

Abstract
Data workflow systems (DWFSs) enable bioinformatics researchers to combine components for data access and data analytics, and to share the final data analytics approach with their collaborators. Increasingly, such systems have to cope with large-scale data, such as full genomes (about 200 GB each), public fact repositories (about 100 TB of data) and 3D imaging data at even larger scales. As moving the data becomes cumbersome, the DWFS needs to embed its processes into a cloud infrastructure, where the data are already hosted. As the standardized public data play an increasingly important role, the DWFS needs to comply with Semantic Web technologies. This advancement to DWFS would reduce overhead costs and accelerate the progress in bioinformatics research based on large-scale data and public resources, as researchers would require less specialized IT knowledge for the implementation. Furthermore, the high data growth rates in bioinformatics research drive the demand for parallel and distributed computing, which then imposes a need for scalability and high-throughput capabilities onto the DWFS. As a result, requirements for data sharing and access to public knowledge bases suggest that compliance of the DWFS with Semantic Web standards is necessary. In this article, we will analyze the existing DWFS with regard to their capabilities toward public open data use as well as large-scale computational and human interface requirements. We untangle the parameters for selecting a preferable solution for bioinformatics research with particular consideration to using cloud services and Semantic Web technologies. Our analysis leads to research guidelines and recommendations toward the development of future DWFS for the bioinformatics research community.
Keywords: data workflow system, Semantic Web, linked data, cloud computing, genome sequencing, drug discovery
Introduction
Scientific workflow systems (SWFSs) efficiently support the analysis of large-scale data in transcriptome data analysis, medical genomics, bioimage informatics, drug discovery and proteomics often using cloud infrastructures and related services [i.e. IaaS, PaaS and Software as a Service (SaaS)]. The workflow systems enable researchers to perform their in silico experiments as a follow-up to their classical experiments in the laboratory, hence enabling the researcher to act as a data scientist to avoid becoming neither a software developer nor a scripting language expert [1]. Owing to the data-intensive nature of bioinformatics research, SWFSs nowadays transform into data workflow systems (DWFSs) that have to cope with the data deluge resulting from the numerous bioinformatics projects in general and the human genome projects in particular (or other data types, e.g. imaging). In addition, the transformation of the numerical data into meaningful information based on fact repositories, such as UniProtKB, and semantic sources, such as the Gene Ontology, puts additional requirements on the DWFS to enable efficient drug discovery and translational medicine based on experimental and numerical data [2].
Workflow technologies were introduced for the optimization of business processes, and specific languages [3] in combination with Web services are used to achieve flow control [4]. After that, the workflow systems have been adapted for scientific computations (i.e. SWFS), but not necessarily for large-scale data analytics nor the integration of semantic technologies. In particular, complex analyses are solved through combinations of modules [5–7], and data-intensive scientific analyses have been optimized for parallel and distributed computing infrastructures anticipating cloud-based services for large-scale data analytics. The integration of data from public fact repositories, e.g. Semantic Web data, is yet another important step, which should enable the sharing of data and the analytics pipelines across research teams, domains and geographic locations.
Bioinformatics research based on experimental and conceptual data with DWFS
Here, we distinguish observational data (i.e. experimental data) from conceptual or symbolic data (aka. ‘Semantic data’) often represented with Semantic Web technologies. The latter comprises not only concepts and labels, e.g. from ontologies but also axioms or facts in knowledge bases (KBs), and is used to add meaning to experimental data for human consumption but also to track the provenance of findings. Both types of data are increasingly analyzed in a joint approach in bioinformatics research and thus lead to innovative contributions to core bioinformatics research as well as drug discovery and translational medicine.
The human genome is composed of 3.2 billion base pairs resulting to ∼200 GB of whole-genome sequencing data. At a larger scale, the experimental data of several individuals or the analysis of the full genome of several cells leads to terabytes of data, which should rather be delivered and analyzed in a central repository; at best using tools like DWFSs are required for extracting useful information out of massive amounts of data [8, 9]. This is in contrast to shuffling the data within a computing cluster or shipping it between different computing centers [9], which would unnecessarily extend the time needed for the analysis because of limits in bandwidth, especially in infrastructure-poor environments. Similar computational challenges for large-scale data analytics (i.e. on experimental data), which have been solved with a DWFS, cover a wide range of problems and approaches, which include, for example large-scale NGS [9–11], gene-expression profiling [12] and peptide and protein identification [13], the analysis of single-nucleotide polymorphism (SNP), phenotype association studies [14] and copy number variation (CNV) analysis [15]. The next generation sequencing (NGS) sequencing platforms and their expected throughputs, error types and rates have been summarized in [16].
Bioinformatics for drug discovery research analyzes the properties of lead compounds and the drug–target interactions for optimal drug activities as well as reduced side effects through optimal selectivity. This research leads into new domains such as pharmacogenomics, which combines pharmacology and genomics to identify how the genotype affects a person’s response to a drug [17]. Specialized DWFS can play an important role in the productivity of such domains [18–22] in developing effective and safe medications tailored to a person’s genetic conditions with considerable successes. Bioinformatics research for drug discovery combines different kinds of data including semantic data to identify inhibitors of a receptor, to find novel drugs affecting specific pathways [23] and to conduct cheminformatics analyses for pharmacogenomics research [24]. Biomedical approaches comprise protocol-based medical treatment [25] and neuroimaging data analysis [26, 27] among others.
DWFS for analyzing large-scale data for bioinformatics research
The DWFS provides data analysis components and an interactive working environment with a number of advantages: automation of workflows through scripting and batch processing, real-time data processing, efficient interpretation of results through data visualization and integration and along with the automated update of newly available or modified analytical results [28]. Thus, experts from heterogeneous backgrounds without special IT skills can still use the systems efficiently as a shared platform for data processing [28, 29]. Ultimately, they can publish and share their workflows over the Web, thereby increasing research collaborations and scientific openness, scientific reproducibility and reusability supported by data provenance across workflows for error backtracking and resolution.
Altogether, the researcher faces the challenging task of identifying the most suitable workflow solutions, and therefore, our review will give an overview of available tools. It will assess the requirements for biomedical large-scale data (i.e. large-scale genome sequencing) and semantics-driven solutions (i.e. for drug discovery). Core questions of the analysis (Table 1) are concerned with the large-scale data analysis in the cloud infrastructure, benefits from Semantic Web technologies, reproducibility of results, Web-based approaches and next-generation workflow systems. Our investigations will focus on genomics, large-scale data analysis and drug discovery as the two contrasting core parts to bioinformatics research. In addition, Appendix describes the review methodology and exposes the filtering of the reference literature.
Table 1.
Questions that arise for the DWFS for large-scale data analytics for bioinformatics research
| Questions | Objective | Do DWFSs reach state of the art? | How important is the answering? |
|---|---|---|---|
| Q1 | Do the current solutions enable large-scale data analysis in a cloud environment? | Yes | Important and need some special care too, for large-scale data analytics using DWFSs |
| Q2 | Do existing solutions align well with the Semantic Web technologies for large-scale data analytics in bioinformatics research? | Mostly not | Bioinformatics research is now dependent on more data-intensive computing; therefore, existing solutions need to be aligned using the benefits of the Semantic Web technologies |
| Q3 | Is reproducibility of a computational analysis ensured over a long period using computational resources? | Mostly not | Reproducibility is one of the most important requirements for a DWFS, so that scientific experiments are more repeatable and transparent to others based on the given infrastructures and associated technologies |
| Q4 | Are current DWFS efficient and lightweight (workflow management and execution) enough for data analytics for bioinformatics research over the Web? | Mostly not | We need to deploy an efficient and lightweight data analytics approach on the cloud or data server without moving the data location |
| Q5 | Can we design a next-generation DWFS with Semantic Web and cloud computing technologies based on existing DWFS? | Yes | Important and our primary objective. However, this mostly depends on the right consideration, research and technical expertise |
The rest of the article is structured as follows: the ‘Semantic Web and cloud services in action’ section is focused on the ongoing trends and possible future outcomes for bioinformatics workflow systems by incorporating Semantic Web and Cloud computing services. The ‘Data workflow systems for bioinformatics research’ section discusses the use of different DWFS and their limitations based on the two use cases. The ‘Advancing DWFS through Semantic Web and cloud technologies’ section provides research and technological guidelines toward the development of a new DWFS. The ‘Conclusions’ section elaborates on anticipated future outcomes and achievements.
Semantic Web and cloud services in action
In this section, we show how the Semantic Web and cloud services improve the usability and performance of existing DWFS. Table 1 summarizes the objectives and our assessment of the relevance of the current DWFS.
Large-scale data management in the cloud for bioinformatics research
Tasks associated with bioinformatics research such as searching, downloading, visualization and analysis are mainly performed on the scientist’s desktops using DWFS. This essentially limits the potential for large-scale data analytics (e.g. for high nucleotide precision [23]) and leading into failures because of ever-increasing amounts of data, time-consuming data downloads and other constraints in terms of data volume and variety [30, 31]. The ‘4 Ms’ in data management, i.e. move, manage, merge and munge, are not sufficiently performant for large-scale data [31]. Furthermore, more complex problems in data representation and data usage have to be addressed for bioinformatics research to make use of data sharing in the cloud [32].
High-throughput technologies, such as NGS, require the bioinformaticians’ expertise to carry out data management and analytics at scale using DWFS, as well as access to high-performance computing infrastructures to mount data resources from distributed hosting infrastructures [33]. Therefore, interoperable data at a central site with efficient cloud-based processing units would form the right setup for DWFS including advancements in data reproducibility. To this end, robust, scalable and effcient data management tools are required for large-scale scientific discoveries including visualizations [30–32, 34–36].
A number of parallel and distributed approaches to workflow creation and management have been suggested to address above challenges [37]. Although existing DWFS can already perform in parallel and distributed environments for high-performance data analysis [31, 38–42], fewer solutions have been migrated to the Cloud as a Service [43–45]. Remember that migrating into the cloud [46] requires careful planning of data management, task dependencies, job scheduling, execution and provenance tracking. However, local plug-in-based architectures (i.e. Eclipse) would offer even better options for researchers [28].
In addition, data provenance based on an abstract specification of workflows and its specific operations [30, 31] is a key element for transforming engineering reproducibility into scientific reproducibility, e.g. in human genomics analysis [47–51]. Specific solutions (e.g. in virtualization technologies) allow result replication step by step [5] and in particular, tools like Docker along with Semantic Web services improve the performance of DWFS in this regard [52]. Scientists may now use the DWFS in combination with cloud infrastructures [e.g. Amazon Web Services (AWS)] [53] and perform data analytics on the database server without knowing the underlying IT infrastructures.
Access to data with open data formats and Semantic technologies
Semantic Web technologies (e.g. linked data, ontologies and execution rules) and KBs connect humans with data and improve workflow systems [30, 54]—by adding human-readable labels to data sets and by providing definitions for concepts (and their labels) and formalizing facts as axiomatic statements.
Bioinformatics solutions already use Semantic Web technologies, if publicly available resources have to be integrated in a transparent way [28], enabling data access in distributed and heterogeneous environments: the bioinformatics domain has embraced linked data as the Life Sciences Linked Open Data (LS-LOD) [24] to deliver its benefits into bioinformatics research [24, 32, 35, 54–67]. Bioinformatics research institutes increasingly provide their data as linked data, for example UniProtKB [63, 68], EMBL-EBI [69] and Data Databank of Japan [70]. Other bioinformatics groups are also contributing such as Bio2RDF [71, 72] that comprises most relevant biomedical data resources such as dbSNP [73, 74], OMIM [75, 76], pathway databases such as KEGG [77, 78], Reactome [79, 80] and Pathway Interaction Database [81]. Correspondingly, the NCBI itself [65, 82] provides its own data repositories in linked data format.
Other existing data resources (on the Web) are enriched with additional metadata and semantic knowledge for efficient reuse, for example as Linked Open Data (LOD) [54]. LOD exposes the data semantics in a machine-readable format including universal identification of data across the World Wide Web [via Uniform Resource Identifiers (URIs)]. The inclusion of semantics into data workflows provides many advantages over traditional architectures [35, 54, 57, 58]. For example, annotating the provenance of data with vocabulary languages (i.e. RDFS and OWL) ensures interpretation of data in an unambiguous way according to the original semantic context [48, 59, 83].
Semantic data integration leads to improved data availability through query access to federated SPARQL end points [84, 85]. More generic solutions support reuse of data among related workflows, and semantically annotated data enable workflow engines to discover the most relevant Web services at runtime, thus achieving data provenance support at low overheads [59]. However, deficiencies in the reuse of available URIs are still a barrier to the accessibility of bioinformatics data [24, 62]. Likewise, broken links hinder progress in the interfacing between various genomic data sources.
Ongoing research targets further improvements in DWFS to advance efficient workflow composition and reuse of workflows, scalability of processes, provenance tracking of data, flexibility in the workflow design, performance tuning and reliability through Web services. However, the existing systems do not yet reach scientists more advanced expectations [35, 57], in particular for embedding the DWFS as a core part of bioinformatics research [86]. Eventually, the researchers seek large-scale data integration for biological phenomena, e.g. biological and biochemical mechanisms and disease biomarkers [28, 87]; however, access to large-scale data from distributed public sources still requires unacceptably high levels of manual data integration, e.g. in drug discovery [19, 88].
Data workflow systems for bioinformatics research
This section gives the analysis of widely used DWFS for the bioinformatics research based on the literature review (see Appendix). Features and their definitions for DWFS are given in Table 2; the features are attributed to three categories, i.e. use of computational sources, human usability and access to public resources, which are again used to judge the DWFS (Table 3), and to provide research recommendations in the subsection ‘Full support for the cloud services and Semantic Web technologies’ (Table 4).
Table 2.
Workflow systems, features and definitions from the scientific literature including [1, 2, 12, 15, 17, 20, 34, 48, 60, 65]
| Features | Definition | Class |
|---|---|---|
| Data set conversion | DWFS enables the users to convert the data for bioinformatics research available in one format to another and helps create the corresponding mapping between different data types, thereafter with ease | IT characteristics |
| Adaptability | DWFS enables users to adopt the workflow system for new or unknown data types or formats | |
| Automation and batch processing | DWFS enables users to configure the workflow environment, workflow editing and submitting the workflow jobs using script-based approach with ease | |
| Workflow scheduling | DWFS enables users to schedule the workflow jobs (in case if the number of workflows to be submitted is enormous) before submitting | |
| Data integration | DWFS enables users to integrate and upload data sets from diverse sources to the workflow data directory | |
| Large-scale data processing | DWFS enables users to handle and process the data sets at scale | |
| System reliability | DWFS ensures that computation will be done successfully and the jobs will not be stalled in between | |
| Workflow specification | DWFS enables users to specify or develop or compose workflows with ease using standard workflow languages | |
| Portability | DWFS enables users to execute a workflow (locally or remotely in platform independent manner) after it has been created somewhere else | Human interface |
| Reproducibility | DWFS enables users to reproduce identical results against claimed results for similar input and computational approaches in elsewhere | |
| Data provenance | DWFS enables users to track experimental steps, parameter settings and intermediate input/outputs and experimental data lineage | |
| Computational transparency | DWFS enables users to share the experimental steps and workflows to the research communities who will be reusing the similar approach | |
| Reusability | DWFS enables users to reuse useful components further for similar experiments iteratively | |
| Ease of use | DWFS enables users to use the DWFS with little or no training overheads | |
| Scalability | DWFS processes data at different extents of data size and numbers of processing modules using available physical and software resources | Public resources |
| Extensibility | DWFS incorporates new modules or tools to the workflow system (when necessary) in the experimental steps | |
| Interoperability | DWFS integrates mergeable components from different DWFSs together | |
| Platform independence | DWFS operates on any operating system or platform (i.e. LINUX, Mac OS and Windows) | |
| Cloud integration support | DWFS migrates the whole workflow system on the cloud to be used as SaaS | |
| Open data and open-source design | DWFS is open to the research community so that they can configure the local copy on their machine or cloud and even contribute by adding new modules/tools or bug fixing, etc., to the next stable release |
Table 3.
Workflow systems and their scoring based on supported features
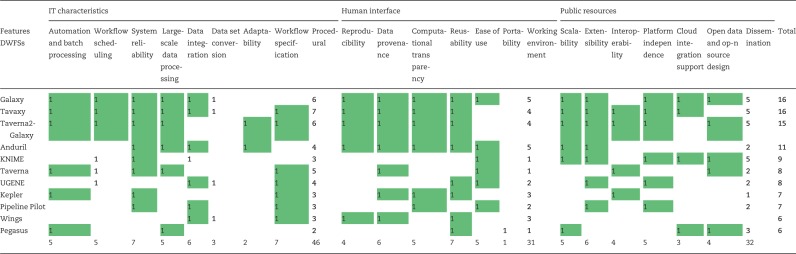
|
Note: Based on our extensive review of the literature, the scoring was marked 1 if the feature is supported by the workflow system and blank otherwise. ‘IT characteristics’ stand for the core processing capabilities of the DWFS, ‘human interface’ for user-friendliness and ‘public resources’ for alignment with publicly available data resources. Supported features are summarized based on our extensive review from literature [1, 2, 4, 11–13, 18, 19, 22, 23, 25, 33, 36, 37, 39, 43, 55, 56, 58, 59].
Table 4.
Features, definitions and their significance to cloud computing, linked data and open data
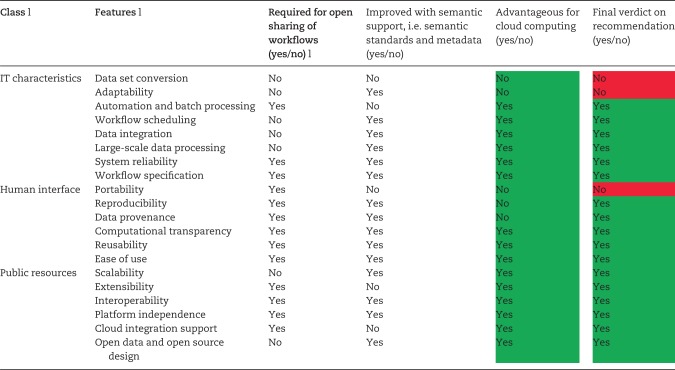
|
Note: These definitions and outcomes have been summarized based on our systematic review including [1, 2, 4, 11–13, 18, 19, 22, 23, 25, 33, 36, 37, 39, 43, 55, 56, 58, 59, 61, 63, 90, 92, 95, 96, 98, 106–110]. The last column signifies that the combined use of DWFS along with Semantic Web and cloud services could help to ensure the availability of (most) the features needed in a DWFS. Based on the review outcome, if the count of yes is at least 2 (of 3), the verdict goes to yes (with green color), no (in red color) otherwise.
In principle, we distinguish solutions that have been designed for the workflow-based integration of heterogeneous data sources and processes. For example, Taverna [67, 89, 111], Anduril [87], Taverna2-Galaxy [106], Konstanz Information Mine (KNIME) extension [107], Tavaxy, LONI [26, 27], SNAPR [90], Graphical Pipeline for Computational Genomics (GPCG) [91], Google Genomic Cloud, Pegasus [57, 58, 112], USC Epigenome Centre collaboration [10], Galaxy [92], GG2P [12] and Unipro UGENE NGS Pipeline [9, 108] that are linked to NGS, drug discovery and large-scale bioinformatics data analytics.
Table 5 shows the DWFS for the bioinformatics area and their use cases along with limitations according to their Web site information and related literature [4, 28, 54, 93, 100, 113]. In addition to these reviews, several solutions for processing NGS data based on shell scripts or graphical workflow environments have been suggested to improve data processing tasks such as high-throughput genome sequencing, data manipulation and visualization [39, 87, 89, 92, 94, 95, 114].
Table 5.
Some widely used DWFS and their potential use cases with limitations summarized from their Web site and other literature including [4, 28, 54, 98–100]
| DWFS | Potential use cases | Technologies | Limitations |
|---|---|---|---|
| Tavaxy | Personalized medicine and NGS (short DNA reads, DNA segments, phylogenetic and taxonomical analyze, EMBOSS, SAMtools, etc.) | SCUFL, JSON, hierarchical workflow structure, asynchronous protocol and DAG style in workflow creation and execution |
|
| Taverna2- Galaxy | Life Sciences (e.g. eukaryotic genome biology) | SCUFL 2 (experimental), Semantics, RDF, OWL and DAG |
|
| Galaxy | NGS (QC and manipulation, Deep Tools, Mapping, RNA Analysis, SAMtools, BAM Tools, Picard, VCF Manipulation, Peak Calling, Variant Analysis, RNA Structure, Du Novo, Gemini, FASTA Manipulation, EMBOSS, etc.) | Python, JavaScript, Shell script, OS: Linux and Mac OS X |
|
| KNIME | Pharma and healthcare (virtual high-throughput screening, chemical library enumeration, outlier detection in BioMed data and NGS analysis with KNIME Extension [107] | Java/Eclipse, KNIME SDK and Spotfire (supports Python ad Perl scripts) |
|
| Taverna | Domain-independent (bioinformatics, cheminformatics, gravitational wave analysis) | WSDL, Java and DAG |
|
| Wings | Multi-omics analysis and cancer omics |
|
|
| Anduril | Cancer research and molecular biology, DNA, RNA and ChIP-seq, DNA and RNA microarrays, cytometry and image analysis |
|
|
| Unipro UGENE | NGS: sequencing, annotationsMultiple alignments, phylogenetic trees, assemblies, RNA/ChIP-seq, raw NGS, local sequence alignment, protein sequencing, plasmid, variant calling, evolutionary biology and virology |
|
|
| Pipeline Pilot | NGS: gene expression and sequence data analysis, imaging, Pharma: drug–chemical material analysis, cheminformatics, ADMET, polymer properties synthesis, data modeling |
|
|
DWFS as a platform for processing genomics data
The workflow representation in a DWFS is mostly a directed acyclic graph (DAG), which excludes cycles in the workflow execution; however, other specifications comprise BigDataScript [109], RDF pipeline [14], PilotScript [6, 24] or SCUFL 2 notations, which enable operational flow control based on decision, forking and joining nodes [84]. Often, the DWFS provides a graphical user interface (GUI) for generating workflows prior executing them and input data and processing tasks to be assigned to the physical resources by the workflow engine. As an alternative, scripting and batch processing help to automate a DWFS, thus avoiding unnecessary human interaction [43], and the Kepler workflow system [34, 95, 110, 115] is a good example of a sophisticated run-time workflow engine that offers a GUI and automatic processing.
Galaxy is a comprehensive, well-established and widely used platform for interactive genomic analysis, reuse and sharing, offering an NGS computational framework for a single processing unit. It is well described with characteristics such as high usability, simplicity, accessibility and reproducibility of the computational results. It supports various sequence file formats like Text, Tabular, FASTA and FASTQ. Galaxy also provides special quality control (QC) by filtering the data sets by a quality score and solving specific gene sequence-related tasks. In addition, it provides full statistical support on user data sets showing the traits scoring and distribution functions.
On the other side, Galaxy lacks the proper interlinking of pipeline functionalities from one module into subsequently dependent modules. It is often not suitable for workflows containing loops and does not support any control-flow operations or remote services [100]. Additionally, it does not use a workflow language but instead uses a relational database (i.e. PostgreSQL). The libraries for available Galaxy routines also require advanced IT knowledge for developing new tools. Although the XML wrappers specify the inputs and outputs for the different tools, so that from a user perspective only, the suitable data formats are given in the drop-down options.
The LONI pipeline system is formed around a core pipeline engine for accessing distributed data sources, Web services and heterogeneous software tools focused on NGS data analysis [26]. The GPCG is also dedicated to NGS data analytics, which includes sequence alignment, SNP analysis, CNV identification, annotation, visualization and analysis of the results. Anduril is a workflow platform for analyzing large data sets—i.e. high-throughput data in biomedical research. The platform is fully extensible by third parties and supports data visualization, microarray analysis and cytometry and image analysis. Unipro UGENE provides the NGS pipelines for SAMtools, Tuxedo pipeline for RNA sequencing (RNA-seq) data analysis and Cistrome pipeline for chromatin immunoprecipitation sequencing (ChIP-seq) data analysis as an integrated platform in the Unipro UGENE desktop toolkit [9].
Other solutions deliver dedicated pipelines for specific data analytics tasks without following the ambition to form a platform. The SNAPR [90, 116] has been developed as a bioinformatics pipeline for effcient and accurate RNA-seq alignment and analysis [91]. The USC Epigenome Centre uses the Pegasus system as a computerized sequencing pipeline to conduct genome-wide epigenetic analyses [93, 100, 112]. GG2P supports seamless integration of various SNP genotype data sources like dbSNP [12, 73], and the discovery of indicative and predictive genotype-to-phenotype association. Recently, the KNIME has even been extended to NGS data analysis and processes NGS data formats like FASTQ, SAM, BAM and BED.
DWFS in drug discovery based on conceptual data
In bioinformatics for drug discovery, the DWFSs combine content from distributed databases to automate the reconstruction of biological pathways and the inference of relationships, for example finding the relationships between genes, proteins and metabolites to relevant knowledge about drugs. Solutions for drug discovery research use public data from fact repositories compliant with Semantic Web technologies and KBs that are contrasted by data from screening experiments for the profiling of chemical entities.
These tools not only help in workflow generation but also support mechanisms for tracing provenance and other methodologies fostering reproducible science. The tight coupling of myExperiment [96] with Taverna enables the Taverna workflow system to access a network of shared workflows for data processing [9]. Stevens et al. [97] proposed to share myExperiment-based bioinformatics-related workflow for facilitating the drug discovery process. In this respect, Pipeline Pilot eases the cheminformatics analysis and the progress in a data pipelining environment by combining the Pipeline Pilot and KNIME [98] leading into an efficient high-level GUI for bioinformatics tasks.
Chem2Bio2RDF [32] is a semantic workflow framework for linking the chemogenomic data to Bio2RDF and the LOD project [69]. It demonstrates the utility in investigating the polypharmacology identification of potential multiple pathway inhibitors and the association of pathways with adverse drug reactions. The customized version of the Kepler system for drug discovery and molecular simulations was proposed by Chichester et al. [99]. However, it is not scalable for large-scale drug-related data resources.
Advancing DWFS through Semantic Web and cloud technologies
This section examines usability improvements through data sharing, uploading, processing and analyzing with a focus on cloud infrastructures and semantics technologies. Table 4 lists characteristic features of DWFS (introduced in Table 2) and their relevance for cloud computing, semantic representation and open data access, respectively.
Increasing usability, reproducibility and data provenance
Scientists are often domain experts—not IT experts—and therefore require that the DWFSs expose high usability (and good documentation). Usability advances by hosting the services in a cloud infrastructure for ready access and by using semantic technologies for improved human–machine interaction through standardized semantic labeling of data. Furthermore, scientists profit from reproducibility of scientific work (i.e. repeatability of experiments and access to open data), which is supported by capturing workflow versioning and provenance information, again achieved with Semantic Web technologies [55, 83]. The data provenance for DWFS is managed by tracking the data management infrastructure, data lineage analysis and visualization [49]. Certainly, any data conversion has to preserve the data semantics.
Semantic Web technologies and KBs in this regard allow integration of LS-LOD at scale [56]. A good example is Wings [57, 58], which is based on the semantic representation for the design and validation of workflows, choice of experimental parameters, selection of appropriate dynamic models suitable for the scientific data and scientist’s requirements. This leads toward automatic workflow generation with sufficient detail to determine the provenance of the data.
As discussed before, provenance—as metadata information for data resources and workflow components—increases reproducibility and usability at a large scale [35, 103]. However, a uniform provenance standard is required to share the metadata in an explicit way [55], the Open Provenance Model could be further improved to this end, or the next release of SCFUL2 may bring semantics into the DWFS. Kepler Archive [115] and myExperiment are two repositories that facilitate the re-execution of workflows in a platform-independent manner by importing them in DWFS directly [104]. The last column in Table 4 signifies that the combined use of DWFS along with Semantic Web and cloud computing could help to ensure the availability of most of the features needed in a DWFS. Where, based on the review outcome, the overall verdict is yes if the count of yes responses is at least 2 of 3, and no otherwise.
Improving performance through data and workflow sharing in the cloud
A workflow engine has to scale according to the number of used resources, services and the volume of data leading to a difficult dependency between scalability and performance [28]. This dependency exposes the workflow engine as the core component solving the performance bottleneck [3]. Furthermore, computing infrastructures may restrict the deployment of workflow applications, and large data resources may only be transferred with significant overheads.
An effcient policy-based data placement bolsters the performance of a DWFS [49] such as known from the Swift workflow system for cloud-based computation [36, 43–45]. Other examples of DWFS in the cloud can be found from Deelman et al. [105]. The Wings DWFS enables large scientific workflows based on semantic representations that expose the provenance of scientific experimentations and the connections to other useful data. The structure and content of the data provenance record can be complex, as it has to correctly represent the data derivations, multiple source origins, multistaged processing and diverse analysis activities.
Finally, platform independence is important in bioinformatics research to share workflows across available platforms. Optimally, the DWFS would provide a browser-based user interface; the Taverna suite is a prime example as an open source, domain- and platform-independent workflow system. Interpreted programming languages like Perl, Python or PHP contribute to platform independence. Moreover, workflows should be easy to exchange, evolve and reusable and open source so that everybody can contribute to producing meaningful scientific results.
Toward fully integrated DWFS for analyzing large-scale data
The analytical overhead of genome sequencing data imposes restrictions to the research performed on NGS research overall [87]. Similarly, modern data-driven drug discovery requires integrated resources and pipeline solutions to support decision-making and enable new discoveries [101]. Data integration in bioinformatics requires resolving data sources heterogeneity when they use on large genomics and pharmacogenomics data sets in a distributed way [41].
The workflow presented in Figure 1 computationally integrates data from four different sources. The drug-related compounds are extracted from PubChem, bioassay from Bio2RDF, gene-related data from ClinVar and HGNC (or from the NCBIGene data set) and the pathway-related data set from Reactome and KEGG. The whole pipeline can be represented in RDF/XML, N3 or Turtle format. According to the literature [14], it is a decentralized approach with no central controller. Furthermore, it is data and programming language agnostic, where each node (rectangle) can be made live using an updater and a wrapper. The former has to be written using the same technology as the DWFS, but the latter could be of any programming language. However, in this regard, we would argue for using the SPARQL query language.
Figure 1.

Workflow for finding the pathways affecting particular drugs by finding the number of inhibitors communicating signals from a receptor using RDF pipeline notation [14]. This helps us in data integration, processing and querying that can be used by a number of collaborative experts together (i.e. practitioners like medical doctors, pharmacologist, chemist and IT experts). This workflow is conceptually adapted from the RDF pipeline by Booth et al. [14].
Workflow systems like Galaxy and KNIME are particularly suitable to bring all the combined genomic data (i.e. numerical or sequence data) and drug-related data (i.e. facts data and KBs) to the scientist. Then, these data can be processed as a DWFS as a Service in the cloud [44]. These approaches have been applied recently for large-scale biological sequence alignments [37, 102] along with the bioKepler [110]. The Tavaxy serves as an interoperable workflow system for analyzing large-scale genomic sequencing.
KNIME [98] has been tailored to drug discovery but could be augmented by incorporating Semantic Web technologies and then be attached to the Open PHACTS platform to query the RDFized drug compound-related data using SPARQL (as shown in Figure 1 as RDF pipeline notation [14]). This access to structured data gives input to questions concerned with the number of drug compounds having specific effects on pathways in the DNA regulation or with the side effects of a drug known from a drug–gene pathway.
However, Galaxy has emerged as the leading open-source workflow platform for data analytics (e.g. NGS data) and for the benchmarking of bioinformatics components because of its high flexibility and extensibility standards [99]. Semantic Web tools can be incorporated into the Galaxy workflow system just like any data analysis tools for processing, job monitoring, workflow creation and delivery of ready workflows to the research communities. Beyond these, Semantic Automated Discovery and Integration (SADI)-Galaxy [66] brings semantics support through the SADI framework into the Galaxy workflow system. Moreover, SADI-Taverna has been implemented in Taverna workflow system as well. A similar extension would be the TopFed–Galaxy integration [8] to make cancer genomic data analytics more reproducible, scalable and transparent, where the TopFed distributes the data from ‘The Cancer Genome Atlas’ as LOD for access to genetic mutations responsible for cancer.
Full support for the cloud services and Semantic Web technologies
Once the semantics requirements have been met, DWFS like Galaxy or KNIME would be migrated to the cloud. The best candidates for NGS analysis are Tavaxy and Galaxy because of their high scores (16 each in Table 3). However, Galaxy would be the most suitable candidate because of its widespread distribution and its ease of use for NGS. KNIME, on the other hand, performed best against the pharmaceutical use cases. Altogether, the biomedical or pharmacogenomics researchers can draft their requirements into the workflow specification using BigDataScript, RDF pipeline notation, PilotScript or SCUFL 2 for creating platform-independent workflows with LOD technologies before submitting the jobs.
Research questions can then be formalized as SPARQL queries addressing the data flow (Figure 1) between computational nodes and then can be submitted as workflow jobs (refer to Figure 2 for a generic overview) for execution. Likewise, Semantic Web tools can provide access to related data from heterogeneous sources (i.e. genomic- or drug-related compound data) via SPARQL end points as LOD with dereferenceable URIs, or Semantic Web tools can automatically transform local data sets and upload them to DWFS in RDF formats.
Figure 2.

Solving bioinformatics research problems for two representative use cases (e.g. genome sequencing analysis and drug discovery) by incorporating Semantic Web technologies and cloud services into the DWFS.
The predictors (in a DWFS) learn models from the training drug data and calculate predictions for the entire targets before combining and submitting them to the workflow engine. After submitting the workflow jobs, data then can be processed in a parallel and distributed way in the cloud services (e.g. Amazon AWS as IaaS and PaaS). Even the DWFS itself could be used to work as a SaaS tool. Further improvements result from the use of semantic provenance (and reasoning) to test and validate semantic consistency of the data model, conciseness of results and the reproducibility. Formal ontologies and KBs may contribute in addition. Automated reasoning validates RDFized instances and their compliance with the OWL classes of the data model.
To validate the results during the drug discovery or sequence analysis process, evaluation and validation could be performed on statistically significant drug data or simulated/real genome data. Moreover, validation can be done by matching the expected results with KBs rules. After the results have been evaluated and validated, biomedical scientists can prove their hypothesis based on the outcome.
Conclusions
Representing and developing new workflow systems or integrating sufficient tools in existing workflow system with suitable scalability and extensibility will be a key challenge for bioinformatics research in the future. DWFS in bioinformatics has to evolve toward distributed and scalable infrastructures including ubiquitous computing and integration of Web services, Semantic Web technologies and also domain-specific tools. Data provenance not only has to be ensured for large-scale data but also LOD manageability on the system level. Here, are some key points for this systematic review for bioinformatics research.
Bioinformatics researchers rely on a number of features such as result reproducibility, data provenance, scalability, openness, reusability, abstraction and simplicity. The suggestions provided in this manuscript should help researchers to develop more advanced DWFS. One particular focus will become the approaches of ontology-based formalism and semantic reasoning to achieve shared data representations and knowledge integration based on existing workflow systems (e.g. Galaxy and KNIME). More specifically:
Using a graph-based approach for representing and executing workflow of pathways (e.g. what is done in KNIME).
Making an efficient use of a modular approach (including parallelization) of the workflow job and processes (e.g. what is done in Galaxy).
Making efficient use of specification languages for the pathway (e.g. SCUFL 2) apart from the graphical approach.
Integration of the provenance information as metadata using Semantic Web technologies (e.g. exploiting the FAIR principles that were recently published in Nature).
Integrating the semantic resources (ontologies, fact repositories) and KBs, e.g. either through access to SPARQL end points, BigDataScript, or RDF pipeline notation.
Enabling the transformation of the experimental data into semantic information (e.g. via ML approaches) as available.
Key Points
For processing large-scale data for bioinformatics research requires an infrastructure—preferably a cloud infrastructure—to enable data analytics at scale to address emerging research problems.
The data deluge in bioinformatics research drives the demand for parallel and distributed computing by imposing a need for scalability and high-throughput capabilities onto the DWFS. Emerging requirements for data sharing and access to public resources suggest that compliance of the DWFS using Semantic Web standards is needed, where the data analytics has to be done on the cloud-based infrastructure.
If genome sequencing and drug discovery are considered as two of the most relevant use cases, following requirements must be met by using Semantic Web technologies on cloud-based infrastructure to attain the above outstanding advancements:
a number of capabilities need to be developed in the existing DWFS to prepare workflow creation, management and execution for parallel and distributed computing;
data provenance should be supported to combine engineering and scientific reproducibility based on Semantic Web technologies;
interoperable data (experimental and symbolic data) should be hosted in a secure environment with efficient cloud-based processing through semantic labeling (for scientists); and
the existing DWFSs have to advance into fully integrated DWFS for big data analytics in the cloud.
Acknowledgment
The authors would like to thank Niall OBrolchain, Brendan Smith and John McCrae for critically reviewing this article and Jaynal Abedin for helping them in the systematic review process and João Bosco Jares for helping them in drawing the Figure 2.
Appendix
The methodology
To this point, only a few initiatives have analyzed the DWFS [4, 5, 28, 35], but have been rather restrictive and did not give systematic considerations to Semantic Web and large-scale data-related benefits from DWFS. According to [28], available workflow systems in bioinformatics need to integrate technologies such as Semantic Web, grid and Web services and large-scale data analytical capabilities leading into pervasive approaches for existing Web service solutions and even propagated rule-based execution at runtime [35, 54, 57]. Therefore, a systematic review methodology including search queries, selection (i.e. inclusion) and exclusion criteria and related statistics is a mandate and hence discussed in this appendix.
Article searching criteria
It is well known that systematic reviews of complex evidence cannot rely solely on protocol-driven search strategies. The literature search, therefore, began with the use of search queries with search terms and a Boolean operator such as (“Scientific workflows”[All Fields]) AND (“Genome sequencing “[All Fields]) and combining it with the snowball sampling searches. We mostly used the PubMed, IEEE Digital Library (IDL) and Google Scholar (GS) specifying more recent years (i.e. 2008–17). The reason behind this source selection is that, when we tried to search related articles in the Web of Sciences and Science Direct, we obtained few publications.
Please refer to Table 6 for the statistics of the systematic searching as of 10 March 2017. Please note that while using the protocol-based and snowball sampling-based searching, only one reason was recorded for each record. In some cases, multiple reasons were applicable, but only one was recorded. Table 6 includes full texts from original search, snowball search (i.e. pursuing references of references) and reference list searches.
Table 6.
Article searching queries and related statistics for the systematic review methodology
| Query | Search query | Source | Results | Number of used publication | Section |
|---|---|---|---|---|---|
| Q1 | (“workflows”[All Fields] AND “next generation sequencing”[All Fields]) OR (“workflows”[All Fields] AND “genomics”[All Fields]) OR (“workflows”[All Fields] AND “Bioinformatics”[All Fields]) |
|
|
23 | ‘Introduction’, ‘Data workflow systems for bioinformatics research’ and ‘DWFS as a platform for processing genomics data’ |
| Q2 | (“Workflows”[All Fields] AND “Drug Discovery”[All Fields]) OR (“Workflows”[All Fields] AND “Pharmacogenomics “[All Fields]) |
|
|
22 | ‘Introduction’ and ‘Data workflow systems for bioinformatics research’ |
| Q3 | (“Workflows”[All Fields] AND “Big Data”[All Fields]) OR (“Workflows”[All Fields] AND “Large Scale Data”[All Fields]) OR (“Workflows”[All Fields] AND “Bioinformatics “[All Fields]) |
|
|
48 | ‘Semantic Web and cloud services in action’, ‘Large-scale data management in the cloud for bioinformatics research’ and ‘Access to data with open data formats and Semantic technologies’ |
| Q4 | (“Workflows”[All Fields] AND “Semantic Web “[All Fields]) OR (“Workflows”[All Fields] AND “Semantic”[All Fields]) OR (“Workflows”[All Fields] AND “Bioinformatics”[All Fields]) |
|
|
13 | ‘Advancing DWFS through Semantic Web and cloud technologies’ |
| Q5 | (“Workflows”[All Fields] AND “Provenance”[All Fields]) |
|
|
9 | ‘Semantic Web and cloud services in action’, ‘Data workflow systems for bioinformatics research’ and ‘Advancing DWFS through Semantic Web and cloud technologies’ |
Article inclusion and exclusion criteria
Figure 3 shows the inclusion and exclusion criteria of the literature used for the systematic review, and based on the outcome, we used only selected research papers that were more relevant, recent and highly cited. As a continuation and following the search process using the queries in Table 6, all records were merged, duplicates were removed and a unique ID was assigned to each record. As we reused the word workflows in every search query, we got some overlapping results as well. Please note that books were not eligible for the review. We excluded any manuscripts retrieved that were marked as drafts not to be cited.
Figure 3.

Records in stage of the systematic review for article inclusion and exclusion.
Through the PubMed and IDL database, 688 and 24 articles were found in peer-reviewed journals, respectively, using search Query Q1. Prevalent research areas focused on bioinformatics use cases like next-generation sequencing and drug discovery. However, only four were considered based on relevance [1–16, 24–29, 33] for the ‘Introduction’ section. On the other hand, the most unanticipated research area that tied for second was GS where we found 2420 articles. However, only the most relevant 19 articles were used [8, 10, 12, 14, 25–28, 37, 39, 87–89, 90–95] for the ‘Data workflow systems for bioinformatics research’ and ‘DWFS as a platform for processing genomics data’ sections.
For the Query Q2, the PubMed database returned 91 research articles, whereas GS and IDL returned 472 and 34 articles, respectively. We used only seven related literature [9, 32, 69, 96–99] in the ‘DWFS in drug discovery based on conceptual data’ section. And some of the most relevant (i.e. 15) articles were used [17–22, 29, 32, 88, 89, 98–102] for the ‘Introduction’ and ‘Data workflow systems for bioinformatics research’ sections.
Through the PubMed database, 552 articles were found in peer-reviewed journals using Query Q3. The search query consisted of words Workflows, big data, Large Scale Data and Bioinformatics. We choose these word choices, as the bioinformatics is also entering into the big data area in the most recent literature and some of the literature also used the term large-scale data too. Likewise, as the bioinformatics research nowadays is more data-intensive computing driven, thus, we argued that these terms will reflect and retrieve the relevant research articles to serve our purposes. IDL, on the other hand, returned only 39 publications. Whereas, the search query in GS returns 470 journal articles, with only the most relevant 48 articles were used [19, 23, 24, 28, 30–42, 44–53, 55–70, 83–88] for the ‘Semantic Web and cloud services in action’, ‘Large-scale data management in the cloud for bioinformatics research’ and ‘Access to data with open data formats and Semantic technologies’.
Through the PubMed database, therefore, 570 articles were found in peer-reviewed journals using Query Q4 for the Semantic Web in SWFSs in bioinformatics research. On the other hand, the same query in GS and IDL returns 2600 and 3 journal articles, respectively. The search query consisted of the words Workflows, Semantic Web, linked data or Semantics and Bioinformatics. Meanwhile, one of our main research goals was to review research articles that discussed the use of Semantic Web technologies in bioinformatics (mainly covering bioinformatics) using the DWFS. Therefore, we also included the term Semantics instead of Semantic Web, as some literature, for example [54, 57], contain the title with only the word Semantic. Most relevant 13 kinds of literature were considered only for this query too [35, 49, 55–58, 98, 99, 101–105] in the ‘Advancing DWFS through Semantic Web and cloud technologies’ section.
When we searched the literature using two keywords, workflow and provenance, for the Query Q5, we got significant results from GS and IDL (i.e. 8100 and 9896 publications) and only 25 from the PubMed. But we used only 10 articles [26, 35, 36, 47–49, 51, 59, 85, 92] in the ‘Semantic Web and cloud services in action’, ‘Data workflow systems for bioinformatics research’ and ‘Advancing DWFS through Semantic Web and cloud technologies’ sections. Note, we conducted the systematic review a few days back; therefore, depending on the contents, addition or deletion to/from the above databases might happen. Consequently, you might receive different results out of the same queries later on.
Md. Rezaul Karim is a PhD researcher at Semantics in eHealth and Life Sciences (SeLS), Insight Centre for Data Analytics, National University of Ireland, Galway. He is working toward developing an abstract method for scientific knowledge discovery workflows with linked data to demonstrate the VALUE from large-scale data for bioinformatics research. His research interests include Semantic Web, machine learning, workflow technologies and bioinformatics. He holds a BSc in Computer Science and an MSc in Computer Engineering. He is a PhD candidate at the National University of Ireland, Galway.
Audrey Michel is a Postdoctoral researcher at School of Biochemistry and Cell Biology, University College Cork, Ireland with expertise in the development of computational resources for the analysis and visualization of ribosome profiling (RiboSeq) and high-throughput gene expression data. She is the coordinator of RiboSeq.Org (http//riboseq.org/).
Achille Zappa is a Postdoctoral researcher at Insight Centre for Data Analytics, National University of Ireland, Galway. His research interests include Semantic Web technologies, semantic data mashup, linked data, big data, knowledge engineering, semantic integration in life sciences and health care and workflow management. He is the World Wide Web Consortium (W3C) Advisory Committee representative for Insight Centre for Data Analytics, National University of Ireland Galway.
Pavel Baranov is a Principal Investigator at the School of Biochemistry and Cell Biology, University College Cork, Ireland. He studies the mechanisms of mRNA translation using high-throughput biochemical methods and phylogenetic approaches.
Ratnesh Sahay is leading the Semantics in eHealth and Life Sciences (SeLS) research unit at the Insight Centre for Data Analytics, National University of Ireland, Galway. Sahay has worked on several European and national (Irish) R&D projects with an emphasis on using semantics for solving key integration/interoperability challenges in the e-health, clinical trial and biomedical domains. He is a member of the Global Alliance for Genomics and Health, Health Level Seven (HL7) Standard and World Wide Web Consortium (W3C) standardization working groups (OWL, HCLS). He previously served as a member of the OASIS SEE Technical Committee, W3C SWS-Challenge working group and CMS Working Group.
Dietrich Rebholz-Schuhmann, PhD, MD, MSc, DSc, is a Medical Doctor and a Computer Scientist. Currently, he has established a chair for Data Analytics at the National University of Ireland, Galway, and the director of the Insight Centre for Data Analytics in Galway. His research is positioned in semantic technologies in the biomedical domain. In his previous research, he has established large-scale on-the-fly biomedical text mining solutions and has contributed to the semantic normalization in the biomedical domain. He is editor-in-chief of the Journal of Biomedical Semantics.
Funding
The Science Foundation Ireland (grant number SFI/12/RC/2289).
References
- 1. McPhillips T, Bowers S, Zinn D, Ludäscher B.. Scientific workflow design for mere mortals. Future Gener Comput Syst 2009;25(5):541–51. [Google Scholar]
- 2. Rebholz-Schuhmann D, Oellrich A, Hoehndorf R.. Text-mining solutions for biomedical research enabling integrative biology. Nat Rev Genet 2012;13(12):829–39. [DOI] [PubMed] [Google Scholar]
- 3. Andrews T, Curbera F, Dholakia H, et al. Business process execution language for web services, version 1.1, 2003.
- 4. Barker A, Van Hemert J. Scientific workflow a survey and research directions. In: Proceedings of the International Conference on Parallel Processing and Applied Mathematics (PPAM). Springer, 2007, 746–53.
- 5. Gil Y, Deelman E, Ellisman M, et al.Examining the challenges of scientific workflows. Computer 2007;40(12):26–34. IEEE [Google Scholar]
- 6. Warr WA. Scientific workflow systems: Pipeline Pilot and KNIME. J Comput Aided Mol Des 2012;26:801–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor IJ, Deelman E, Gannon DB, Shields M.. Workflows for e-Science Scientific Workflows for Grids. Springer Publishing Company, Incorporated, 2014. [Google Scholar]
- 8. Poplawski A, Marini F, Hess M, et al.Systematically evaluating interfaces for RNA-seq analysis from a life scientist perspective. Brief Bioinform 2016;17:213–23. [DOI] [PubMed] [Google Scholar]
- 9. Golosova O, Henderson R, Vaskin Y, et al.Unipro UGENE NGS pipelines and components for variant calling, RNA-seq and ChIP-seq data analyses. PeerJ 2014;2:e644.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torri F, Dinov ID, Zamanyan A, et al.Next-generation sequence analysis and computational genomics using graphical pipeline workflows. Genes 2012;3(3):545–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baylin SB, Jones PA.. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer 2011;11(10):726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koumakis L, Moustakis V, Tsiknakis M, et al.Supporting genotype-to-phenotype association studies with grid-enabled knowledge discovery workflows.Conf Proc IEEE Eng Med Biol Soc 2009;2009:6958–62. [DOI] [PubMed] [Google Scholar]
- 13. Holl S, Mohammed Y, Zimmermann O, et al.Scientific workflow optimization for improved peptide and protein identification. BMC Bioinformatics 2015;16(1):284.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Booth D. The RDF pipeline framework automating distributed, dependency-driven data pipelines, In: International Conference on Data Integration in the Life Sciences (DILS 2013). Springer, 2013, 54–68.
- 15. Yoo J, Ha IC, Chang GT, et al.Cnvas copy number variation analysis system—the analysis tool for genomic alteration with a powerful visualization module. BioChip J 2011;5(3):265–70. [Google Scholar]
- 16. Scholz MB, Lo CC, Chain PS.. Next generation sequencing and bioinformatic bottlenecks the current state of metagenomic data analysis. Curr Opin Biotechnol 2012;23(1):9–15. [DOI] [PubMed] [Google Scholar]
- 17. Ocaña KA, de Oliveira D, Dias, et al. Discovering drug targets for neglected diseases using a pharmacophylogenomic cloud workflow. In: Proceedings of the International Conference on E-Science (e-Science). IEEE, 2012, 1–8.
- 18. Baumeister A, Pow J, Henderson K, et al.On the exploitation of serendipity in drug discovery. Clin Exp Pharmacol 2013;3:e121. [Google Scholar]
- 19. Shon J, Ohkawa H, Hammer J.. Scientific workflows as productivity tools for drug discovery. Curr Opin Drug Discov Devel 2008;11(3):381–8. [PubMed] [Google Scholar]
- 20. Kennedy JP, Williams L, Bridges TM, et al.Application of combinatorial chemistry science on modern drug discovery. J Comb Chem 2008;10(3):345–54. [DOI] [PubMed] [Google Scholar]
- 21. Harnie D, Saey M, Vapirev AE, et al.Scaling machine learning for target prediction in drug discovery using apache-spark. Future Gener Comput Syst, 2016. [Google Scholar]
- 22. Arvidsson S. Automating model building in ligand-based predictive drug discovery using Spark framework, 2015.
- 23. Wiewiórka MS, Messina A, Pacholewska A, et al.SparkSeq fast, scalable, cloud-ready tool for the interactive genomic data analysis with nucleotide precision. Bioinformatics 2014;30(18):2652–3. [DOI] [PubMed] [Google Scholar]
- 24. Hassan M, Brown RD, Varma-OBrien S, et al.Cheminformatics analysis and learning in a data pipelining environment. Mol Divers 2006;10(3):283–99. [DOI] [PubMed] [Google Scholar]
- 25. Greiner U, Mueller R, Rahm E, et al.AdaptFlow protocol-based medical treatment using adaptive workflows. Methods Inf Med 2005;44(1):80–8. [PubMed] [Google Scholar]
- 26. MacKenzie-Graham AJ, Payan A, Dinov ID, et al. Neuroimaging data provenance using the LONI pipeline workflow environment. In: Provenance and Annotation of Data and Processes, Second International Provenance and Annotation Workshop, IPAW 2008. Springer, 2008, 208–20.
- 27. Dinov I, Van Horn J, Lozev K, et al.Efficient, distributed and interactive neuroimaging data analysis using the LONI pipeline. Front Neuroinform 2009;3:22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tiwari A, Sekhar AK.. Workflow based framework for life science informatics. Comput Biol Chem 2007;31(5):305–19. [DOI] [PubMed] [Google Scholar]
- 29. Siepel AC, Tolopko AN, Farmer AD, et al.An integration platform for heterogeneous bioinformatics software components. IBM Syst J 2001;40(2):570–91. [Google Scholar]
- 30. Vahi K, Rynge M, Juve G, et al. Rethinking data management for big data scientific workflows. In: IEEE International Conference on Big Data, 2013. IEEE, 2013, 27–35.
- 31. Aloisioa G, Fiorea S, Foster I, et al. Scientific big data analytics challenges at large scale. In: Proceedings of Big Data and Extreme-scale Computing (BDEC), 2013.
- 32. Cheung KH, Prudhommeaux E, Wang Y, et al.Semantic web for health care and life sciences a review of the state of the art. Brief Bioinform 2009;10(2):111–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spjuth O, Bongcam-Rudloff E, Hernández GC, et al.Experiences with workflows for automating data-intensive bioinformatics. Biol Direct 2015;10(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ludäscher B, Altintas I, Berkley C, et al.Scientific workflow management and the Kepler system. Concurr Comput 2006;18(10):1039–65. [Google Scholar]
- 35. Garijo D, Gil Y. Towards open publication of reusable scientific workflows abstractions, standards and linked data, internal project report, 2012.
- 36. Zhao Y, Hategan M, Clifford B, et al. Swift fast, reliable, loosely coupled parallel computation. In: IEEE International Workshop on Scientific Workflows, 2007. IEEE, 2007, 199–206.
- 37. Chua CL, Tang F, Lim YP, et al.Implementing a bioinformatics workflow in a parallel and distributed environment In: Parallel and Distributed Computing Applications and Technologies. Springer, 2004, 1–4. [Google Scholar]
- 38. Von Laszewski G, Hategan M, Kodeboyina D.. Workflows for e-Science Scientific Workflows for Grids. Springer, 2007. [Google Scholar]
- 39. Yu J, Buyya R.. A taxonomy of scientific workflow systems for grid computing. ACM SIGMOD Rec 2005;34(3):44–9. [Google Scholar]
- 40. Lathers A, Su MH, Kulungowski A, et al. Enabling parallel scientific applications with workflow tools. In: Proceedings of Challenges of Large Applications in Distributed Environments (CLADE) IEEE, 2006, 55–60.
- 41. Bux M, Leser U. Parallelization in scientific workflow management systems, preprint arXiv1303.7195, 2013.
- 42. Ostrowski K, Birman K, Dolev D.. An extensible architecture for high-performance, scalable, reliable publish-subscribe eventing and notification. Int J Web Serv Res 2007;4:18. [Google Scholar]
- 43. Wu Q, Zhu M, Lu X, et al. Automation, and management of scientific workflows in distributed network environments. In: IEEE International Symposium on Parallel & Distributed Processing, Workshops and PhD Forum (IPDPSW), 2010. IEEE, 2010, 1–8.
- 44. Zhao Y, Li Y, Tian W, et al. Scientific-workflow-management-as-a-service in the cloud. In: Proceedings of the International Conference on Cloud and Green Computing (CGC), 2012. IEEE, 2012, 97–104.
- 45. Zhao Y, Li Y, Raicu I, et al.A service framework for scientific workflow management in the cloud. IEEE Trans Serv Comput 2015;8(6):930–44. [Google Scholar]
- 46. Zhao Y, Li Y, Lu S, et al. Devising a cloud scientific workflow platform for big data. In: Proceedings of the 2014 IEEE World Congress on Services IEEE, 2014, 393–401.
- 47. Luo R, Yang P, Lu S, et al. Analysis of scientific workflow provenance access control policies. In: Proceedings of IEEE International Conference on Services Computing, SCC 2012. IEEE, 2012, 266–73.
- 48. Buneman P, Chapman A, Cheney J. Provenance management in curated databases. In: Proceedings of the 2006 ACM SIGMOD International Conference on Management of Data. ACM, 2006, 539–50.
- 49. Davidson SB, Freire J. Provenance and scientific workflows challenges and opportunities. In: Proceedings of the 2008 ACM SIGMOD International Conference on Management of Data. ACM, 2008, 1345–50.
- 50. Ames DP, Quinn NW, Rizzoli AE. Intelligent Workflow Systems and Provenance-Aware Software.
- 51. Buneman P, Khanna S, Wang-Chiew T. Why and where: a characterization of data provenance. In: Proceedings of International Conference on Database Theory. Springer, 2001, 316–30.
- 52. Engaña Aranguren M, Wilkinson M.. Enhanced reproducibility of SADI web service workflows with Galaxy and Docker. Gigascience 2015;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Juve G, Deelman E, Vahi K, et al. Scientific workflow applications on Amazon EC2. In: Proceedings of the IEEE International Conference on E-Science Workshops, 2009. IEEE, 2009, 59–66.
- 54. Zhao Z, Paschke A. A survey on semantic scientific workflow, Semantic Web J. IOS Press, 2012, 1–5.
- 55. Samwald M, Giménez JAM, Boyce RD, et al.Pharmacogenomic knowledge representation, reasoning and genome-based clinical decision support based on OWL 2 DL ontologies. BMC Med Inform Decis Mak 2015;15(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rehman MA, Jablonski S, Volz B, An ontology-based approach to automating data integration in scientific workflows. In: Proceedings of International Conference on Frontiers of Information Technology. ACM, 2009, 44.
- 57. Gil Y, Kim J, Ratnakar V, et al. Wings for Pegasus: a semantic approach to creating very large scientific workflows. In: Proceedings of the OWLED*06 Workshop on OWL: Experiences and Directions, Athens, Georgia, USA. 2006.
- 58. Gil Y, Ratnakar V, Deelman E, et al. Wings for Pegasus creating large-scale scientific applications using semantic representations of computational workflows. In: Conference on Innovative Applications of Artificial Intelligence (IAAI-07). AAAI Press; MIT Press, Menlo Park, CA; Cambridge, MA; London, 2007, 1767–74.
- 59. Bonatti PA, Hogan A, Polleres A, et al.Robust and scalable linked data reasoning incorporating provenance and trust annotations. J Web Semantics 2011;9(2):165–201. [Google Scholar]
- 60. Lin C, Lu S, Lai Z, et al. Service-oriented architecture for VIEW a visual scientific workflow management system. In: IEEE International Conference on Services Computing, 2008. IEEE, 2008, 335–42.
- 61. Gil Y, Szekely P, Villamizar S, et al. Mind your metadata exploiting semantics for configuration, adaptation, and provenance in scientific workflows. In: Proceedings of International Semantic Web Conference (ISWC). Springer, 2011, 65–80.
- 62. Hasnain A, Dunne N, Rebholz-Schuhmann D. Processing Life Science Data at Scale using Semantic Web Technologies.
- 63. Jain E, Bairoch A, Duvaud S, et al.Infrastructure for the life sciences design and implementation of the UniProt website. BMC Bioinformatics 2009;10(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kosuge T, Mashima J, Kodama Y, et al.DDBJ progress reports a new submission system for leading to a correct annotation. Nucleic Acids Res 2014;42:D44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Maloney C. RESTful API to NCBIs Entrez Utilities (E-utilities), in Editor Book RESTful API to NCBIs Entrez Utilities (E-utilities) (edn.), pp.
- 66. Aranguren ME, González AR, Wilkinson MD.. Executing SADI services in Galaxy. J Biomed Semantics 2014;5(1):42.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wilkinson MD, Vandervalk B, McCarthy L.. The Semantic Automated Discovery and Integration (SADI) web service design-pattern, API and reference implementation. J Biomed Semantics 2011;2(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schneider M, Lane L, Boutet E, et al.The UniProtKB/Swiss-Prot knowledgebase and its plant proteome annotation program. J Proteomics 2009;72(3):567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jupp S, Malone J, Bolleman J, et al.The EBI RDF platform linked open data for the life sciences. Bioinformatics 2014;30(9):1338–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miyazaki S, Sugawara H, Gojobori T, et al.DNA data bank of Japan (DDBJ) in XML. Nucleic Acids Res 2003;31(1):13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Belleau F, Nolin MA, Tourigny N, et al.Bio2RDF towards a mashup to build bioinformatics knowledge systems. J Biomed Inform 2008;41(5):706–16. [DOI] [PubMed] [Google Scholar]
- 72. Nolin MA, Ansell P, Belleau F, et al. Bio2RDF network of linked data.Citeseer, 2008.
- 73. Sherry ST, Ward M, Sirotkin K.. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 1999;9(8):677–9. [PubMed] [Google Scholar]
- 74. Smigielski EM, Sirotkin K, Ward M, et al.dbSNP a database of single nucleotide polymorphisms. Nucleic Acids Res 2000;28(1):352–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hamosh A, Scott AF, Amberger JS, et al.Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33(Suppl 1):D514–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hamosh A, Scott AF, Amberger J, et al.Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2002;30(1):52–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kanehisa M. The KEGG database In: ‘In Silico’ Simulation of Biological Processes 247. 2002, 91–103. [Google Scholar]
- 78. Posma JM, Robinette SL, Holmes E, Nicholson JK.. MetaboNetworks, an interactive Matlab-based toolbox for creating, customizing and exploring sub-networks from KEGG. Bioinformatics 2014;30(6):893–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Joshi-Tope G, Gillespie M, Vastrik I, et al.Reactome a knowledgebase of biological pathways. Nucleic Acids Res 2005;33(Suppl 1):D428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schmidt E, Birney E, Croft D, et al. Reactome–a knowledgebase of biological pathways. In: OTM Confederated International Conferences On the Move to Meaningful Internet Systems. Springer, 2006, 710–19.
- 81. Schaefer CF, Anthony K, Krupa S, et al.PID the pathway interaction database. Nucleic Acids Res 2009;37(Suppl 1):D674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. NCBI RC. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2013;41:D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Santana-Perez I, Pérez-Hernández MS.. Towards reproducibility in scientific workflows: an infrastructure-based approach. Sci Program 2015;2015:243180. [Google Scholar]
- 84. Yu J, Buyya R.. Scheduling scientific workflow applications with a deadline and budget constraints using genetic algorithms. Sci Program 2006;14(3–4):217–30. [Google Scholar]
- 85. Chebotko A, Chang S, Lu S, et al. Scientific workflow provenance querying with security views. In: International Conference on Web-Age Information Management, 2008. IEEE, 2008, 349–56.
- 86. Deelman E, Gannon D, Shields M, et al.Workflows and e-science: an overview of workflow system features and capabilities. Future Gener Comput Syst 2009;25(5):528–40. [Google Scholar]
- 87. Ovaska K, Laakso M, Haapa-Paananen S, et al.Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med 2010;2(9):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kanterakis A, Potamias G, Zacharioudakis G, et al.Scientific discovery workflows in bioinformatics: a scenario for the coupling of molecular regulatory pathways and gene expression profiles. Stud Health Technol Inform 2009;160(Pt 2):1304–8. [PubMed] [Google Scholar]
- 89. Oinn T, Addis M, Ferris J, et al.Taverna a tool for the composition and enactment of bioinformatics workflows. Bioinformatics 2004;20(17):3045–54. [DOI] [PubMed] [Google Scholar]
- 90. Magis AT, Funk CC, Price ND.. SNAPR a bioinformatics pipeline for efficient and accurate RNA-Seq alignment and analysis. IEEE Life Sci Lett 2015;1(2):22–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dinov ID, Torri F, Macciardi F, et al.Applications of the pipeline environment for visual informatics and genomics computations. BMC Bioinformatics 2011;12(1):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Goecks J, Nekrutenko A, Taylor J; Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010;11(8):R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Curcin V, Ghanem M. Scientific workflow systems can one size fit all? In: Cairo International Biomedical Engineering Conference. IEEE, 2008, 1–9.
- 94. Abouelhoda M, Issa SA, Ghanem M.. Tavaxy integrating Taverna and Galaxy workflows with cloud computing support. BMC Bioinformatics 2012;13(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ieong PU, Sørensen J, Vemu PL, et al.Progress towards automated Kepler scientific workflows for computer-aided drug discovery and molecular simulations. Procedia Comput Sci 2014;29:1745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Goble CA, Bhagat J, Aleksejevs S, et al.myExperiment a repository and social network for the sharing of bioinformatics workflows. Nucleic Acids Res 2010;38(Suppl 2):W677–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kell DB. Systems biology, metabolic modeling and metabolomics in drug discovery and development. Drug Discov Today 2006;11(23):1085–92. [DOI] [PubMed] [Google Scholar]
- 98. Mazanetz MJ, Marmon RBT, Reisser C, et al.Drug discovery applications for KNIME an open source data mining platform. Curr Top Med Chem 2012;12(18):1965–79. [DOI] [PubMed] [Google Scholar]
- 99. Chichester C, Digles D, Siebes R, et al.Drug discovery FAQs workflows for answering multidomain drug discovery questions. Drug Discov Today 2015;20(4):399–405. [DOI] [PubMed] [Google Scholar]
- 100. Achilleos KG, Kannas CC, Nicolaou CA, et al. Open source workflow systems in life sciences informatics. In: IEEE International Conference on Bioinformatics and Bioengineering (BIBE), 2012. IEEE, 2012, 552–8.
- 101. Yeh SH, Yeh HY, Soo VW.. A network flow approach to predict drug targets from microarray data, disease genes and interactome network case study on prostate cancer. J Clin Bioinforma 2012;2(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhao G, Ling C, Sun D. Sparksw scalable distributed computing system for large- scale biological sequence alignment. In: IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing. IEEE, 2015, 845–52.
- 103. Aoki-Kinoshita KF, Kinjo AR, Morita M, et al.Implementation of linked data in the life sciences at BioHackathon 2011. J Biomed Semantics 2015;6(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Brooks C, Lee EA, Liu X, et al. Ptolemy II-heterogeneous concurrent modeling & design in Java, 2005.
- 105. Juve G, Deelman E.. Scientific workflows in the cloud In: Grids, Clouds, and Virtualization. Springer, 2011, 71–91. [Google Scholar]
- 106. Wolstencroft K, Haines R, Fellows D, et al.The Taverna workflow suite designing and executing workflows of web services on the desktop, web or in the cloud. Nucleic Acids Res 2013;41:W557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jagla B, Wiswedel B, Coppée JY.. Extending KNIME for next-generation sequencing data analysis. Bioinformatics 2011;27(20):2907–9. [DOI] [PubMed] [Google Scholar]
- 108. Fursov M, Oshchepkov D, Novikova O. UGENE interactive computational schemes for genome analysis. In: Proceedings of the Moscow International Congress on Biotechnology, 2009, 14–15.
- 109. Cingolani P, Sladek R, Blanchette M.. BigDataScript a scripting language for data pipelines. Bioinformatics 2015;31(1):10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Altintas I. Distributed workflow-driven analysis of large-scale biological data using bio Kepler. In: Proceedings of International Workshop on Petascal Data Analytics: Challenges and Opportunities. ACM, 2011, 41–2.
- 111. Oinn T, Greenwood M, Addis M, et al.Taverna lessons in creating a workflow environment for the life sciences. Concurr Comput 2006;18(10):1067–100. [Google Scholar]
- 112. Deelman E, Singh G, Su MH, et al.Pegasus: a framework for mapping complex scientific workflows onto distributed systems. Sci Program 2005;13(3):219–37. [Google Scholar]
- 113. Talia D. Workflow systems for science concepts and tools. ISRN Softw Eng 2013;2013:404525. [Google Scholar]
- 114. MacKenzie-Graham A, Payan A, Dinov I, et al. Neuroimaging data provenance using the LONI pipeline workflow environment. In: Provenance and Annotation of Data International Provenance and Annotation Workshop, IPAW 2008. University of Utah, Salt Lake City, UT, 2008.
- 115. Altintas I, Berkley C, Jaeger E, et al. Kepler: an extensible system for design and execution of scientific workflows. In: Proceedings of the International Conference on Scientific and Statistical Database Management. IEEE, 2004, 423–4.
- 116. Sontag E, Singh A.. Exact moment dynamics for feedforward nonlinear chemical reaction networks. IEEE Life Sci Lett 2015;1(2):26–9. [Google Scholar]