

ABSTRACT
We report long-term clinical outcomes and immune responses observed from a phase 1 trial of agonist CD40 monoclonal antibody (mAb) and blocking CTLA-4 mAb in patients with metastatic melanoma. Twenty-four patients previously untreated with checkpoint blockade were enrolled. The agonistic CD40 mAb CP-870,893 and the CTLA-4 blocking mAb tremelimumab were dosed concomitantly every 3 weeks and 12 weeks, respectively, across four dose combinations. Two patients developed dose-limiting grade 3 immune-mediated colitis that led to the definition of the maximum tolerated dose (MTD). Other immune-mediated toxicity included uveitis (n = 1), hypophysitis (n = 1), hypothyroidism (n = 2), and grade 3 cytokine release syndrome (CRS) (n = 1). The estimated MTD was 0.2 mg/kg of CP-870,893 and 10 mg/kg of tremelimumab. In 22 evaluable patients, the objective response rate (ORR) was 27.3%: two patients (9.1%) had complete responses (CR) and four (18.2%) patients had partial responses (PR). With a median follow-up of 45 months, the median progression-free survival (PFS) was 3.2 months (95% CI, 1.3–5.1 months) and median overall survival (OS) was 23.6 months (95% CI, 11.7–35.5 months). Nine patients are long-term survivors (> 3 years), 8 of whom subsequently received other therapy including PD-1 mAb, surgery, or radiation therapy. Elevated baseline soluble CD25 was associated with shorter OS. Immunologically, treatment was associated with evidence of T cell activation and increased tumor T cell infiltration that was accomplished without therapeutic PD-1/PD-L1 blockade. These results suggest opportunities for immune activation and cancer immunotherapy beyond PD-1.
Keywords: CD40, CTLA4, melanoma
Introduction
T lymphocytes are prime mediators of tumor immuno-surveillance. In patients with melanoma, T cell tumor infiltration predicts clinical outcome, and immunotherapeutic strategies such as antibodies blocking cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) can have remarkable efficacy.1 Nevertheless, many patients still fail to respond clinically to these approaches, yet for PD-1 antibody, immune modulation is observed in nearly every patient.2 A key goal is to understand the potential of immune modulation that goes beyond CTLA-4 and PD-1.
Here, we report the clinical and immunological impact of simultaneously activating CD40 and blocking CTLA-4 in patients with metastatic melanoma who had not previously received CTLA-4, PD-1 or programmed cell death ligand 1 (PD-L1) antibodies. Both CD40 and CTLA-4 are critical regulators of the cancer immune response that can be exploited therapeutically.3,4 CD40 is a cell-surface receptor that mediates activation of antigen presenting cells (APC) and plays an important role in immunological “licensing”, particularly for enabling tumor immunity.3 CTLA-4 is a negative regulator of T cells, and blockade of the CD80/86-CTLA-4 pathway with CTLA-4 monoclonal antibody (mAb), such as ipilimumab, enhances anti-tumor T cell responses and leads to tumor rejection.5 In mice, combination therapy with agonist CD40 mAb and blocking CTLA-4 mAb – without PD-1 or PD-L1 blockade – enhances the induction of tumor-specific T cells and tumor rejection.6
In this study, we evaluated the combination of the agonist CD40 mAb CP-870,893 with the blocking CTLA-4 mAb tremelimumab for patients with metastatic melanoma. Although each mAb has been tested separately and shown single-agent activity in patients with advanced melanoma,7,8 the combination has not. Tremelimumab is a fully human IgG2 anti-CTLA-4 mAb that produced clinical activity8-10 similar to ipilimumab, although it did not improve OS when compared to chemotherapy in a randomized phase III study.8 CP-870,893 is a fully human IgG2 agonist mAb evaluated in a single-dose study, a weekly infusion study, and studies combining CP-870,893 with chemotherapy.7,11–15 The most common side effect with CP-870,893 is transient, grade 1–2 cytokine release syndrome. We and others have shown that CP-870,893 activates human B cells and dendritic cells in vitro which in turn can induce T cell proliferation and cytokine production.16-18 Here, we report that the combination of CD40 and CTLA-4 mAbs results in reinvigoration of T cells and is associated with clinical activity and acceptable toxicity in metastatic melanoma.
Results
Patient characteristics
Between March 2010 and December 2012, 24 patients with metastatic melanoma were enrolled (Table 1). Median age was 60 (range 34–80 years) and 54% of patients were women. Fifteen patients (62.5%) had been previously treated for metastatic melanoma, and nine (37.5%) of those had had previous immunotherapy (none with anti-CTLA4 or anti-PD1/PDL1 agents). Twelve (50%) of the patients were BRAF wild-type; however, 5 of the patients (21%) were not tested at the time of the trial. Two patients were not evaluable for response (due to rapid, symptomatic progression of disease) and were replaced, although included in toxicity analysis and swimmer plot analysis.
Table 1.
Patient characteristics.
| Median age (range) | 60 years (34–80) |
| Male sex – no.(%) | 11 (45.8) |
| ECOG score- no.(%) | |
| 0 | 17 (70.8) |
| 1 | 7 (29.2) |
| BRAF V600E Mutation Status | |
| Present | 7 (29.2) |
| Not Present | 12 (SO) |
| Unknown | 5 (20.8) |
| Stage at diagnosis- no (%) | |
| IV-a | 6 (25) |
| IV-b | 7 (29.2) |
| IV-c | 11(45.8) |
| LDH at initiation- no (%) | |
| Within normal range | 17 (70.8) |
| Greater than ULN | 7(29.2) |
| Treated as first line – no. (%) | 9 (37.5) |
| Median no. prior tx (range) | 1(0–5) |
| Previous Treatments | n (%) |
| Any | 15 (62.5) |
| # with previous chemo1 | 8 (33.3) |
| #with previous targeted tx2 | 6 (25) |
| #with previous immune tx3 | 9 (37.5) |
| # with previous anti-CTLA-4 | 0 (0) |
| #with previous anti-PD-1/PD-Ll | 0 (0) |
| Treatment details: 1dacarbazine, temozolamide, carboplatin/paclitaxel,nab-paclitaxel, 2 imatinib,nilotinib, vemurafenib, 3 sargramostim, HD interferon, interleukin-2, multiepitope peptide vaccine, other vaccine | |
Treatment and safety
Patients were treated with a median of four doses of CP-870,893 (range, 1 to 16 doses) and one dose of tremelimumab (range, 1 to 4 doses) over a median of 12 weeks (range, 1 to 48 weeks). Two patients received all four cycles of treatment (i.e., 20 doses of study agents, including 16 doses of CP-870,893 and 4 doses of tremelimumab) allowed by the protocol and one patient received 19 of 20 doses. There were 2 DLTs during the dose-escalation phase – each of which was grade 3 diarrhea which met the protocol definition of DLT. These 2 patients with DLT were the only 2 patients treated at the fourth dose level. Another grade 3 event of diarrhea did not meet the definition of DLT. Consequently, the MTD of the combination was estimated to be dose level 3 (0.2 mg/kg CP-870,893 and 10 mg/kg tremelimumab) on which, after enrollment expansion, a total of 16 patients were treated. Thus, enrollment per dose level was: 3 patients on level 1, 3 patients on level 2, 16 patients on level 3 (MTD) and 2 patients on level 4.
Adverse events (AE) in cycle 1 are listed in Table 2. The most common treatment-related AE was transient, grade 1–2 CRS within 12 hours of CP-870,893 administration, which occurred in 20 (83.3%) patients. CRS manifested as fevers, chills, myalgias, arthralgias, or headache and was self-limited, resolving within 36 hours of CP-870,893 administration. Control of mild CRS symptoms was achieved with acetaminophen and ibuprofen taken during the 36-hour span. One patient with grade 3 CRS also developed grade 3 hypotension and required hospitalization for less than 24 hours, an event that was considered a treatment-related serious adverse event (SAE). There were no other treatment-related SAEs. The other most common AEs were fatigue (9%), constipation (7%), and headache (5%). Overall, 3 patients developed grade 3 diarrhea, two of whom improved with steroids alone and a third patient was refractory to steroids but rapidly responded to anti-TNFα treatment. Two of these episodes met the definition of DLT. One patient developed hypophysitis and hypopituitarism, responded symptomatically to dexamethasone, and required long-term thyroid and corticosteroid replacement. One patient developed grade 2 uveitis, fully responsive to topical steroids. Potential autoimmune effects were further monitored by a panel of autoantibodies (anti-nuclear antibody, anti-dsDNA antibody, anti-liver/kidney microsomal, and anti-islet cell antibody). Of these, one patient developed a positive ANA (1:160 compared to undetectable at baseline), deemed clinically insignificant. Other grade 3 toxicities were upper gastrointestinal bleeding (n = 1) and lower gastrointestinal bleeding (n = 1). While still on study, one patient died of progressive disease.
Table 2.
Adverse events in cycle 1.
| Any Grade (% of all events) |
Grade 3 or higher (% of all events) |
|
|---|---|---|
| ALLERGY/IMMUNOLOGY | ||
| Autoimmune reaction-uveitis (anterior) | 1 (1) | 0 |
| AUDITORY/EAR | ||
| Hearing (no baseline audiogram) | 1 (1) | 0 |
| CARDIAC ARRHYTHMIA | ||
| Supraventricular and nodal arrhythmia- Sinus tachycardia | 1 (1) | 0 |
| CARDIAC GENERAL | ||
| Bilateral Pedal Edema | 1 (1) | 0 |
| Hypertension | 1 (1) | 0 |
| Hypotension | 3 (2) | 1 (6) |
| CONSTITUTIONAL | ||
| Rigors/chills | 4 (3) | 0 |
| Fatigue | 12 (9) | 0 |
| DEATH | ||
| Death | 1 (1) | 1 (6) |
| DERMATOLOGY/SKIN | ||
| Alopecia | 1 (1) | 0 |
| Bruising (in absence of thrombocytopenia) | 1 (1) | 0 |
| Hyperpigmentation | 1 (1) | 0 |
| Pruritus | 5 (4) | 0 |
| Rash | 6 (4) | 0 |
| Ulceration | 1 (1) | 0 |
| Urticaria | 1 (1) | 0 |
| ENDOCRINE | ||
| Hypothyroidism | 2 (2) | 0 |
| Other (hypopituitarism) | 1 (1) | 1 (6) |
| GASTROINTESTINAL | ||
| Abdominal cramps | 1 (1) | 0 |
| ALT- elevated | 7 (5) | 2 (13) |
| AST- elevated | 7 (5) | 1 (6) |
| Anorexia | 1 (1) | 0 |
| Constipation | 9 (7) | 0 |
| Dehydration | 1 (1) | 0 |
| Diarrhea | 4 (3) | 3 (19) |
| Dry mouth (xerostomia) | 1 (1) | 0 |
| Mucositis – Oral cavity | 2 (2) | 0 |
| Nausea | 2 (2) | 0 |
| Taste alteration (dysgeusia) | 1 (1) | 0 |
| Vomiting | 2 (2) | 0 |
| Weight loss | 1 (1) | 0 |
| GENITOURINARY | ||
| Other- Shortened menstrual peirod | 1 (1) | 0 |
| HEMORRHAGE/BLEEDING | ||
| Hemorrhage, GI – Colon | 1 (1) | 1 (6) |
| Hemorrhage, GI – Stomach | 1 (1) | 1 (6) |
| INFECTION | ||
| With normal ANC or Gr 1–2 neutrophils- Upper airway NOS |
2 (2) | 0 |
| With normal ANC or Gr 1–2 neutrophils- Bladder | 2 (2) | 0 |
| LYMPHATICS | ||
| Other- Lymphadenopathy | 1 (1) | 0 |
| MUSCULOSKELETAL | ||
| Muscle Weakness – Trunk | 1 (1) | 0 |
| Other- Chondroid lesion, humerus | 1 (1) | 0 |
| NEUROLOGY | ||
| Dizziness (vertigo) | 1 (1) | 0 |
| Insomnia | 1 (1) | 0 |
| Mood alteration- anxiety | 1 (1) | 0 |
| Mood alteration- irritability | 1 (1) | 0 |
| Mood alteration- depression | 1 (1) | 0 |
| Neuropathy- sensory | 1 (1) | 0 |
| Sensitive to light | 1 (1) | 0 |
| Tinnitus | 1 (1) | 0 |
| OCCULAR/VISUAL | ||
| Other- Ptosis, unilateral | 1 (1) | 0 |
| Uveitis | 1 (1) | 0 |
| PAIN | ||
| Back | 2 (2) | 0 |
| Chest | 1 (1) | 0 |
| Headache | 7 (5) | 0 |
| Joints | 1 (1) | 0 |
| Extremity-limb | 1 (1) | 0 |
| Muscle | 1 (1) | 0 |
| Throat | 1 (1) | 0 |
| Tumor | 3 (2) | 0 |
| PULMONARY | ||
| Dyspnea | 1 (1) | 0 |
| Plural effusion | 1 (1) | 0 |
| Sinusitis | 1 (1) | 0 |
| SYNDROMES | ||
| Cytokine release syndrome | 20 (15) | 1 (6) |
| TOTAL | 143 (100) | 12 (100) |
Liver function test abnormalities were observed on day 8, fully resolving in each patient by day 22. These abnormalities never met the definition of DLT. One patient had grade 3 elevations in AST and ALT and a second patient had grade 3 elevation of ALT only. Five patients had grade 1 AST and 1 patient had grade 2 AST elevations. Four patients had grade 1 ALT and one patient had grade 2 ALT elevation. There were no grade 1 or higher elevations in total bilirubin, amylase, or lipase on day 8, except for one grade 1 amylase abnormality.
Efficacy
Clinical activity of treatment is illustrated by waterfall, spider, and swimmer plots in Figure 1a-c. For 22 patients evaluable for response, the ORR was 27.3% (95% CI 8.7 – 45.9%) including 2 CRs and 4PRs. Of these 6 responders, 4 were at the MTD (Figure 1a). With a median potential follow-up of 45 months, 14 deaths were observed. The median PFS was 3.2 months (95% CI, 1.3–5.1 months) and median OS was 23.6 months (95% CI, 11.7–35.5 months). One-year and 2-year OS were 72.7% (95% CI, 54.1 – 91.3%) and 48.7% (95% CI, 27.3 – 70.1%). Patients with M1a or M1b disease had a substantially higher ORR than patients with M1c disease, although the difference did not reach statistical significance (45.5% vs 9.1%, Fisher’s exact p = 0.15); however, M1a or M1b patients achieved a significantly longer OS than M1c patients (median OS not reached vs 6.3 months, respectively; log rank p = 0.004).
Figure 1.
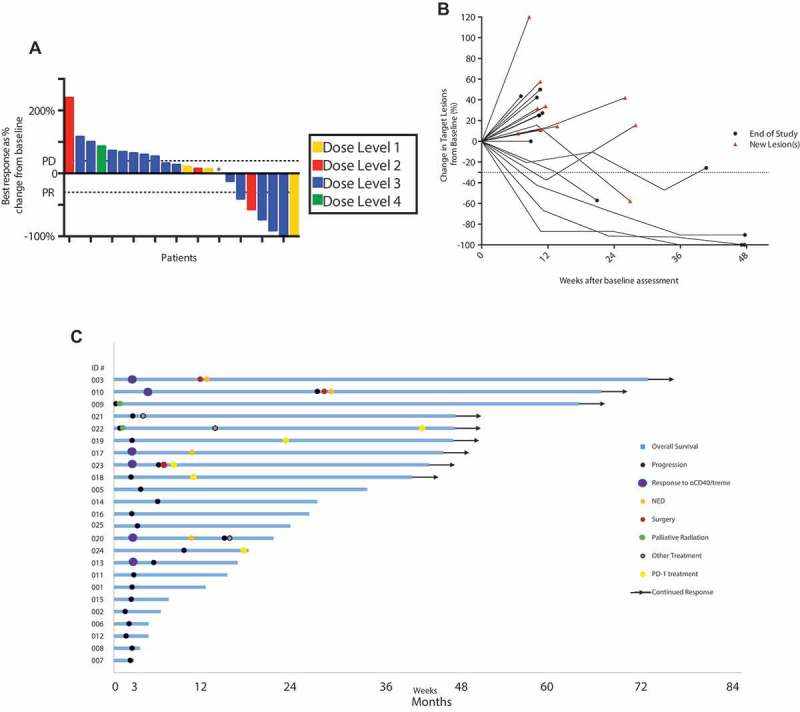
Clinical activity of CP-870,893 and tremelimumab in metastatic melanoma. Data are illustrated as (a) waterfall plot showing RECIST responses per dose level, * patient came off study before a second scan. (b) spider plot showing tumor measurements for each patient, and (c) swimmer plot showing long-term clinical course for each patient.
Among all patients treated, there were 9 patients alive beyond 36 months (Figure 1c). One patient among the long-term survivors developed a sustained CR and received no other therapy after CP-870,893. All other long-term survivors received additional therapy after this trial. Among these, 4 patients received PD-1 mAb (either nivolumab or pembrolizumab) once completing this study, with 3 PRs and 1 CR (Figure 1c) after PD-1 initiation. Of the 15 patients who lived less than 36 months, only one received PD-1 mAb and this was given in the final days before death from progressive disease. At the time, use of PD-1 mAb in melanoma was an emerging investigational strategy; as such, no patient in our study had previously received PD-1 mAb.
Two other patients who were alive beyond 36 months had developed rapid clinical deterioration and tumor progression before the end of the first cycle and were removed from this study; each patient then received palliative radiation therapy (Figure 1c). One patient, without further therapy beyond radiation, developed a CR and remains NED at 6+ years (Supplemental Figure 1). The second patient received another therapy a year after radiation and subsequently received PD-1 mAb and is alive at 3+ years.
Three of the long-term survivors underwent surgical resection of tumor deposits. In two cases, patients were NED after surgery and remained in CR and alive at 3+ years. In the other case, the patient received PD-1 mAb, as noted above. A final patient underwent treatment with tumor-infiltrating lymphocytes for progressive disease.
Analysis of cytokine and inflammatory biomarkers
Serum CRP and 30 cytokines were measured at baseline and throughout cycle 1. No marker was predictive of objective response. However, in addition to tumor stage, baseline values of LDH (p = 0.006), CRP (p = 0.006), and soluble CD25 (sCD25) (p = 0.01) were significantly associated with OS in univariate Cox regression analysis. Using CART analysis, cut points of 110 pg/mL for sCD25 and 7 mg/dL for CRP were identified. CART-defined variables denoted sCD25high/low (> 110 vs. ≤ 110, HR = 13.38, 95% CI 3.17 – 56.50) and CRPhigh/low (> 7 vs. ≤ 7, HR = 4.14, 95% CI 1.39 – 12.33) were highly associated with OS (Figure 2a-b). These CART-defined variables were strongly associated with one another (Fisher’s exact p = 0.004). CRPhigh/low was not associated with IL-6. Interestingly, sCD25high/low remained associated with OS (HR = 11.12, 95% CI 2.42 – 51.12) after adjusting for LDH (p = 0.05). Median OS was 33.8 months for 16 patients with sCD25 ≤ 110 and was 4.6 months for 6 patients with sCD25 > 110.
Figure 2.
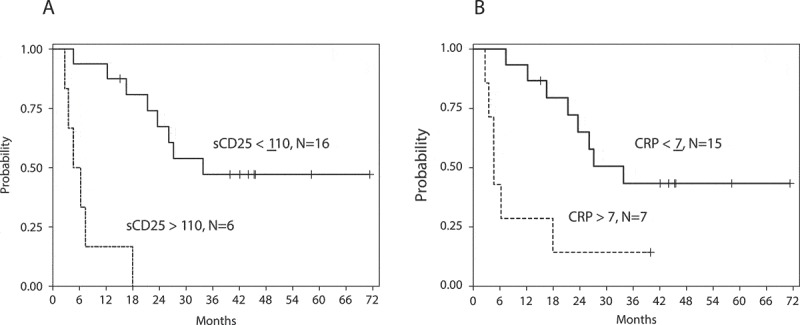
Probability of OS. Data for baseline (a) sCD25 and (b) CRP, grouped according to classification and regression tree (CART) analysis. For sCD25high/low, HR = 13.38, 95% CI 3.17 – 56.50. For CRPhigh/low, HR = 4.14, 95% CI 1.39 – 12.33.
Analysis of peripheral t cell activation
We examined peripheral blood T lymphocytes before, during, and after treatment using multi-parameter flow cytometry. There were no changes in the overall frequency of CD3, CD4 or CD8 T cell subsets during the course of therapy. We detected an upregulation of Ki67 in CD4, CD8, and FoxP3+ Treg T cell subsets after cycle 1 of therapy, and this effect extinguished in subsequent cycles (Figure 3a). Change in phenotype as well as the kinetics of this effect on T cells mirrored our previous observations for melanoma patients treated with PD-1 mAb in a separate clinical trial.2 We found that Ki67+ CD8 T cells after cycle one of therapy were enriched for a phenotype consistent with T cell reinvigoration: Tbet- Eomes± and CD45RA- CD27+, as well as expression of PD-1 and granzyme B (Figure 3b). These data indicate that a systemic CD8 T cell response similar to that observed in response to PD-1 blockade in melanoma patients 2 can also be generated using combination of CD40 activation and CTLA-4 blockade.
Figure 3.
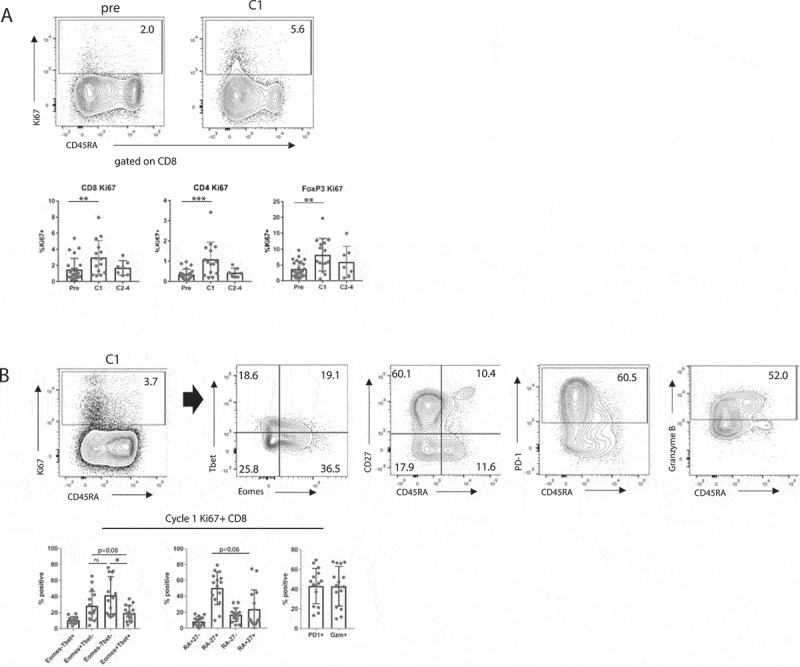
Immune analysis of peripheral lymphocytes. (a) Ki67 expression in CD8 T cells, CD4 T cells, and Tregs at serial time points. ** p < 0.01, *** p < 0.001 by linear regression. (b) Expression of Eomes versus Tbet, CD45RA versus CD27, PD-1, and Granzyme B in Ki67+ CD8 T cells at cycle 1. Eomes versus Tbet analyzed using Student’s paired t-test, CD45A versus CD27 analyzed using Wilcoxon matched pairs signed rank test, * p < 0.05.
Inspection of tumor microenvironment
Tumor biopsies were obtained from 8 patients at baseline and from 7 patients at various times after therapy. In 2 cases, paired pre/post samples were obtained. Immunohistochemistry for CD8 and PD-L1 was performed (Figure 4a). Overall, there was a statistically significant increase in CD8 infiltration when baseline and post-treatment samples were compared (mean ± SE 31 ± 12 cells/HPF vs. 167 ± 61 cells/HPF, respectively, p = 0.009) (Figure 4b). PD-L1 expression was negative in all but one baseline sample, yet was more prominent post-therapy with moderate to extensive expression in 4 of 7 samples. The difference between PD-L1 expression in pre- and post-treatment samples approached statistical significance (median 0 vs median 2, respectively, p = 0.072) (Figure 4c). In the post-treatment samples, higher PD-L1 expression correlated with higher CD8 infiltration (Spearman’s ρ = 0.82 and p = 0.022) (Figure 4d). One patient who was biopsied at the time of progression after achieving a PR to CP-870,893/tremelimumab demonstrated tumor CD8 infiltration and PD-L1 on tumor cells. After subsequent anti-PD1 mAb therapy, the patient responded and is a long-term survivor.
Figure 4.
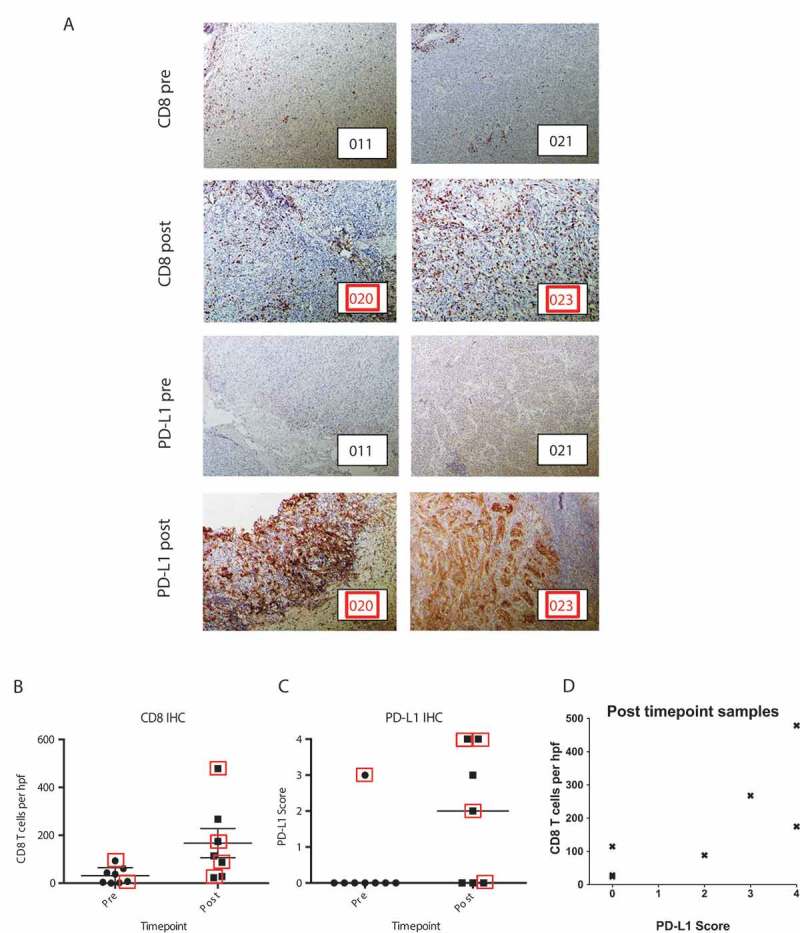
Immunohistochemistry analysis of tumor. (a) Representative examples of tumor biopsies obtained at baseline and post-treatment, each stained for CD8 (top) and PD-L1 (bottom). Patient identifying numbers are shown in bottom right of each photomicrograph, corresponding to Figure 1c. (b) Quantification of CD8 cells. Mean of five high-powered fields (hpf) are shown for each sample. Whisker plots indicate mean and standard error of the group. Group differences were determined by linear regression, p = 0.009. (c) Quantification of PD-L1 expression; group differences were determined by ordinal logistic regression, p = 0.072. Lines indicate median. (d) Correlation of PD-L1 and CD8 IHC in post-treatment tumor samples. The x-axis indicates PD-L1 score as in (c) and the y-axis indicates mean CD8 cells of 5 hpf for each patient. Spearman’s correlation coefficient ρ = 0.82 with p = 0.022. Red squares indicate patients with objective responses.
T cell receptor sequencing
To examine the changes in patients’ T cell repertoire with therapy, TCRβ deep sequencing was performed on post-treatment tumor biopsies and PBMC from 4 patients (003, 012, 023, 024) for which responses were 2 PR, 1 SD, and 1 PD. To focus on the clonotypes most likely to be relevant to an anti-tumor adaptive immune response, we identified the clonotypes that were expanded within the tumor. Tumor expanded clones (TEC) were defined as the most frequent clones for which the running sum accounted for 25% of the entire intratumoral clonal repertoire. TEC identified in the tumor were then tracked in PBMC (Supplemental Table 1). For each patient, we found that TEC were more prevalent in post-treatment PBMC than baseline PBMC (mean fold increase 3.3; 95% CI, 1.03 to 5.57), representing an average half-log increase in the circulating prevalence of tumor-infiltrating T cell clones in response to CP-870,893/tremelimumab.
Discussion
Findings from preclinical tumor models have long provided a convincing rationale for pursuing CD40 activation in cancer immunotherapy.19 CD40/CD40L interactions critically control adaptive immunity.3,20 The CD40 pathway is therapeutically tractable with mAb that activate and “license” APC upon binding, thereby upregulating numerous immunoglobulin-like and TNF superfamily receptors that trigger immune activation via downstream pathways.3 This mechanism of immune activation contrasts with that of mAb against CTLA-4, PD-1, PD-L1 that block inhibitory pathways. Prior clinical trials of single-agent agonistic CD40 mAb, or CD40 mAb in combination with chemotherapy, have demonstrated clinical activity.7,11–13,15,21 In a previous trial, 4 patients among 15 with advanced melanoma had a PR to a single dose of the CD40 mAb CP-870,893.7 Potential synergy of CD40 activation and CTLA-4 blockade has been established in mouse tumor models,6 but this study is the first to test this hypothesis in patients. This trial also represents a first attempt to combine an agonistic mAb with a checkpoint inhibitor for immunotherapy of cancer.
We found promising clinical activity with an acceptable toxicity profile using CP-870,893 (CD40) and tremelimumab (CTLA-4) to treat patients with metastatic melanoma. Our clinical follow up is extensive as patients were enrolled 5–7 years ago; as such, no patient had received PD-1/PD-L1 mAb. ORR was 27.3% including 2 CRs and 4 PRs. Median OS was 23.6 months, 1-year OS was 72.0%, and 2-year OS was 48.7%. Elevated baseline soluble CD25 appeared to be associated with shorter OS, although the small number of patients precludes a definitive conclusion; however, in a larger series of patients treated with ipilimumab, baseline serum concentrations of sCD25 represented an independent indicator of OS, with high levels predicting resistance to therapy.22 A mechanistic basis for this observation relates to findings that decoy sCD25 in mouse studies inhibits anti-tumor effects of CTLA4 blockade.22 Thus, it appears that our findings regarding baseline sCD25 reflect the use of tremelimumab in the treatment schema (more likely than a relation to CD40 mAb) and indicates that baseline sCD25 as a negative predictor of survival following treatment with ipilimumab or tremelimumab may be a class effect. Additional studies are needed to see if sCD25 may be a useful biomarker in patients with cancers other than melanoma and treated with CTLA-4 inhibitors.
One patient who completed the 4 maximum cycles of therapy had a CR and remains disease-free at more than 4 years without further therapy. For the 8 other long-term survivors, remission was achieved with additional therapy including PD-1 mAb, metastasectomy, radiation and cellular therapy for progressive disease, indicating a utility of sequentially delivering immune and standard therapies to achieve remarkable clinical outcomes.
Randomized studies will be important to discern the relative contributions of CP-870,893 and tremelimumab. By comparison, in patients with treatment-naive, unresectable stage IIIc or IV melanoma, single-agent tremelimumab led to an ORR of 10.7% and median OS was 12.6 months .8 In a pooled analysis of patients with advanced melanoma, treatment with single-agent ipilimumab produced a median OS of 11.4 months.23 In CheckMate 067, objective response to ipilimumab in patients with advanced melanoma was 19%.24
Combination treatment did not exacerbate the toxicity of the single agents. As in previous studies of CP-870,893 – either alone or in combination with chemotherapy7,11–15 – mild to modest and transient CRS was the most common adverse event. Published concerns of clinical unsuitability or lethal hepatotoxicity from intravenous agonistic CD40 mAb based on murine studies25,26 were once again not realized in this clinical trial. CP-870,893 at the MTD has been found to be a feasible combination partner with multiple types of chemotherapy and now with CTLA-4 blockade. Our findings from long-term observations also suggest that CD40/CTLA-4 followed by PD-1 is also clinically feasible. Rates of grade 3 or higher diarrhea/colitis in this study were similar to the rates reported in the phase II study of single-agent tremelimumab at a higher dose (15mg/kg). Interestingly, 10mg/kg was the MTD identified in the first-in-human study of tremelimumab9 and ultimately this dose was found to be more tolerable when dosed at a frequency of every 3 months.10 We found no evidence that CP-870,893 aggravated tremelimumab-associated toxicity or vice versa, nor did a new spectrum of autoimmunity emerge.
Immune analysis revealed evidence for activation of adaptive immunity both systemically and in the tumor. Phenotypic changes in CD8 T cells in the blood after one cycle of therapy were consistent with T cell reinvigoration and nearly identical in scope to that observed in melanoma patients treated with pembrolizumab.2 In the tumor, there was an overall increase in CD8 density post-treatment compared to baseline. This change often paralleled upregulation of PD-L1, possibly representing a resistance mechanism. Of note, 4 of 5 patients who progressed on CD40/CTLA-4 and went on to receive anti-PD-1 mAb are long-term survivors. From TCR deep sequencing, we observed expansion in the circulation of T cells clones that were found in relatively high prevalence in post-treatment tumors. Thus, increased tumor T cell infiltration, T cell reinvigoration, and T cell clonal expansion were observed.
In summary, combination treatment with CP-870,893 and tremelimumab was well- tolerated, triggered immune activation, and did so without PD-1 or PD-L1 blockade. A promising rate and depth of anti-tumor clinical responses was observed. CP-870,893 and tremelimumab appeared to cooperate clinically and mechanistically with other therapies such as PD-1 mAb and radiation delivered subsequently to patients who progressed on this study. Additional studies are highly warranted.
Materials and methods
Study design and patient population
This investigator-initiated and investigator-sponsored trial was conducted at the Abramson Cancer Center, University of Pennsylvania (Philadelphia, PA). The clinical protocol (NCT01103635) was approved by the University of Pennsylvania Institutional Review Board and an Investigator New Drug application (IND 107,547) was approved by the United States Food and Drug Administration. The primary objectives of this study were to assess the safety of CP-870,893 and tremelimumab given together and to determine a maximum tolerated dose (MTD) for the combination. Secondary objectives were to measure clinical response, PFS and OS and to assess immune pharmacodynamics of this combination. Immune parameters included activation of circulating T lymphocytes, serum cytokines, intratumoral T cell inspection by immunohistochemistry, and T lymphocyte clonal repertoire by T cell receptor (TCR) deep-sequencing.
Eligibility criteria included patients 18 years or older with histologically proven metastatic melanoma and Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1. Patients were required to have measurable disease as defined by RECIST 1.1 and adequate bone marrow, kidney and liver function. Patients were ineligible if they had received prior treatment with CD40 or CTLA4 targeted therapy or had central nervous system metastases. Other exclusion criteria included concomitant illnesses such as autoimmune diseases (excluding vitiligo), or a history of gastrointestinal diseases causing diarrhea, diverticulitis, thrombotic disease such as stroke, transient ischemic attack or symptomatic coronary artery disease, deep venous thrombosis, or coagulopathies. Patients with continuous or planned corticosteroid use were not eligible for enrollment in this study. All enrolled patients provided written informed consent.
Study treatment
Patients were assigned to dose levels of CP-870,893 and tremelimumab following a standard 3 + 3 escalation design. Tremelimumab was infused every 12 weeks starting at a dose of 6 mg/kg whereas CP-870,893 was given every 3 weeks starting at a dose of 0.1 mg/kg. A single dose of tremelimumab and 4 doses of CP-870,893 constituted one cycle. At the start of each cycle, tremelimumab was given on day 1 and CP-870,893 given on day 2. Dose levels were: 0.1 mg/kg CP-870,893 and 6 mg/kg tremelimumab (dose level 1); 0.1 mg/kg CP-870,893 and 10 mg/kg tremelimumab (dose level 2); 0.2 mg/kg CP-870,893 and 10 mg/kg tremelimumab (dose level 3); 0.2 mg/kg CP-870,893 and 15 mg/kg tremelimumab (dose level 4). The first patient at each dose level was monitored for 6 weeks prior to additional patients being enrolled at that dose level. Escalation to the next higher dose level occurred if all patients on the current dose level had been observed for at least 6 weeks and either no patients out of 3 or ≤ 1 patient out of six experienced dose limiting toxicity (DLT). DLT was defined as the following toxicities occurring during the first cycle: (i) any grade 4 treatment-related toxicity except lymphopenia lasting < 10 days, (ii) grade 3 hypersensitivity or grade 2 hypersensitivity in which symptoms reappear after infusion restarted, (iii) grade 2 or higher retinopathy or uveitis, (iv) grade 3 or higher elevations in aspartate aminotransferase (AST) or alanine aminotransferase (ALT) associated with symptoms, (v) grade 3 or higher diarrhea requiring infliximab or corticosteroids for more than 10 days, (vi) grade 3 or higher neurotoxicity, cardiac toxicity, pulmonary toxicity, or thrombotic events; or (vii) any other grade 3 treatment-related toxicity that fails to revert to grade 1 or baseline within 3 weeks, despite adequate medical therapy. The MTD was defined as the highest dose level at which one or fewer of six patients experienced DLT in the first 12 weeks and at least two patients treated at the next higher dose level experienced DLT. Once defined, an additional 8 patients were treated at the MTD. A maximum of four total cycles were allowed, in the absence of DLT or disease progression.
Clinical endpoints
Toxicity assessment was performed by clinical history, exam and laboratory evaluation on days 2, 8, 22, 43, and 64 of the first cycle. Toxicity assessments were identical in subsequent cycles, except day 8 was omitted. All patients had baseline imaging within 4 weeks of their first infusion of tremelimumab to define measurable target lesions. Radiographic assessments were performed every 12 weeks unless earlier assessment was indicated clinically. Comparisons to baseline were made by RECIST 1.1. The objective response rate (ORR) was defined as the proportion of response-evaluable patients who achieved a CR or PR. Patients were evaluable for response if they completed one full cycle of treatment. A radiologist at the University of Pennsylvania audited all RECIST measurements and the corresponding imaging studies. PFS was defined from the initiation of treatment to first date of documented disease progression, death or last patient contact alive and progression-free. Patients who had not progressed, were censored on the most recent date that documented their progression-free status (i.e., date of scan or clinic visit). OS was defined from the initiation of treatment to date of death or last patient contact alive.
Serum cytokine analysis
With a cytokine magnetic 30-plex panel (Life Technologies, Carlsbad, CA), the following analytes were analyzed: IL-1RA, FGF-Basic, MCP-1, G-CSF, IFN-γ, IL-12, IL-13, IL-7, GM-CSF, TNF-α, IL-1β, IL-2, IL-4, IL-5, IL-6, IFN-α, IL-15, IL-10, MIP-1α, IL-17, IL-8, EGF, HGF, VEGF, MIG, RANTES, Eotaxin, MIP-1β, IP-10, soluble CD25. Serum samples obtained at baseline and serially were cryopreserved at −80°C. Upon thawing, batched samples were analyzed using FlexMAP 3d instrument (Luminex), as previously described.27 C-reactive protein (CRP) and lactate dehydrogenase (LDH) were measured at the same times via the clinical laboratory of the Hospital of the University of Pennsylvania.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tumor samples were stained for CD8 and PD-L1 as previously described.28 CD8 positive cells were counted in 5 random high powered fields (HPF, 40x) and averaged per sample. PD-L1 staining was scored 0 to 4, as previously described.29
Flow cytometric analysis
Whole blood was collected from patients at baseline and approximately every 3 weeks, prior to each dose of CP-870,893. Peripheral blood mononuclear cells (PBMC) were separated from whole heparinized blood using Ficoll Paque (GE Healthcare Life Sciences) and cryopreserved for batch analysis. As previously described,2 flow cytometry of PBMC was performed with the following antibodies specific for CD3 (OKT-3), CD8 (RPA-T8), CD4 (RPA-T4), CD45RA (MEM-56), CD27 (O323), CD14 (M5E2), CD16 (3G8), CD19 (HIB19) and PD-1 (EH12.2H7) in FACS buffer (HBSS containing 1% FBS and 0.02% sodium azide) at RT for 20 min. Dead cells were excluded by staining for LIVE/DEAD stain (Life Technologies). Cells were fixed, permeabilized and stained for Eomesodermin (WD1928), FoxP3 (PCH101), granzyme B (GB11) and Ki-67 (B56) using the fixation/permeabilization kit from eBiosciences according to the manufacturer’s instructions. Samples were acquired on an LSR instrument (Becton-Dickinson) and data were analyzed using FlowJo (FlowJo, LLC).
Tcrβ sequencing
Genomic DNA was isolated from paraffin embedded tumor samples, as previously described14 and from thawed PBMC samples using the DNeasy kit (Qiagen). TCRβ deep sequencing was then performed by ImmunoSEQ, Adaptive Biotechnologies (Seattle, WA). ImmunoSEQ files were further analyzed using R base functions.
Statistical analysis
Descriptive statistics included mean, median, standard deviation, and range for continuous variables, and frequency and proportion for categorical variables. Natural log transformation was employed to normalize continuous distributions prior to statistical testing, when necessary. PFS and OS were estimated by the Kaplan-Meier method. Median potential follow-up (i.e., the length of time the study was able to observe study patients) was calculated by the reverse Kaplan-Meier method.30 Associations between OS and both clinical variables (e.g., stage, LDH) and immune markers were determined. The log rank test was employed to determine significance of categorical variables. Significance of continuous variables was determined by the Wald test from the Cox proportional hazards regression model. The magnitude of the association was measured by the hazard ratio and 95% confidence interval (CI) derived from Cox regression. Immune markers which were significant on univariate analysis were further examined by classification and regression tree (CART) analysis. This analysis approach identifies an optimal cut point, from all possible cut points of a continuous variable, which dichotomizes patients into lower risk and higher risk subgroups for an outcome of interest, which in this setting was OS. A p value < 0.05 was defined to be statistically significant. These statistical analyses were performed using either IBM SPSS v23 or STATA v14.
Linear regression was used to evaluate post-treatment changes in Ki67 in CD4, CD8 and FoxP3 + T cell subsets. Time (coded pretreatment, after cycle 1 or after cycles 2–4) was a class variable modeled using indicator variables. Due to incomplete data (i.e., cycle 1 and/or cycles 2–4 were missing), we employed the STATA procedure regress with the vce(cluster) option. This regression procedure allowed all observations to contribute to the model, while the cluster option addressed correlation among observations within patients. Natural log transformation was applied to all variables before modeling. For other immunological analyses, statistical analysis was performed using PRISM v7.03. For data that passed the D’Agostino & Pearson normality test, Student’s paired t-test was performed, otherwise a non-parametric Wilcoxon matched-pairs test was performed.
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI) [R01 CA158186];HHS | NIH | National Cancer Institute (NCI) [P30 CA016520];HHS | NIH | National Cancer Institute (NCI) [P50 CA174523];
Supplementary material
Supplemental data for this article can be accessed here.
Acknowledgments
This research was supported by National Institutes of Health grants NCI R01 CA158186, P50 CA174523, and P30 CA016520.
Author Contributions
Concept and design: DLB, RM, RHV. Acquisition of data: DLB, MJR, ACH, BS, FC, JJM, SFL, XX, TCG, RKA, LMS, RHV. Analysis and interpretation of data: DLB, RM, MJR, ACH, BS, LPR, DAT, JJM, SFL, XX, EJW, TCG, RKA, LMS, RHV. Writing, review and/or revision of the manuscript: DLB, RM, MJR, ACH, BS, LPR, DAT, SMG, ES, GC, JJM, SFL, XX, EJW, TCG, RKA, LMS, RHV. Administrative, technical, or material support: EJW, LMS, RHV.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
References
- 1.Sharma P, Allison JP.. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 2.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19:1035–1043. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377–3383. doi: 10.1172/JCI80012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito D, Ogasawara K, Iwabuchi K, Inuyama Y, Onoe K. Induction of CTL responses by simultaneous administration of liposomal peptide vaccine with anti-CD40 and anti-CTLA-4 mAb. J Immunol. 2000;164:1230–1235. [DOI] [PubMed] [Google Scholar]
- 7.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 8.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–622. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, Comin-Anduix B, Reuben JM, Seja E, Parker CA, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23:8968–8977. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- 10.Camacho LH, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, Redman B, Pavlov D, Bulanhagui C, Bozon VA, Gomez-Navarro J, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol. 2009;27:1075–1081. doi: 10.1200/JCO.2008.19.2435. [DOI] [PubMed] [Google Scholar]
- 11.Ruter J, Antonia SJ, Burris HA 3rd, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–993. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, Tolcher AW, Hamid O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033. doi: 10.4161/onci.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajor DL, Xu X, Torigian DA, Mick R, Garcia LR, Richman LP, Desmarais C, Nathanson KL, Schuchter LM, Kalos M, et al. Immune activation and a 9-year ongoing complete remission following CD40 antibody therapy and metastasectomy in a patient with metastatic melanoma. Cancer Immunol Res. 2014;2:1051–1058. doi: 10.1158/2326-6066.CIR-14-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak AK, Cook AM, McDonnell AM, Millward MJ, Creaney J, Francis RJ, Hasani A, Segal A, Musk AW, Turlach BA, et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann Oncol. 2015;26:2483–2490. doi: 10.1093/annonc/mdv387. [DOI] [PubMed] [Google Scholar]
- 16.Hunter TB, Alsarraj M, Gladue RP, Bedian V, Antonia SJ. An agonist antibody specific for CD40 induces dendritic cell maturation and promotes autologous anti-tumour T-cell responses in an in vitro mixed autologous tumour cell/lymph node cell model. Scand J Immunol. 2007;65:479–486. doi: 10.1111/j.1365-3083.2007.01927.x. [DOI] [PubMed] [Google Scholar]
- 17.Carpenter EL, Mick R, Ruter J, Vonderheide RH. Activation of human B cells by the agonist CD40 antibody CP-870,893 and augmentation with simultaneous toll-like receptor 9 stimulation. J Transl Med. 2009;7:93. doi: 10.1186/1479-5876-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gladue RP, Paradis T, Cole SH, Donovan C, Nelson R, Alpert R, Gardner J, Natoli E, Elliott E, Shepard R, et al. The CD40 agonist antibody CP-870,893 enhances dendritic cell and B-cell activity and promotes anti-tumor efficacy in SCID-hu mice. Cancer Immunol Immunother. 2011;60:1009–1017. doi: 10.1007/s00262-011-1014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13:1083–1088. doi: 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- 20.Grewal IS, Flavell RA. The CD40 ligand. At the center of the immune universe? Immunol Res. 1997;16:59–70. [DOI] [PubMed] [Google Scholar]
- 21.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hannani D, Vetizou M, Enot D, Rusakiewicz S, Chaput N, Klatzmann D, Desbois M, Jacquelot N, Vimond N, Chouaib S, et al. Anticancer immunotherapy by CTLA-4 blockade: obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015;25:208–224. doi: 10.1038/cr.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen T-T, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergmann S, Pandolfi PP. Giving blood: a new role for CD40 in tumorigenesis. J Exp Med. 2006;203:2409–2412. doi: 10.1084/jem.20061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res. 2011;17:2270–2280. doi: 10.1158/1078-0432.CCR-10-2888. [DOI] [PubMed] [Google Scholar]
- 27.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411. doi: 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Controlled Clinical Trials. 1996;17:343–346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.