and promote CD8 T cell-dependent anti-tumor immunity in mice bearing EL4 tumor.31 In that study, activated p53 was able to eliminate immunosuppressive MDSCs. However, reactivation of endogenous p53 via nutlin-3a requires tumor cells harboring wtp53 and many tumor cells would be expected to be unresponsive to nutlin-3a. In contrast, our tumor-targeted gene therapy approach to restore functional p53 and subsequently induce tumor cell immunogenicity and anti-tumor immunity would not be expected to be dependent on the p53 status of the tumor. Indeed, in human tumor cell lines, we have observed that SGT-53 can push cells harboring either wtp53 or mutated p53 readily into apoptotic death.27,28
Despite clinical success of immunotherapy based on blockade of the PD1/PD-L1 axis, only a subset of patients exhibit durable responses.5 Moreover, therapeutic resistance can also develop. A recent study found that mutations that affected the antigen presentation and the sensitivity of tumor cells to T cell-derived IFNs could cause acquired resistance to anti-PD1 therapy.4,48 To overcome, or potentially prevent, the development of acquired immunotherapeutic resistance, a very large number of trials are now underway that combine checkpoint blockade with a wide range of other agents.10 In many cases, the combination under study appears to be lacking any strong mechanistic rationale for that particular combination. Although the elements involved in tumor response are complex,10 studies seeking biomarkers that might be used to predict response to anti-PD1 antibody have found that tumor expression of PD-L1 is the single feature most highly correlated with response.49,50 It has been suggested that cancer patients who do not respond to treatment with anti-PD1 antibodies are those having tumors with relatively low expression of PD-L1.2, 50–52 Our experiments revealed that SGT-53 treatment up-regulated PD-L1 expression in cultured 4T1 cells and in mouse syngeneic breast tumors in vivo. This PD-L1 up-regulation has also been observed in other mouse syngeneic tumor models (e.g., MOC1 head and neck cancer,21 GL261 glioblastoma and LL2 lung cancer, data not shown). Thus, it is our hypothesis that SGT-53 treatment will also elevate expression of PD-L1 on human tumors and expand the fraction of patients who respond to anti-PD1 antibodies. The ability of SGT-53 to elevate tumor PD-L1 in multiple syngeneic mouse models together with the fact that the treatment with SGT-53 plus the checkpoint inhibitor results in enhanced infiltration of the tumors by TILs provides a clear rationale for a trial involving this combination. This notion is supported by our observation in four syngeneic tumor models (4T1 breast cancer, LL2 lung cancer, and GL261 glioblastoma shown here plus MOC1 head and neck cancer21). These observations supports the contention that the combination of SGT-53 and anti-PD1 antibody could prove more efficacious as an immunotherapy regimen than the checkpoint blockade alone and thereby improve outcomes for cancer patients.
In addition to inhibiting tumor growth, SGT-53 plus anti-PD1 reduced metastases. Mice bearing 4T1, a highly metastatic breast tumor, experience metastases to the lung akin to what is observed in some breast cancer patients. Strikingly, in the lungs of 4T1 tumor bearing mice, SGT-53 alone was able to reduce substantially metastatic tumor nodules, whereas anti-PD1 antibody treatment alone was essentially ineffective in blocking lung metastases. In mice treated with the anti-PD1/SGT-53 combination, 4T1 lung nodules were essentially not detected. Although further studies are warranted, these data would support a combination clinical trial of an anti-PD1 antibody plus SGT-53.
Although PD1/PD-L1 checkpoint blockade is normally well-tolerated, three patients receiving nivolumab died from pneumonitis while participating in a trial.53 We observed that BALB/c mice bearing 4T1 tumors were killed when given 4–5 injections of anti-PD1 antibody (clone RMP1-14; rat IgG). Deaths from the anti-PD1 treatment of mice occurred prior to their tumors having grown large enough to be fatal. Hypersensitivity leading to death after repeated injections of xenogeneic anti-PD1 (clone J43; hamster IgG) or anti-PD-L1 (clone 10F.9G2; rat IgG) has also been observed by others using the 4T1 tumor model in BALB/c mice.22 Increased accumulation of neutrophils in the lung and the neutrophil-induced anaphylaxis were identified as the cause of death.22 We saw an abnormal lung infiltration of neutrophils only with anti-PD1 antibody monotherapy but not with either SGT-53 alone or with the combination of anti-PD1 and SGT-53 treatment. Likewise, mortality of mice seen after treatment with anti-PD1 antibody alone was prevented when SGT-53 was given in conjunction with the checkpoint inhibitor. Gene expression profiling of 4T1 tumors using the NanoString nCounter® analysis revealed a set of candidate genes linked to our observations. Many of these candidate genes are known to be involved in cellular immune responses including neutrophil priming and activation. The involvement of these candidate genes in the fatal xenogenic response warrants further study as does the mechanism by which death is averted by exposure to SGT-53. Nonetheless, our data suggest that SGT-53, when added to anti-PD1 immunotherapy, may augment checkpoint blockade not only in terms of rendering checkpoint inhibitors more effective but also in making them safer for patients.
In summary, we describe that SGT-53 not only increased the immunogenicity of tumor cells and the number of tumor-infiltrating immune cells but also alleviated immunosuppression and improved anti-tumor activity when used in combination with an anti-PD1 antibody. This improved efficacy of the combination therapy was observed in three mouse syngeneic tumor models (4T1 breast cancer, LL2 non-small cell lung cancer and GL261 glioblastoma). A similar enhancement of anti-tumor activity was also observed in mouse syngeneic model of MOC1 head and neck cancer.21 Given that SGT-53 could alleviate fatal hypersensitivity associated with an anti-PD1 antibody in 4T1 breast cancer, this nanomedicine may be able to reduce immune-related adverse events that are sometimes seen with cancer immunotherapies. Collectively, our data suggest that SGT-53, representing tumor-targeted p53 gene therapy, has potential to augment significantly immune checkpoint blockade agents for improved outcomes in a variety of malignancies. It is possible that the SGT-53 would not only improve outcomes in patients that already respond to checkpoint blockade, but also increase the percentage of patients who respond. SGT-53 has completed a first-in-man Phase I and Ib trials with favorable safety profiles54,55 and is now being evaluated in multiple Phase Ib and II trials as combination therapy with currently approved chemotherapeutic agents. Our data here provide a strong mechanistic rationale for combining SGT-53 and PD1/PD-L1-based immune checkpoint blockade in a clinical trial setting.
Materials and methods
Cell lines
4T1 and LL2 were obtained from American Type Culture Collection. GL261 was obtained from National Cancer Institute. Cells were maintained at 37°C in a 5% CO2 atmosphere in modified RPMI-1640 (Mediatech; 4T1) or DMEM (Mediatech; LL2 and GL261) supplemented with 10% heat-inactivated FBS (Sigma), 2 mM L-glutamine (Mediatech), and 50 μg/mL each of penicillin, streptomycin, and neomycin (Gibco).
Preparation of SGT-53 nanocomplex
The cationic liposome consisting of 1,2-dioleoyl-3-trimethylammonium propane (Avanti Polar Lipids, 890890) and dioleolylphosphatidyl ethanolamine (Avanti Polar Lipids, 850725), referred to as Lip, was prepared using the ethanol injection method as described previously.56 TfRscFv/Lip/p53 (SGT-53, scL encapsulating plasmid DNA encoding human wtp53) complex was prepared by simple mixing of Lip with TfRscFv and plasmid DNA as previously described.57 For in vitro experiments, SGT-53 was further diluted with serum-free media (SFM). For animal injections, 5% dextrose (Hospira) was added to the SGT-53 preparation. The size and zeta potential of SGT-53 were determined by dynamic light scattering at 25°C with a Zetasizer Nano ZS System (Malvern Instruments). The mean particle size of SGT-53 in water was 114.4 ± 8.4 nm. The mean zeta potential of SGT-53 was 28.2 ± 1.2 mV.
Expression of p53 by SGT-53 in vitro
4T1 cells were plated at 6.0 × 105 cells per 10 cm dish for 24 h before transfection. SGT-53 was prepared as described above and added in SFM to the dishes (7 μg of DNA/dish). The p53 expression plasmid pCMV-p53 contains the 1.7 kb human wtp53 cDNA under the control of the CMV promoter, followed by the SV40 polyadenylation signal.29 As a control treatment, scL-GFP or scL-vec nanocomplexes were prepared using either GFP expression plasmid pCMV-GFP or empty plasmid pCMV, respectively. After incubation for 4 h at 37°C, the medium was replaced with complete medium and the cells were further incubated. At the indicated time after treatment, cells were collected for analysis.
HMGB1 release assay
Concentration of the HMGB1 protein in the cell culture medium was measured according to the manufacturer’s instructions using the HMGB1 ELISA kit (Aviva Systems Biology, OKEH00424).
ATP assay
Level of extracellular ATP in the cell culture medium was measured by luciferin-based ENLITEN ATP Assay (Promega, FF2000) following the manufacturer’s instructions using Gen-probe Leader 50 luminometer (MGM Instruments).
Animal models
All animal experiments were performed in accordance with and under approved Georgetown University GUACUC protocols. For the syngeneic 4T1 breast tumor model, 5–6 week old female BALB/c mice (Envigo) were s.c. inoculated with 4T1 cells in SFM (0.5 × 106 cells/site). For the syngeneic LL2 lung cancer or GL261 glioblastoma models, 5–6 week old female C57BL/6 mice (Envigo) were s.c. inoculated with either LL2 cells (0.5 × 106 cells/site) or GL261 cells (1.0 × 106 cells/site), respectively. Mice were systemically injected with either SGT-53 [30 μg DNA/injection/mouse, intravenous (i.v.)], anti-PD1 antibodies [200 μg/injection/mouse, intraperitoneal (i.p.), RMP1-14, BioXCell] or the combination of both, following the indicated treatment schedules in Figure 3A.
Quantitative RT-PCR assay
Total RNAs were extracted from either cell pellet or tumor tissues using PureLink RNA Mini Kit (Ambion, 12183018A) according to the manufacturer’s protocol. One microgram of extracted RNA was reverse transcribed in 20 μL reaction volume with Superscript IV VILO Master Mix (Life Technologies, 11766050) with ezDNase enzyme, which removes genomic DNA, following the manufacturer’s protocol. PCR was performed using TaqMan Fast Advanced Master Mix (Life Technologies, 4444557) and TaqMan gene expression assays (Life Technologies) for human p53 (Hs01034249_m1), mouse IFNα1 (Mm03030145_gH), mouse CCL2 (Mm00441242_m1), mouse CXCL1 (Mm04207460_m1), mouse IL15 (Mm00434210_m1), mouse DEC1 (Mm00478593_m1), mouse PD-L1 (Mm00452054_m1), and mouse GAPDH (Mm99999915_g1) with StepOnePlus RT-PCR system (Life Technologies). Relative mRNA expression was analyzed using StepOne Software v2.3 via the ΔΔCt method with normalization to GAPDH mRNA. Samples were assayed in triplicate.
Fluorescence-activated cell sorting analysis
Tumors were harvested, weighed, and cells were dissociated by enzymatic digestion in Hank’s balanced solution containing 1 mg/mL collagenase D (Roche, 1108888200) and 2 mM DNase I (Sigma, D4263) for 1 h at 37°C. Cells collected from both in vitro and in vivo studies were stained with antibodies against FAS (152606), PD-L1 (124308), CD80 (104716), CD86 (105037), ICAM1 (116120), H-2Kd/H-2Dd (114708), I-A/I-E (107632), CD45 (103146), CD31 (102424), CD3 (100218), CD4 (100449), CD8a (100708), CD11c (117310), F4/80 (123110), CD107a (121608), Gr1 (108452), FoxP3 (126419), GzmB (515406), IFNγ (505839, all from BioLegend), CD11b (BD Biosciences, 552850), and CRT (Novus Biologicals, NBP1-47518APC). For the cells isolated from the tumor tissue, they were labelled with Zombie-NIR viability dye (BioLegend, 423105) prior to the staining with antibodies according to the manufacturer’s instructions. To assess the level of apoptosis, cells were stained with Annexin V apoptosis detection kit with 7-AAD (BioLegend, 640926). Cells were analyzed by LSRFortessa flow cytometer (BD Biosciences).
Ihc
Mice were euthanized after completing treatment (day 17) or when the control animals developed excessive tumor burden. Harvested tumor, lung, and liver were fixed in 10% neutral buffered formalin, paraffin embedded, and sectioned at 5 μm. Tumor sections were stained for Casp3 (Cell Signaling Technology, 9661) or Ki-67 (Dako, M7240) antibodies according to manufacturer’s instructions. TUNEL staining was performed using the ApopTag peroxidase in situ apoptosis detection kit (Millipore, S7100) according to manufacturer’s instructions. H&E (Surgipath, Leica Biosystems) staining was performed on lung and liver sections. Images were captured using Olympus DP70 camera on Olympus BX61 microscope. The percentages of Casp3, TUNEL, or Ki-67 positive cells were counted using the ImmunoRatio, an automated cell counting software (http://153.1.200.58:8080/immunoratio/) at least five fields of view from three tumor sections.
Elisa
Collected serum samples were assayed using mouse GM-CSF ELISA kit (BioLegend, 432204) or TNFα ELISA kit (eBioscience, 88732422) according to the manufacture’s protocols.
NanoString analysis
Two commercially available gene panels (mouse PanCancer Pathways and mouse PanCancer Immune profiler) containing total 1330 unique genes were used (NanoString Technologies).58 RNA was isolated as described above from 4T1 tumor tissues and hybridized with probes according to the manufacture’s protocols. The resulting RNA complexes were subsequently immobilized and counted on an nCounter® analyzer (NanoString Technologies). Raw data were normalized based on the geometric mean of negative controls, internal housekeeping genes, and positive controls in nSolver 3.0 software (NanoString Technologies). Normalized counts from genes included in both panels were analyzed further using nSolver 3.0.
Statistical analysis
Data represent mean ± SEM. The statistical significance was determined by one-way ANOVA or by t tests. P values of <0.05 were considered significant. All graphs and statistical analysis were prepared using SigmaPlot (Systat Software, San Jose, CA).
Funding Statement
This study was supported in part by NCI grant 5R01CA132012-02 (EHC), National Foundation for Cancer Research grant HU0001 (EHC), and a research grant from SynerGene Therapeutics Inc. This study was conducted in part at the Lombardi Comprehensive Cancer Center Preclinical Imaging Research Laboratory, Microscopy & Imaging Shared Resource, Genomics & Epigenomics Shared Resource, Tissue Culture Shared Resource, Flow Cytometry & Cell Sorting Shared Resource, and Animal Core Facilities which are supported by NIH/NCI grant P30-CA051008 and U.S. Public Health Service grant 1S10RR15768-01. This investigation was performed in part in a facility constructed with support from Research Facilities Improvement grant C06RR14567 from the National Center for Research Resources, NIH.
Acknowledgments
We thank K. Creswell and D. Xun, Flow Cytometry Shared Resource, Georgetown University Medical Center, for their technical assistance with flow cytometry. We also thank R. Stephens, SynerGene Therapeutics, for her technical assistance.
Disclosure statement
Dr. Chang is one of the inventors of the described technology, for which several patents owned by Georgetown University have been issued. The patents have been licensed to SynerGene Therapeutics for commercial development. Dr. Chang owns an equity interest in SynerGene Therapeutics and serves as a non-paid scientific consultant to SynerGene Therapeutics. Dr. Kim is salaried employee of SynerGene Therapeutics. Dr. Harford serves as salaried President & CEO of SynerGene Therapeutics and owns stock in same.
Supplementary matierials
Supplementary matierials for this article can be accessed here.
References
- 1.Mellman I, Coukos G, Dranoff G.. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Donnell JS, Smyth MJ, Teng MW. Acquired resistance to anti-PD1 therapy: checkmate to checkpoint blockade? Genome Med. 2016;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med. 2016;213:2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassel JC, Heinzerling L, Aberle J, Bahr O, Eigentler TK, Grimm MO, Grunwald V, Leipe J, Reinmuth N, Tietze JK, et al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): evaluation and management of adverse drug reactions. Cancer Treat Rev. 2017;57:36–49. [DOI] [PubMed] [Google Scholar]
- 7.Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 10.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 11.Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 12.El-Deiry WS. Insights into cancer therapeutic design based on p53 and TRAIL receptor signaling. Cell Death Differ. 2001;8:1066–1075. doi: 10.1038/sj.cdd.4400943. [DOI] [PubMed] [Google Scholar]
- 13.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valente JF, Queiroz JA, Sousa F. p53 as the focus of gene therapy: past, present and future. Curr Drug Targets. 2018;19. doi: 10.2174/1389450119666180115165447. [DOI] [PubMed] [Google Scholar]
- 15.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol. 2013;25:85–92. doi: 10.1097/CCO.0b013e32835b6386. [DOI] [PubMed] [Google Scholar]
- 17.Cui Y, Guo G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int J Mol Sci. 2016;17:E1942. doi: 10.3390/ijms17111942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SS, Rait A, Rubab F, Rao AK, Kiritsy MC, Pirollo KF, Wang S, Weiner LM, Chang EH. The clinical potential of targeted nanomedicine: delivering to cancer stem-like cells. Mol Ther. 2014;22:278–291. doi: 10.1038/mt.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5:403–416. doi: 10.18632/oncotarget.v5i2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu K, Wang J, Zhu J, Jiang J, Shou J, Chen X. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene. 1999;18:7740–7747. doi: 10.1038/sj.onc.1203235. [DOI] [PubMed] [Google Scholar]
- 21.Moore EC, Sun L, Clavijo PE, Friedman J, Harford JB, Saleh AD, Van Waes C, Chang EH, Allen CT. Nanocomplex-based TP53 gene therapy promotes anti-tumor immunity through TP53- and STING-dependent mechanisms. OncoImmunology. 2018;7:e1404216. doi: 10.1080/2162402X.2017.1404216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mall C, Sckisel GD, Proia DA, Mirsoian A, Grossenbacher SK, Pai CS, Chen M, Monjazeb AM, Kelly K, Blazar BR, et al. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology. 2016;5:e1075114. doi: 10.1080/2162402X.2015.1075114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuire HM, Shklovskaya E, Edwards J, Trevillian PR, McCaughan GW, Bertolino P, McKenzie C, Gourlay R, Gallagher SJ, Fazekas de St Groth B, et al. Anti-PD-1-induced high-grade hepatitis associated with corticosteroid-resistant T cells: a case report. Cancer Immunol Immunother. 2018;67:563–573. doi: 10.1007/s00262-017-2107-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudlak K, Demuro JP, Hanna AF, Brem H. Acute lung injury following the use of granulocyte-macrophage colony-stimulating factor. Int J Crit Illn Inj Sci. 2013;3:279–281. doi: 10.4103/2229-5151.124168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Descotes J. Immunotoxicity of monoclonal antibodies. MAbs. 2009;1:104–111. doi: 10.4161/mabs.1.2.7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22:7486–7495. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 27.Kim SS, Rait A, Kim E, Pirollo KF, Nishida M, Farkas N, Dagata JA, Chang EH. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano. 2014;8:5494–5514. doi: 10.1021/nn5014484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim SS, Rait A, Kim E, Pirollo KF, Chang EH. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine. 2015;11:301–311. doi: 10.1016/j.nano.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Pirollo KF, Tang WH, Rait A, Chang EH. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum Gene Ther. 1999;10:2941–2952. doi: 10.1089/10430349950016357. [DOI] [PubMed] [Google Scholar]
- 30.Muñoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16:741–750. doi: 10.1038/nri.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo G, Yu M, Xiao W, Celis E, Cui Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017;77:2292–2305. doi: 10.1158/0008-5472.CAN-16-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaur P, Asea A. Radiation-induced effects and the immune system in cancer. Front Oncol. 2012;2:191. doi: 10.3389/fonc.2012.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H, Forveille S, Sauvat A, Yamazaki T, Senovilla L, Ma Y, Liu P, Yang H, Bezu L, Müller K, et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 2016;7:e2134. doi: 10.1038/cddis.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017;77:839–850. doi: 10.1158/0008-5472.CAN-15-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lettau M, Paulsen M, Schmidt H, Janssen O. Insights into the molecular regulation of FasL (CD178) biology. Eur J Cell Biol. 2011;90:456–466. doi: 10.1016/j.ejcb.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Braun MW, Iwakuma T. Regulation of cytotoxic T-cell responses by p53 in cancer. Transl Cancer Res. 2016;5:692–697. doi: 10.21037/tcr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang B, Niu D, Lai L, Ren EC. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun. 2013;4:2359. doi: 10.1038/ncomms3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiery J, Abouzahr S, Dorothee G, Jalil A, Richon C, Vergnon I, Mami-Chouaib F, Chouaib S. p53 potentiation of tumor cell susceptibility to CTL involves Fas and mitochondrial pathways. J Immunol. 2005;174:871–878. doi: 10.4049/jimmunol.174.2.871. [DOI] [PubMed] [Google Scholar]
- 40.Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/MCB.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meslin F, Thiery J, Richon C, Jalil A, Chouaib S. Granzyme B-induced cell death involves induction of p53 tumor suppressor gene and its activation in tumor target cells. J Biol Chem. 2007;282:32991–32999. doi: 10.1074/jbc.M705290200. [DOI] [PubMed] [Google Scholar]
- 42.Ben Safta T, Ziani L, Favre L, Lamendour L, Gros G, Mami-Chouaib F, Martinvalet D, Chouaib S, Thiery J. Granzyme B-activated p53 interacts with Bcl-2 to promote cytotoxic lymphocyte-mediated apoptosis. J Immunol. 2015;194:418–428. doi: 10.4049/jimmunol.1401978. [DOI] [PubMed] [Google Scholar]
- 43.Hirayama Y, Gi M, Yamano S, Tachibana H, Okuno T, Tamada S, Nakatani T, Wanibuchi H. Anti-PD-L1 treatment enhances antitumor effect of everolimus in a mouse model of renal cell carcinoma. Cancer Sci. 2016;107:1736–1744. doi: 10.1111/cas.2016.107.issue-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harter PN, Bernatz S, Scholz A, Zeiner PS, Zinke J, Kiyose M, Blasel S, Beschorner R, Senft C, Bender B, et al. Distribution and prognostic relevance of tumor-infiltrating lymphocytes (TILs) and PD-1/PD-L1 immune checkpoints in human brain metastases. Oncotarget. 2015;6:40836–40849. doi: 10.18632/oncotarget.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 46.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. 2015;13:412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 INHIBITORS: from bench to bedside. Cancer Res. 2017;77:6795–6811. doi: 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 51.Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, Hodi FS, Joshua AM, Kefford R, Hersey P, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34:4102–4109. doi: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jing W, Li M, Zhang Y, Teng F, Han A, Kong L, Zhu H. PD-1/PD-L1 blockades in non-small-cell lung cancer therapy. Onco Targets Ther. 2016;9:489–502. doi: 10.2147/OTT.S94993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gangadhar TC, Vonderheide RH. Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol. 2014;11:91–99. doi: 10.1038/nrclinonc.2013.245. [DOI] [PubMed] [Google Scholar]
- 54.Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, Nunan R, Pirollo KF, Rait A, Chang EH. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21:1096–1103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pirollo KF, Nemunaitis J, Leung PK, Nunan R, Adams J, Chang EH. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: a phase 1b study. Mol Ther. 2016;24:1697–1706. doi: 10.1038/mt.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, Cruz I, Xiang LM, Pirollo KF, Chang EH. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther. 2002;1:337–346. [PubMed] [Google Scholar]
- 57.Yu W, Pirollo KF, Yu B, Rait A, Xiang L, Huang W, Zhou Q, Ertem G, Chang EH. Enhanced transfection efficiency of a systemically delivered tumor-targeting immunolipoplex by inclusion of a pH-sensitive histidylated oligolysine peptide. Nucleic Acids Res. 2004;32:e48. doi: 10.1093/nar/gnh049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bresler SC, Min L, Rodig SJ, Walls AC, Xu S, Geng S, Hodi FS, Murphy GF, Lian CG. Gene expression profiling of anti-CTLA4-treated metastatic melanoma in patients with treatment-induced autoimmunity. Lab Invest. 2017;97:207–216. doi: 10.1038/labinvest.2016.126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.