

Abstract
We describe two unrelated patients, a 12-yr-old female and a 6-yr-old male, with congenital contractures and severe congenital muscular atrophy. Exome and genome sequencing of the probands and their unaffected parents revealed that they have the same de novo deletion in BICD2 (c.1636_1638delAAT). The variant, which has never been reported, results in an in-frame 3-bp deletion and is predicted to cause loss of an evolutionarily conserved asparagine residue at position 546 in the protein. Missense mutations in BICD2 cause autosomal dominant spinal muscular atrophy, lower-extremity predominant 2 (SMALED2), a disease characterized by muscle weakness and arthrogryposis of early onset and slow progression. The p.Asn546del clusters with four pathogenic missense variants in a region that likely binds molecular motor KIF5A. Protein modeling suggests that removing the highly conserved asparagine residue alters BICD2 protein structure. Our findings support a broader phenotypic spectrum of BICD2 mutations that may include severe manifestations such as cerebral atrophy, seizures, dysmorphic facial features, and profound muscular atrophy.
Keywords: absent speech, arthrogryposis multiplex congenita, decreased muscle mass, flexion contracture, fractures of the long bones, frontal bossing, generalized cerebral atrophy/hypoplasia, generalized muscle weakness, infantile spasms, prominent ear helix, relative macrocephaly, severe muscular hypotonia, skeletal myopathy, thick eyebrow
CASE PRESENTATION
Here we report two unrelated patients with muscular atrophy and arthrogryposis who ultimately were found to have the same molecular diagnosis. Their clinical features are compared in Table 1; additional clinical data are included in the supplement.
Table 1.
Clinical features
| HPO term | Patient 1—Female, 12 yr | Patient 2—Male, 6 yr |
|---|---|---|
| Decreased fetal movement | + | + |
| Polyhydramnios | unk | + |
| Arthrogryposis multiplex congenita | + | + |
| Skeletal muscle atrophy | + | + |
| Muscle weakness | + | + |
| Recurrent fractures | + | + |
| Thoracolumbar kyphoscoliosis | + | − |
| Feeding difficulties | + | + |
| Macrocephaly | + | + |
| Frontal bossing | + | − |
| Abnormality of the ear | + | − |
| Open mouth | + | + |
| Downturned corners of mouth | + | + |
| Tapered fingers | + | + |
| EEG abnormality | + | + |
| Seizures | + | + |
| Cerebral cortical atrophy | + | + |
| Weak cry | unk | + |
| Absent speech | + | + |
| Left ventricular hypertrophy | + | − |
| Mitral regurgitation | + | − |
Human Phenotype Ontology (HPO) terms are listed with an indication of whether each patient was positive (+), negative (−), or unknown (unk) for each feature.
Patient 1 is a 12-yr-old girl with history of decreased fetal movement, bilateral femur fractures, and contractures of the ankles, digits, and wrists from birth. She required gastrostomy feeding and a tracheostomy, which was removed at age 8 yr. She had severe kyphoscoliosis and underwent multiple orthopedic procedures and spinal fusion surgery. On examination, she had relative macrocephaly with frontal bossing, thick eyebrows, prominent ears with hypoplastic outer helices and prominent anthelix, and mild midface hypoplasia with upturned nose and pointed nasal tip. Her extremities were thin, with muscle wasting and discoloration. She was nonverbal and nonambulatory with a history of “rolandic” seizures in the past. Brain MRI showed volume loss with thinning of the cortex.
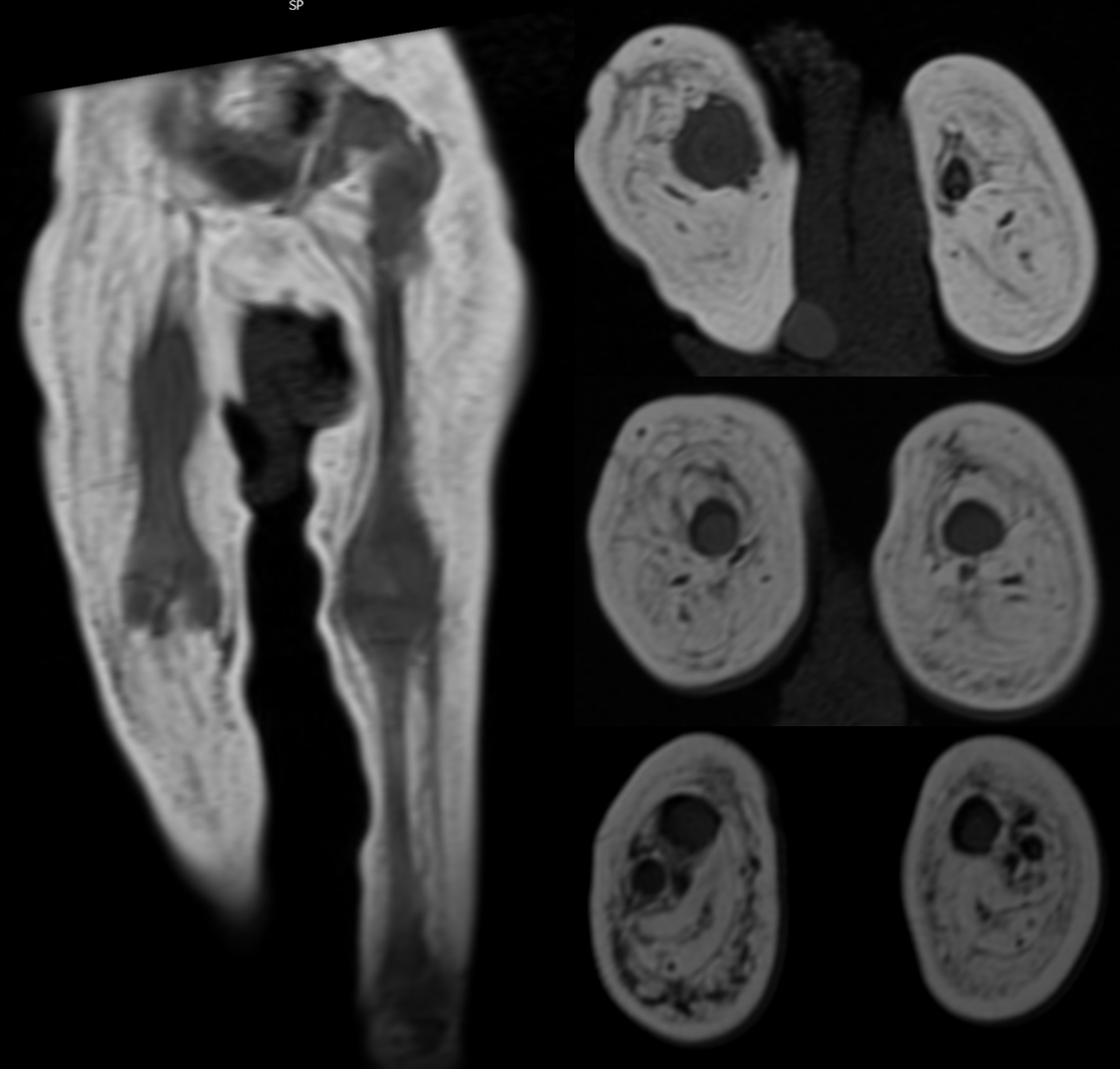
Patient 2 is a 6-yr-old boy born at 38 wk gestation after a pregnancy noted to have severe polyhydramnios, fetal macrocephaly, and marked decreased fetal movement, particularly of the arms and legs. At birth he had dysmorphic features, bilateral symmetric facial weakness, micrognathia, weak cry, absent gag, and markedly decreased muscle bulk/tone with severe contractures (elbows, wrists, fingers, ankles in flexion, knees extended, hips internally rotated). Bone survey on day of life 1 showed right femur fracture. Neonatal brain MRI was consistent with very severe in utero hypoxic ischemic injury. Whole-body muscle MRI including the pelvis and thighs revealed marked absence of the musculature with fatty replacement (Supplemental Fig. 1). He developed intractable epilepsy and hydrocephalus requiring shunt placement at 6 mo of age. Follow-up brain MRI at 3.5 yr showed near complete absence of cerebral white matter and cortex with subsequent enlargement of the ventricular system.
VARIANT INTERPRETATION
Patient 1
Whole-exome sequencing (WES) revealed a de novo, previously unreported, heterozygous deletion in BICD2 (Table 2). The c.1636_1638delAAT variant results in an in-frame 3-bp deletion and is predicted to cause loss of an evolutionarily conserved Asparagine residue at position 546 in the protein, denoted as p.Asn546del. The c.1636_1638delAAT variant was not observed in large population cohorts (Lek et al. 2016). GeneDx interpreted the c.1636_1638delAAT variant as a likely pathogenic variant and submitted it to ClinVar in October 2016 (SCV000571877.3). The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/).
Table 2.
Genomic findings and variant interpretation
| Genomic location | HGVS cDNA | HGVS protein | Zygosity | Origin | Interpretation |
|---|---|---|---|---|---|
| 9: 95481289 ATT/− | NM_001003800: c.1636_1638delAAT | BICD2: p.Asn546Del | Het | De novo | Likely pathogenic |
Both Patient 1 and Patient 2 were found to have the same de novo mutation in BICD2. Genomic coordinates reflect build GRCh37.
Patient 2
Whole-genome sequencing (WGS) revealed a heterozygous deletion in BICD2 (Table 2), which we interpreted according to ACMG guidelines (Richards et al. 2015). Sanger sequencing of the patient and both parents confirmed its de novo status. The c.1636_1638delAAT variant has not observed in the gnomAD populations, and is predicted to cause a deletion of a highly conserved asparagine residue in a nonrepeat region. The ClinVar report from GeneDx of the same de novo mutation in a patient with similar features provided further supporting evidence. Nationwide Children's interpreted the variant as pathogenic and submitted it to ClinVar in April 2018 (SCV000715101.1).
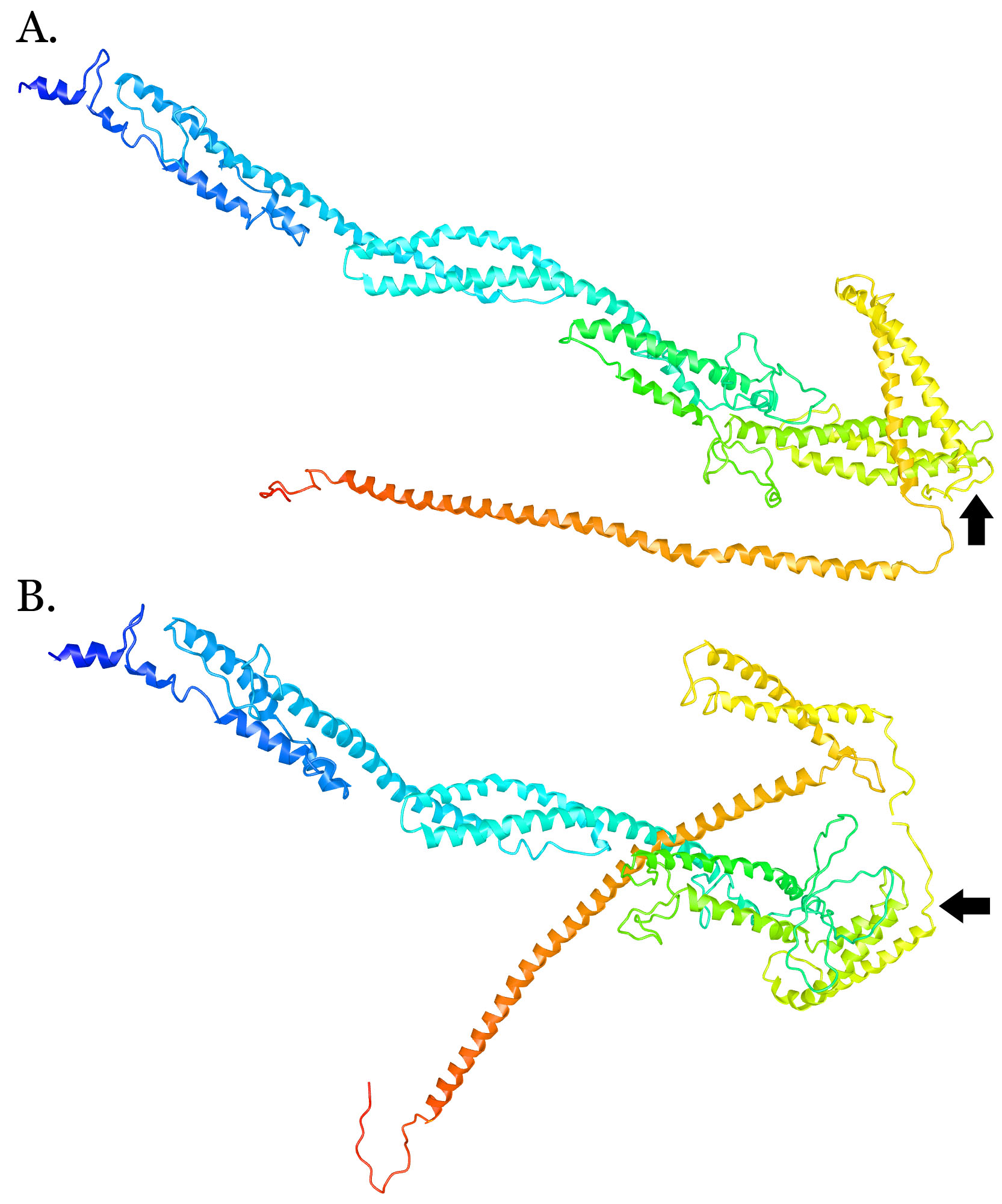
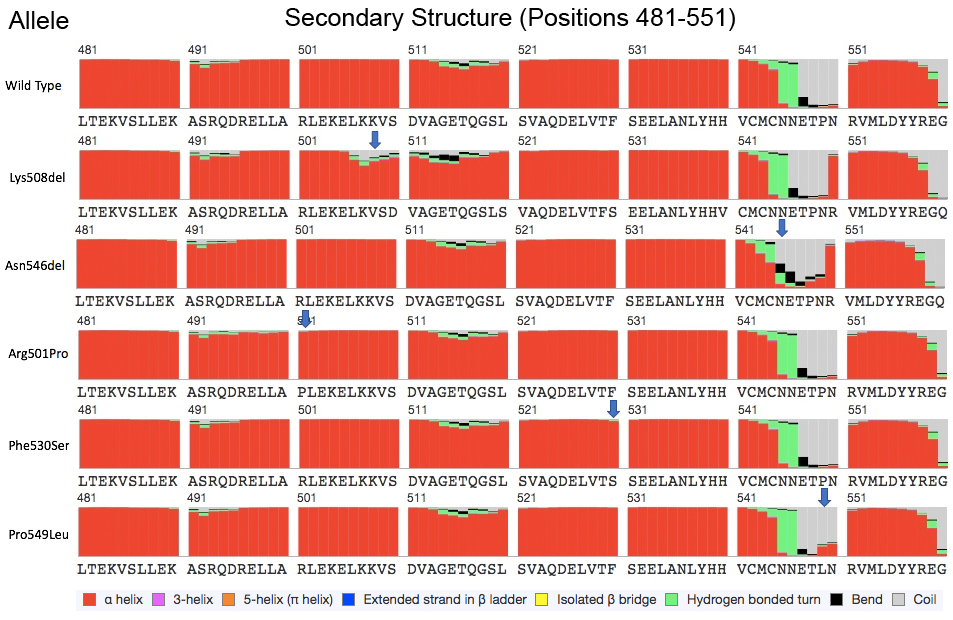
The p.Asn546Del variant maps just outside one of the coiled-coil domains that harbor several reported BICD2 mutations (Fig. 1A), but within a region that (in mice) interacts with molecular motor kinesin-1 (Splinter et al. 2010). A nearby variant, p.Arg501Pro, appears to increase the binding affinity of the BICD2 protein for the dynein–dynamin complex (Oates et al. 2013). Protein modeling suggests that the p.Asn546del variant alters protein structure (Supplemental Fig. 2), and that the changes to secondary structure are more striking for the p.Asn546del than for nearby missense mutations (Supplemental Fig. 3).
Figure 1.

Location and conservation of the p.Asn546del variant. (A) Pathogenic and likely pathogenic variants reported in the ClinVar database as of July 3, 2018. Blue dots represent missense variants. The p.Asn542del variant is shown in red. BICD2 protein structure, coiled-coil domains (gold), and KIF5 interaction region (red bar) were taken from UniProtKB entry Q8TD16. (B) UCSC Genome Browser screenshot for the region harboring the p.Asn542del variant.
SUMMARY
The BicD gene was discovered in Drosophila and named bicaudal D (“having two tails”), because mutant embryos showed abnormal body patterning in which the head, thorax, and anterior abdominal segments are replaced with a mirror image of the posterior body segments and tail (Bullock and Ish-Horowicz 2001). The two mammalian homologs of BicD (BICD1 and BICD2) encode motor adaptor proteins that interact with the dynein–dynactin complex and facilitate transport of mRNAs as well as other cellular cargoes (Vazquez-Pianzola et al. 2017).
Heterozygous missense changes in BICD2 cause autosomal dominant spinal muscular atrophy with lower extremity predominance (SMALED2, MIM #615290) (Neveling et al. 2013; Oates et al. 2013; Peeters et al. 2013), a disease characterized by early-childhood or congenital onset of muscle weakness and atrophy. To date, 11 pathogenic missense variants in BICD2 have been reported to the ClinVar database (Fig. 1A). The c.1636_1638delAAT variant is the only reported in-frame deletion in ClinVar; however, during preparation of this manuscript, Trimouille et al. (2018) published an in-frame deletion segregating in a family with variable disease severity.
There is a growing appreciation of the wide phenotypic spectrum of BICD2 mutations, which can range from asymptomatic to lethal congenital manifestations (Ravenscroft et al. 2016; Storbeck et al. 2017). A study of a four-generation Australian kindred with dominant spinal muscular atrophy (Oates et al. 2012) found that the clinical presentation of this disease can vary widely even within a single family. The manifestations in our patients are at the severe end of the spectrum, including lack of ambulation, dysmorphic craniofacial features, seizures, and cerebral atrophy. Patient 2 in particular had a striking lack of skeletal muscle, although there was also evidence of ischemic brain injury by MRI that could be a contributing factor. Even so, the substantial clinical overlap between our two patients, and the fact that they share the same de novo mutation, support the p.Asn546del mutation as a likely molecular basis for disease.
METHODS
Patient 1
Using genomic DNA from the proband and parents, the exonic regions and flanking splice junctions of the genome were captured using the Clinical Research Exome kit (Agilent Technologies) by genetic testing provider GeneDx. Massively parallel (NextGen) sequencing was performed using an Illumina system with 100-bp paired-end reads. Additional sequencing technology and variant interpretation protocols have been previously described (Tanaka et al. 2015).
Patient 2
The proband and his parents underwent WGS (2 × 100 bp) performed using the Illumina HiSeq 2500 instrument. Reads were mapped to the GRCh37 reference sequence and secondary data analysis was performed using Churchill (Kelly et al. 2015). Our approach to variant annotation and prioritization has already been described (Koboldt et al. 2018).
More details on the sequencing and analysis can be found in the Supplemental Analysis and Methods section. Sequencing metrics are provided in Supplemental Table 1. The mutation diagram in Figure 1A was generated with Lollipops v1.3.2 (Jay and Brouwer 2016) using information from UniProt (entry #Q8TD16) and the ClinVar (Landrum et al. 2018) database (accessed March 22, 2018). Protein modeling was performed using the Phyre2 (Kelley et al. 2015) and RaptorX (Källberg et al. 2012) web servers.
ADDITIONAL INFORMATION
Data Deposition and Access
The variants and their interpretations have been submitted to the ClinVar database (ID #422408) (https://www.ncbi.nlm.nih.gov/clinvar/). Submitted reports are available under accession numbers SCV000571877.3 (Patient 1) and SCV000715101.1 (Patient 2). Raw sequencing data was not deposited because of lack of patient consent.
Ethics Statement
For Patient 1, written consent for GeneDx exome sequencing was obtained as part of the patient's clinical evaluation at Mount Sinai Medical Center. For Patient 2, written consent was obtained enrolling subjects into a research protocol approved by the Institutional Review Board at Nationwide Children's Hospital (IRB11-00215 Study: Using Genome Sequencing to Identify Causes of Rare Birth Defects and Rare Disorders).
Acknowledgments
We thank the patients and their families for participation in this research.
Author Contributions
All authors contributed to scientific discussion, variant interpretation, and manuscript review.
Funding
This work was supported by The Research Institute at Nationwide Children's Hospital.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Bullock SL, Ish-Horowicz D. 2001. Conserved signals and machinery for RNA transport in Drosophila oogenesis and embryogenesis. Nature 414: 611–616. [DOI] [PubMed] [Google Scholar]
- Jay JJ, Brouwer C. 2016. Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS One 11: e0160519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7: 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BJ, Fitch JR, Hu Y, Corsmeier DJ, Zhong H, Wetzel AN, Nordquist RD, Newsom DL, White P. 2015. Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol 16: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Mihalic Mosher T, Kelly BJ, Sites E, Bartholomew D, Hickey SE, McBride K, Wilson RK, White P. 2018. A de novo nonsense mutation in ASXL3 shared by siblings with Bainbridge–Ropers syndrome. Cold Spring Harb Mol Case Stud 4: a002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. 2018. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46: D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveling K, Martinez-Carrera LA, Hölker I, Heister A, Verrips A, Hosseini-Barkooie SM, Gilissen C, Vermeer S, Pennings M, Meijer R, et al. 2013. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 92: 946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates EC, Reddel S, Rodriguez ML, Gandolfo LC, Bahlo M, Hawke SH, Lamandé SR, Clarke NF, North KN. 2012. Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells. Brain 135: 1714–1723. [DOI] [PubMed] [Google Scholar]
- Oates EC, Rossor AM, Hafezparast M, Gonzalez M, Speziani F, MacArthur DG, Lek M, Cottenie E, Scoto M, Foley AR, et al. 2013. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 92: 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters K, Litvinenko I, Asselbergh B, Almeida-Souza L, Chamova T, Geuens T, Ydens E, Zimoń M, Irobi J, De Vriendt E, et al. 2013. Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am J Hum Genet 92: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft G, Di Donato N, Hahn G, Davis MR, Craven PD, Poke G, Neas KR, Neuhann TM, Dobyns WB, Laing NG. 2016. Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria. Neuromuscul Disord 26: 744–748. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter D, Tanenbaum ME, Lindqvist A, Jaarsma D, Flotho A, Yu KL, Grigoriev I, Engelsma D, Haasdijk ED, Keijzer N, et al. 2010. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol 8: e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storbeck M, Horsberg Eriksen B, Unger A, Hölker I, Aukrust I, Martínez-Carrera LA, Linke WA, Ferbert A, Heller R, Vorgerd M, et al. 2017. Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet 25: 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, et al. 2015. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 97: 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimouille A, Obre É, Banneau G, Durr A, Stevanin G, Clot F, Pennamen P, Perez JT, Bailly-Scappaticci C, Rouanet M, et al. 2018. An in-frame deletion in BICD2 associated with a non-progressive form of SMALED. Clin Neurol Neurosurg 166: 1–3. [DOI] [PubMed] [Google Scholar]
- Vazquez-Pianzola P, Schaller B, Colombo M, Beuchle D, Neuenschwander S, Marcil A, Bruggmann R, Suter B. 2017. The mRNA transportome of the BicD/Egl transport machinery. RNA Biol 14: 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}