

Abstract
Recent evidence has implicated EFL1 in a phenotype overlapping Shwachman–Diamond syndrome (SDS), with the functional interplay between EFL1 and the previously known causative gene SBDS accounting for the similarity in clinical features. Relatively little is known about the phenotypes associated with pathogenic variants in the EFL1 gene, but the initial indication was that phenotypes may be more severe, when compared with SDS. We report a pediatric patient who presented with a metaphyseal dysplasia and was found to have biallelic variants in EFL1 on reanalysis of trio whole-exome sequencing data. The variant had not been initially reported because of the research laboratory's focus on de novo variants. Subsequent phenotyping revealed variability in her manifestations. Although her metaphyseal abnormalities were more severe than in the original reported cohort with EFL1 variants, the bone marrow abnormalities were generally mild, and there was equivocal evidence for pancreatic insufficiency. Despite the limited number of reported patients, variants in EFL1 appear to cause a broader spectrum of symptoms that overlap with those seen in SDS. Our report adds to the evidence of EFL1 being associated with an SDS-like phenotype and provides information adding to our understanding of the phenotypic variability of this disorder. Our report also highlights the value of exome data reanalysis when a diagnosis is not initially apparent.
Keywords: congenital thrombocytopenia; exocrine pancreatic insufficiency; hepatic bridging fibrosis; hypercalciuria; intellectual disability, mild; portal fibrosis; short stature; spondylometaphyseal dysplasia
INTRODUCTION
Shwachman–Diamond syndrome (SDS; OMIM 260400) is an autosomal recessive condition that is characterized by a triad of exocrine pancreatic insufficiency, bone marrow dysfunction, and metaphyseal dysostosis (Bodian et al. 1964; Shwachman et al. 1964; Burke et al. 1967). Most patients present as infants with growth failure due to pancreatic insufficiency and/or recurrent infections due to neutropenia (Dror et al. 2011), whereas the metaphyseal changes leading to short stature typically become evident in childhood or later (Levin et al. 2015). The pancreatic dysfunction varies in severity, and a subset of patients may show improvement in pancreatic function in late childhood (Ginzberg et al. 1999). Hematologic abnormalities in patients with SDS vary in type of lineage affected, as well as severity (Dror et al. 2011). Neutropenia and anemia are the most common bone marrow abnormalities, but thrombocytopenia and pancytopenia may also be seen (Dror et al. 2011). Metaphyseal chondrodysplasia seen in SDS is present in all patients but shows variability, even within the same family (Levin et al. 2015). Skeletal findings are typically symmetrical and are more severe in the lower limbs. By age 2 yr, metaphyseal changes seen include broadening of the metaphyses with characteristic lucent and sclerotic changes, which disappear when the physes fuse at puberty (Levin et al. 2015).
Approximately 90% of patients with SDS phenotypes have been found to carry pathogenic variants in the SBDS gene (Warren 2017). The SBDS protein promotes the activation of Elongation factor-like 1 (EFL1) in order for EFL1 to remove the antiassociation factor eIF6 and subsequently facilitate cytoplasmic maturation of the large (60S) ribosomal subunit (García-Márquez et al. 2015). EFL1 variants have recently been implicated in an SDS-like phenotype (OMIM 617941) (Stepensky et al. 2017). In the cohort of six patients reported with EFL1 variants, half the patients presented with severe failure to thrive presumably due to untreated pancreatic insufficiency and died prior to age 15 mo (Stepensky et al. 2017). The other three patients (ages 15 mo to 6 yr) manifested the hallmarks of SDS, including neutropenia (in isolation or with other bone marrow abnormalities), pancreatic insufficiency, and metaphyseal dysostosis (Stepensky et al. 2017). In addition to EFL1, the DNAJC21 protein also plays a role in 60S ribosome maturation (Meyer et al. 2010) and has been associated with bone marrow failure (OMIM 617052; [Tummala et al. 2016]), as well as an SDS phenotype (Dhanraj et al. 2017). Thus, it is evident that ribosomopathies can include a spectrum of abnormalities with overlapping findings in organ systems such as the bone marrow, skeleton, and pancreas. Recently, variants in SRP54, which encodes a signal recognition particle involved in the translocation of nascent polypeptides, have also been implicated in the SDS phenotype (Carapito et al. 2017).
We report a 14-yr-old patient who was referred to the Undiagnosed Diseases Network (UDN; https://undiagnosed.hms.harvard.edu/) with a radiologic diagnosis of spondylometaphyseal dysplasia, corner fracture type, who also had a history of thrombocytopenia, growth failure, liver fibrosis, scoliosis, and learning difficulties. She had extensive clinical and molecular evaluations in the previous 12 years, including trio whole-exome sequencing (WES), without a unifying diagnosis. Exome reanalysis as part of the UDN revealed a homozygous missense variant in EFL1, which likely explains her phenotypic features and adds to the evidence of EFL1 being associated with an SDS-like syndrome.
RESULTS
Clinical Presentation and Family History
The patient is a 14-yr-old Caucasian female enrolled in the UDN with a previous clinical diagnosis of spondylometaphyseal dysplasia—corner fracture type and other multiple manifestations with no unifying diagnosis. There is a paternal family history of short stature. Her father is 5′4″, paternal grandmother was 4′11″, and paternal great-grandmother was 4′6″. The family history was otherwise noncontributory; the parents are not consanguineous. The patient was born at 39 wk gestational age following a pregnancy complicated by intrauterine growth restriction. Her birthweight was 1956 grams (<5th percentile). Neonatal complications included thrombocytopenia (initial platelet count 82 × 109/l, normal 140–300 × 109/l) which improved to 108 × 109/l on day of life 4. She first presented to endocrinology at 22 mo because of failure to thrive (height and weight below the third percentile) despite appropriate nutrition. A comprehensive endocrinology workup detected transaminitis and urine organic acids profile revealed a nonspecific pattern concerning for a possible mitochondrial disorder. Workup for mitochondrial disorder, including muscle and liver biopsies, was inconclusive. However, the liver biopsy did demonstrate moderate portal, periportal bridging, and centrilobular fibrosis (Fig. 1). Transient elevated transaminases were present at age 2–4 yr with no identified cause for the liver disease. Short stature failed to respond to growth hormone therapy.
Figure 1.

Liver biopsy showing decrease in liver fibrosis with age. Liver biopsy obtained at age 2 yr stained with (A) hematoxylin and eosin (H&E) and (B) Masson trichrome stain, and a liver needle biopsy obtained at age 14 yr stained with (C) H&E and (D) Masson trichrome stain. All 100× magnification.
Thrombocytopenia was mild and intermittent between the ages of 2 and 5 yr, but became persistent at age 6 yr when her platelet counts ranged from 45 to 136 × 109/l. However, there were no clinical manifestations related to thrombocytopenia. She has never been neutropenic or anemic except for an episode of pancytopenia at age 9 yr with a fever and presumed viral illness. Bone marrow biopsy performed during this illness showed low cellularity (10%, normal for age ∼60%–70%) but no other specific abnormalities. Prior to age 2 yr, she had four episodes of pneumonia, but none required hospitalization, and she has not had recurrent infections since.
A skeletal survey at age 6 yr (Fig. 2) demonstrated mild S-shaped scoliosis, irregularity of the vertebral body endplates (Fig. 2A), metaphyseal irregularity of the femoral necks (Fig. 2B) and the left proximal humerus (Fig. 2C), irregularity of the bony acetabular roofs and iliac crests, and metaphyseal cupping involving the metacarpals of the hands (not shown), later classified as spondylometaphyseal dysplasia—corner fracture type, after review by the International Skeletal Dysplasia Registry (ISDR). She developed restrictive lung disease secondary to worsening scoliosis. The patient underwent surgery at age 13 yr to correct left genu varum.
Figure 2.

Skeletal anomalies on radiography. The three images on the top row were obtained at age 6 yr, and the three images on the lower row were obtained at age 14 yr. (A) Arrows show vertebrae with irregularity and scalloping subjacent to the disc spaces that are more pronounced in the radiograph (D) obtained at age 14 yr. (B) There is bilateral coxa vara and arrows demonstrate metaphyseal abnormalities of bilateral proximal femurs of mixed sclerosis and lucency. (E) Hip imaging at age 14 yr shows abnormal configuration (flattening) of the femoral heads, with continued metaphyseal heterogeneity in proximal and distal femurs (arrows). (C) Arrow on left proximal humerus shows heterogeneity in the ossification in the metaphysis (right humerus was normal; not shown). (F) Similar abnormal mixed sclerosis and lucency found in both humeri at age 14 yr (arrows).
The patient presented with hematuria at age 4 yr, which recurred at age 7 yr, with hypercalciuria on laboratory testing (24 h urine calcium 6.2 mg/kg/day, normal <4 mg/kg/day). She was started on hydrochlorothiazide with resolution of urine calcium levels. Renal ultrasound examinations were normal. There were no developmental concerns in early childhood. Learning disabilities were diagnosed in the second grade, for which she received an Individualized Education Plan. She also had multiple dental caries that required full oral rehabilitation.
At age 14 yr, she was referred to the UDN and reanalysis of the previous WES data completed prior to her evaluation reported a homozygous variant in EFL1. Because of this finding, the patient's UDN evaluation was subsequently tailored to look for manifestations described in the prior report of EFL1-associated SDS-like phenotype (Stepensky et al. 2017). At age 14 yr, her height and weight were below the first percentile. A repeat skeletal survey at age 14 yr demonstrated progression of the previously identified metaphyseal abnormalities (Fig. 2). Upon review by the ISDR and skeletal dysplasia experts at the Baylor School of Medicine within the UDN, the metaphyseal changes were thought to be consistent with SDS, but with greater severity than typically observed in these patients. A repeat bone marrow biopsy at age 14 yr demonstrated a hypocellular bone marrow (50%, normal for age ∼60%–70%) with trilineage hematopoiesis (Fig. 3) and no cytogenetic abnormalities. There was no history of intractable diarrhea. Evaluation for pancreatic insufficiency revealed high spot fecal fat (25%, normal <20%), low serum amylase (18 U/l, normal 31–119 U/l) with normal stool pancreatic elastase and normal fat-soluble vitamin levels. Sample collection for 72-h fecal fat was not completed because of lack of patient compliance. The pancreas was not well visualized on abdominal ultrasound. Although her pancreatic phenotype was not completely delineated, she appeared to have mild pancreatic insufficiency based on history of failure to thrive and available laboratory assessments. A repeat liver biopsy showed regenerative changes, improved fibrosis with no inflammation or steatosis (Fig. 1), and liver enzymes and liver synthetic function were normal. Cognitive testing revealed a borderline IQ of 73 (4th %) on the Wechsler Abbreviated Scale of Intelligence Second Edition and specific impairments in perceptual-motor functioning and visual memory. She remains in a regular classroom with accommodations.
Figure 3.

Bone marrow biopsy at age 14 yr. Hypocellular bone marrow (50%) at (A) 40× and (B) 200× magnification, showing trilineage hematopoiesis (H&E stain). (C) Touch preparation showing normal maturation of the myeloid and erythroid lineages (500× magnification, Wright–Giemsa stain).
Genomic Analysis
Trio WES had been performed on a research basis (Zhu et al. 2015) at age 10 yr and was negative, with no variants of interest. On reanalysis of the WES data through the UDN in 2017, a homozygous missense variant (c.379 A>G; p.Thr127Ala) in EFL1 was identified (Table 1). The variant results in a nonconservative amino acid substitution in the GTP binding domain, which is located in the amino terminus of the protein (Fig. 4). The variant is absent from internal controls (13,119 samples) as well as the Exome Aggregation Consortium (ExAC) database (60,706 samples) (Lek et al. 2016) and Exome Variant Server (EVS) database (6503 samples) (http://evs.gs.washington.edu/EVS/). One individual was heterozygous for this variant in gnomAD (1 allele out of 240,260), but it is absent in a homozygous state. The site is strongly conserved with the Genomic Evolutionary Rate Profiling (GERP++RS) score of 4.01 (Davydov et al. 2010). EFL1 is intolerant to homozygous loss of function variants with a pREC score of 0.966 (Lek et al. 2016). Multiple in silico tools, including CADD (25.9) (Kircher et al. 2014), PolyPhen (0.993) (Adzhubei et al. 2010), SIFT (0) (Kumar et al. 2009), and MutationTaster (D,1.0) (Schwarz et al. 2010), predicted that the variant was damaging. Parental testing confirmed carrier status for each parent. No pathogenic variants were identified in the SBDS gene.
Table 1.
EFL1 variant information
| Gene | Chr: position GRCh37(hg19) | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | Genotype | ClinVar accession | Inheritance |
|---|---|---|---|---|---|---|---|---|
| EFL1 | Chr15: 82532896 | NM_024580.5 c.379A>G | p.Thr127Ala | Substitution | Missense | Homozygous | SCV000746595.2 | Maternal and paternal |
Figure 4.
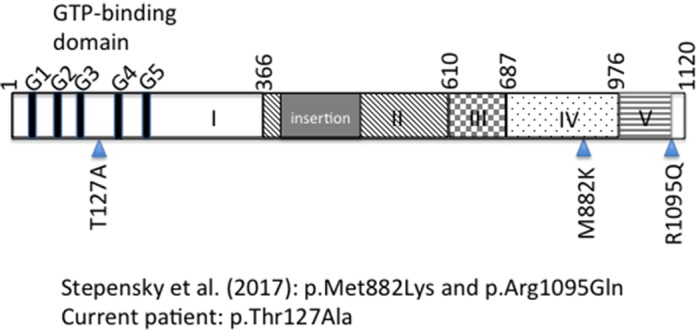
Schematic of the EFL1 protein highlighting the GTP-binding domain (domain 1) (Finch et al. 2011) in the amino terminus and the variants associated with human disease, as reported by the referenced articles. Numbers refer to amino acid position. G1–G5 refer to conserved motifs important for GTP binding (Wittinghofer and Vetter 2011) (domains labeled according to Asano et al. [2014]).
DISCUSSION
Disorders that affect ribosome production or function are referred to as ribosomopathies, of which SDS is one of the more well-known examples (Nakhoul et al. 2014). For the 10% of patients with manifestations of SDS who have no associated variants in the SBDS gene, next-generation sequencing has begun to uncover pathogenic variants in additional genes with similar functions who present with SDS-like phenotypes. Six patients from three families who presented with SDS-like features were identified by Stepensky et al. (2017) to have homozygous variants in the EFL1 gene. These patients presented with developmental delay, failure to thrive, symptoms of pancreatic insufficiency, and cytopenias of one or more blood lineages (Stepensky et al. 2017). The severity of the disorder was such that mortality was high, with three patients dying in childhood likely because of untreated pancreatic insufficiency. Metaphyseal anomalies were also observed on radiographs for all six patients (Stepensky et al. 2017).
Contrary to the initial cohort of patients (Stepensky et al. 2017) whose primary presentation was similar to SDS with growth failure and pancreatic insufficiency, our patient presented primarily with striking metaphyseal anomalies which worsened with age. Although she did have mild thrombocytopenia, the bone marrow manifestations had not been medically significant, with no documented episodes of neutropenia other than an episode of pancytopenia during a viral illness, nor has she needed transfusions. Additionally, the lack of overt pancreatic insufficiency resulted in lack of clinical recognition that she had an SDS-like phenotype, with the skeletal manifestations becoming the main diagnostic focus. Table 2 compares the features in our patient and those of Stepensky et al. (2017).
Table 2.
Clinical features associated with ELF1 variants
| Stepensky et al. case series | Duke case | |
|---|---|---|
| Stature | −2.6 to −4.8 SD | −3.7 SD |
| Head circumference | Normal in 2/6 patients (other 4 were from consanguineous families) | Normal |
| Radiologic findings | Metaphyseal irregularities in proximal and distal femurs, fibulae, and humeri; anterior rib cupping | Metaphyseal irregularities in proximal and distal femurs, tibia, fibulae, humeri; flattening of hip epiphyses; iliac crest apophyseal irregularity; vertebral body endplate irregularities; scoliosis |
| Pancreatic insufficiency | 6/6 patients (5/6 with diarrhea) | No diarrhea; equivocal pancreatic laboratory results |
| Liver disease | 1 patient with mild hepatomegaly | Transaminitis (ages 2–4 yr) (AST 64–169 U/l; ALT 73–238 U/l)a; moderate portal, periportal bridging, and centrilobular fibrosis (age 2 yr) |
| Anemia | 4/5 patients | Absent; hemoglobin 11.1–12.1 g/dlb,c |
| Neutropenia | 4/5 patients | Absent; ANC 1353–14874 cells/µlc |
| Thrombocytopenia | 4/5 patients | Present; chronic since age 6 yr (platelets 96–136 × 109/l)c,d |
| Development | 6/6 developmental delay | Learning disabilities in school |
| Outcome | 3/6 deceased (ages 7 mo–15 mo); 3/6 alive (ages 15 mo to 6 yr) | Alive (age 14 yr) |
SD, standard deviation; AST, aspartate transaminase; ALT, alanine transaminase; ANC, absolute neutrophil count.
aReference ranges for AST: 10–60 U/l; ALT: 10–45 U/l.
bReference ranges for hemoglobin: normal 11.5–13.5 g/dl ages 2–5 yr; 11.5–15.5 g/dl ages 6–11 yr; 12.0–16.0 g/dl ages 12–17 yr.
cExcluding blood counts obtained during episode of pancytopenia (see text).
dReference range for platelet counts: normal 150–400 × 109/l.
The identification of a homozygous EFL1 variant led to further investigation of pancreatic insufficiency in our patient. The failure to gain weight could be related to pancreatic insufficiency, with laboratory findings of high fecal fat and low amylase, but the otherwise normal pancreatic tests could be indicative of mild insufficiency. Additional testing, including further imaging studies, and direct measurement of pancreatic enzymes are being considered, as well as a trial of enzyme supplementation. Our patient's hypercalciuria could also represent a manifestation of the EFL1-associated SDS phenotype. Renal lithiasis has been reported in patients with SDS (Ginzberg et al. 1999). Dental caries, as well as neurodevelopmental delays, as seen in our patient, are common as well (Dror et al. 2011). Specific impairments in perceptual-motor functioning and visual memory are consistent with prior neurocognitive investigations in children with SDS (Kerr et al. 2010).
Patients with SDS have been reported to present with transaminitis, hyperbilirubinemia, hepatomegaly, and cirrhosis (Toiviainen-Salo et al. 2009). In one longitudinal study of 12 patients with SDS, the transaminitis resolved by age 5 yr, hepatomegaly by age 3 yr, and hyperbilirubinemia by age 4 yr, but elevated bile acids and hepatic microcysts were seen in some adult patients (Toiviainen-Salo et al. 2009). Our patient's mild transaminitis and fibrosis, with no sign of liver dysfunction after age 4 yr, is certainly consistent with the SDS phenotype, although none of the six patients reported previously had liver dysfunction except for one patient with mild hepatomegaly (Stepensky et al. 2017). The diagnosis in our patient has medical management implications such as periodic monitoring for hematological malignancies and possible treatment for mild pancreatic insufficiency to determine if it would help with growth.
Interestingly, all the patients in the prior report had variants associated with the carboxyl terminus of the protein, and Stepensky et al. (2017) observed that these mutant protein variants fail to release eIF6 from the 60S subunit, hence interrupting the formation of mature ribosomes. Our variant is located near the amino terminus of the protein (Fig. 4) and results in a missense, nonconservative amino acid substitution in the EFL1 protein within the GTP-binding domain (Finch et al. 2011). This variant may hence affect the GTPase function of EFL1, although we are not able to confirm the actual functional change conferred by the variant without functional assays. Perhaps the variants being in different regions of the protein result in phenotypic differences in our patient compared with the previously reported patients with EFL1 pathogenic variants (Stepensky et al. 2017), although it is worth noting that there is little genotype–phenotype correlation even for well-studied ribosomopathies such as SDS (Myers et al. 2014).
The features of our patient and the other six patients with EFL1 biallelic variants may be representative of a SDS-like disorder or may be indicative of the genetic heterogeneity of SDS, which until recently has been known to be associated with only one gene. Further literature reports will help clarify if these are distinct ribosomopathies with overlapping features or could be classified as one entity.
Using American College of Medical Genetics and Genomics (ACMG) variant classification guidelines (Richards et al. 2015), the EFL1 variant is denoted as a variant of uncertain significance. Using the ClinGen Gene-Disease Association curation framework (Strande et al. 2017) with protocol updates published on the ClinGen website (www.clinicalgenome.org), the evidence supporting an association between EFL1 and SDS-like syndrome is currently considered “limited.” However, considering the functional interaction of SBDS with EFL1, as well as our patient's overlap with features of SDS and SDS-like phenotypes in unrelated individuals with EFL1 variants (Stepensky et al. 2017), we strongly favor that the homozygous EFL1 variant is a likely diagnosis for our patient. The combination of uncommon clinical features in our patient—that is, both spondylometaphyseal dysplasia (population prevalence estimated to be at 1/100,000; www.orpha.net) and childhood exocrine pancreatic insufficiency (cystic fibrosis tested and excluded; not shown)—also argues for a diagnosis of an SDS-like syndrome. In addition, we note that the ACMG variant classification guidelines are specifically developed to assess pathogenicity in well-characterized disease genes or variants that occur de novo in a patient; both of which are not applicable in this case. We find the ClinGen curation framework useful in assessing gene-disease relationships, and we believe that evidence for EFL1-SDS syndrome association will be stronger with the inclusion of more cases and functional data.
A notable finding with our patient is that the EFL1 variant was not initially prioritized from the exome data obtained in 2013. The EFL1 gene was not associated with human disease at the time, but EFL1 was known to interact physically with SBDS and function epistatically in yeast (Menne et al. 2007) and to release eIF6 in mammalian cells (Finch et al. 2011) in pre-60S ribosomal processing. The research laboratory that performed and analyzed the sequence data utilizes an agnostic approach to the genome, but despite the strong bioinformatics signatures, this variant was not reported. Retrospectively, it is difficult to determine the exact reason for this, but it could be related to the focus of the laboratory at that time on de novo variants. It is certainly very plausible that this variant would also not have been reported by clinical laboratories, given the lack of association with human disease until very recently. Indeed, many of the cases that get resolved with WES reanalyses are due to interim new gene-disease associations (Wenger et al. 2017). Thus, this experience adds credence to the value of WES reanalysis when the initial report is negative.
In conclusion, our report provides further clinical and genomic evidence for EFL1 being associated with an SDS-like phenotype, highlighting the phenotypic variability that can occur. It also illustrates the current conundrum within genomic medicine of bioinformatically compelling variants not being prioritized in WES data, emphasizing the value of WES reanalysis when a diagnosis is not evident initially.
METHODS
Whole-Exome Sequencing
The sequence data were reanalyzed using our established trio sequencing protocols (Zhu et al. 2015) that identifies qualifying variants forming novel genotypes not observed in the parents or in a set of 13,119 controls or external databases provided by the Exome Sequencing Project (ESP6500SI) (NHLBI GO Exome Sequencing Project; evs.gs.washington.edu), and the Exome Aggregation Consortium (Lek et al. 2016). Samples sequenced at the Institute for Genomic Medicine (IGM) (previously the Center for Human Genome Variation at Duke University) were collected as peripheral blood. Exome enrichment was done using the SeqCap EZ Exome v3 (Roche Nimblegen, Madison, WI). Sequencing was performed in the Genomic Analysis Facility on an Illumina HiSeq 2000. Please see Supplemental Table S1 for sequencing coverage.
Our standard bioinformatics pipeline was used to analyze data. Briefly, the pipeline uses GATK best practices, utilizing BWA 0.5.1023, picard tools 1.59, and the Unified Genotyper from GATK 1.6-1124–26. Reads are aligned to the hg19 reference sequence utilized in 1000 genomes phase 2, which includes EBV derived decoy sequences. Variant calls are functionally annotated with SnpEff 3.327 using Ensembl build 73. All samples are processed individually through the bioinformatics pipeline and single sample calling is performed.
Variant calls are loaded into our internal mysql database AnnoDB and further analyzed using our software Analysis Tool for Annotated Variants (ATAV). Newly homozygous calls are made by comparing proband variant calls to parents and ensuring both parents have a high quality heterozygous call at the site.
Members of the Undiagnosed Diseases Network
Mercedes E. Alejandro,14 Mahshid S. Azamian,14 Carlos A. Bacino,14 Ashok Balasubramanyam,14 Bret L. Bostwick,14 Lindsay C. Burrage,14 Shan Chen,14 Gary D. Clark,14 William J. Craigen,14 Shweta U. Dhar,14 Lisa T. Emrick,14 Alica M. Goldman,14 Neil A. Hanchard,14 Fariha Jamal,14 efkothea Karaviti,14 Seema R. Lalani,14 Brendan H. Lee,14 Richard A. Lewis,14 Ronit Marom,14 Paolo M. Moretti,14 David R. Murdock,14 Sarah K. Nicholas,14 Jordan S. Orange,14 James P. Orengo,14 Jennifer E. Posey,14 Lorraine Potocki,14 Jill A. Rosenfeld,14 Susan L. Samson,14 Daryl A. Scott,14 Alyssa A. Tran,14 Tiphanie P. Vogel,14 Hugo J. Bellen,14,15 Michael F. Wangler,14,15 Shinya Yamamoto,14,15 Christine M. Eng,14 Donna M. Muzny,14 Patricia A. Ward,14 Yaping Yang,14 David B. Goldstein,16 Nicholas Stong,16 Heidi Cope,17 Yong-hui Jiang,17 Allyn McConkie-Rosell,17 Loren D.M. Pena,17 Kelly Schoch,17 Vandana Shashi,17 Rebecca C. Spillmann,17 Jennifer A. Sullivan,17 Queenie K.-G. Tan,17 Nicole M. Walley,17 Aday Aaron,18 Alan H. Beggs,18 Gerard T. Berry,18 Lauren C. Briere,18 Cynthia M. Cooper,18 Laurel A. Donnell-Fink,18 Elizabeth L. Fieg,18 Frances High,18 Susan Korrick,18 Joel B. Krier,18 Sharyn A. Lincoln,18 Joseph Loscalzo,18 Richard L. Maas,18 Calum A. MacRae,18 J. Carl Pallais,18 Lance H. Rodan,18 Edwin K. Silverman,18 Joan M. Stoler,18 David A. Sweetser,18 Melissa Walker,18 Chris A. Walsh,18 Cecilia Esteves,18 Emily Glanton,18 Ingrid A. Holm,18 Isaac S. Kohane,18 Alexa T. McCray,18 Matthew Might,18 Kimberly LeBlanc,18 David P. Bick,19 Camille L. Birch,19 Braden E. Boone,19 Donna M. Brown,19 Daniel C. Dorset,19 Angela L. Jones,19 Jozef Lazar,19 Shawn E. Levy,19 Thomas May,19 J. Scott Newberry,19 Elizabeth A. Worthey,19 Gabriel F. Batzli,20 Heather A. Colley,20 Jyoti G. Dayal,20 David J. Eckstein,20 Sarah E. Gould,20 Ellen M. Howerton,20 Donna M. Krasnewich,20 Laura A. Mamounas,20 Teri A. Manolio,20 John J. Mulvihill,20 Tiina K. Urv,20 Anastasia L. Wise,20 Matthew Brush,21 Jean-Philippe F. Gourdine,21 Melissa Haendel,21 David M. Koeller,21 Jennifer E. Kyle,22 Thomas O. Metz,22 Katrina M. Waters,22 Bobbie-Jo M. Webb-Robertson,22 Euan A. Ashley,23 Jonathan A. Bernstein,23 Devon Bonner,23 Terra R. Coakley,23 Jean M. Davidson,23 Annika M. Dries,23 Gregory M. Enns,23 Liliana Fernandez,23 Paul G. Fisher,23 Noah D. Friedman,23 Jason Hom,23 Yong Huang,23 Jennefer N. Kohler,23 Marta M. Majcherska,23 Shruti Marwaha,23 Colleen E. McCormack,23 Jason D. Merker,23 Chloe M. Reuter,23 Jacinda B. Sampson,23 Kevin S. Smith,23 Daryl M. Waggott,23 Matthew T. Wheeler,23 Diane B. Zastrow,23 Chunli Zhao,23 Patrick Allard,24 Hayk Barseghyan,24 Manish J. Butte,24 Esteban C. Dell'Angelica,24 Katrina M. Dipple,24 Naghmeh Dorrani,24 Emilie D. Douine,24 Ascia Eskin,24 Brent L. Fogel,24 Hane Lee,24 Sandra K. Loo,24 Martin G. Martin,24 Julian A. Martínez-Agosto,24 Stan F. Nelson,24 Christina GS. Palmer,24 Jeanette C. Papp,24 Neil H. Parker,24 Rebecca Signer,24 Janet S. Sinsheimer,24 Eric Vilain,24 Jijun Wan,24 Amanda J. Yoon,24 Allison Zheng,24 Babak Behnam,25 Elizabeth A. Burke,25 Precilla D'Souza,25 Mariska Davids,25 David D. Draper,25 Tyra Estwick,25 Carlos Ferreira,25 Rena A. Godfrey,25 Catherine A. Groden,25 Jean M. Johnston,25 C. Christopher Lau,25 Ellen F. Macnamara,25 Valerie V. Maduro,25 Thomas C. Markello,25 Marie Morimoto,25 Jennifer L. Murphy,25 Michele E. Nehrebecky,25 Donna Novacic,25 Barbara N. Pusey,25 Prashant Sharma,25CamiloToro25 Colleen E. Wahl,25 Guoyun Yu,25 Andrea L. Gropman,25,26 Eva Baker,25 David R. Adams,25,27 William A. Gahl,25,27 May Christine V. Malicdan,25,27 Cynthia J. Tifft,25,27 Lynne A. Wolfe,25,27 John Yang,25,27 John H. Postlethwait,28 Monte Westerfield,28 Anna Bican,29 Elly Brokamp,29 Laura Duncan,29 Rizwan Hamid,29 Mary Kozuira,29 John H. Newman,29 John A. Phillips III,29 Lynette Rives,29 Amy K. Robertson,29 Lisa Shakachite,29 Joy D. Cogan,29
ADDITIONAL INFORMATION
Data Deposition and Access
Variant data have been deposited in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) under accession number SCV000746595.1. WES data is not available as patient consent was not obtained for deposition.
Ethics Statement
All applicants provided written informed consent as approved by the National Human Genome Research Institute Institutional Review Board under research protocol 15-HG-0130.
Acknowledgments
We thank Marina DiStefano for assistance with ClinGen gene variant curation. We wish to thank the family for their participation and our UDN consultants for their valuable input.
Author Contributions
Q.K.-G.T. and H.C. drafted the manuscript and analyzed and interpreted the data. R.C.S., N.S., Y.-H.J., M.T.M., J.A.R., M.W.B., D.P.F., R.S.L., B.L., C.A.B., M.J.B., C.M.M., A.A.P., N.W., the Undiagnosed Diseases Network, V.S., and L.D.M.P. acquired, analyzed, and interpreted the data. All authors contributed to critical revision of the manuscript.
Funding
Research reported in this manuscript was supported by the National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) U01HG007672 (Duke University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
Baylor College of Medicine, Houston, Texas 77030, USA
Baylor College of Medicine, Model Organisms Screening Center, Houston, Texas 77030, USA
Columbia University, New York, New York 10027, USA
Duke University, Durham, North Carolina 27708, USA
Harvard University, Cambridge, Massachusetts 02138, USA
HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA
National Institutes of Health, Bethesda, Maryland 20892, USA
Oregon Health and Science University, Portland, Oregon 97239, USA
Pacific Northwest National Laboratory, Richland, Washington 99354, USA
Stanford University, Stanford, California 94305, USA
University of California at Los Angeles, Los Angeles, California 90095, USA
Undiagnosed Diseases Program, NIH Bethesda, Maryland 20892, USA
Children's National Medical Center, Washington, D.C. 20010, USA
National Human Genome Research Institute, Bethesda, Maryland 20892, USA
University of Oregon Model Organisms Screening Center, Eugene, Oregon 97403, USA
Vanderbilt University, Nashville, Tennessee 37235, USA
Contributor Information
Collaborators: Mercedes E. Alejandro, Mahshid S. Azamian, Carlos A. Bacino, Ashok Balasubramanyam, Bret L. Bostwick, Lindsay C. Burrage, Shan Chen, Gary D. Clark, William J. Craigen, Shweta U. Dhar, Lisa T. Emrick, Alica M. Goldman, Neil A. Hanchard, Fariha Jamal, Lefkothea Karaviti, Seema R. Lalani, Brendan H. Lee, Richard A. Lewis, Ronit Marom, Paolo M. Moretti, David R. Murdock, Sarah K. Nicholas, Jordan S. Orange, James P. Orengo, Jennifer E. Posey, Lorraine Potocki, Jill A. Rosenfeld, Susan L. Samson, Daryl A. Scott, Alyssa A. Tran, Tiphanie P. Vogel, Hugo J. Bellen, Michael F. Wangler, Shinya Yamamoto, Christine M. Eng, Donna M. Muzny, Patricia A. Ward, Yaping Yang, David B. Goldstein, Nicholas Stong, Heidi Cope, Yong-hui Jiang, Allyn McConkie-Rosell, Loren D.M. Pena, Kelly Schoch, Vandana Shashi, Rebecca C. Spillmann, Jennifer A. Sullivan, Queenie K.-G. Tan, Nicole M. Walley, Aday Aaron, Alan H. Beggs, Gerard T. Berry, Lauren C. Briere, Cynthia M. Cooper, Laurel A. Donnell-Fink, Elizabeth L. Fieg, Frances High, Susan Korrick, Joel B. Krier, Sharyn A. Lincoln, Joseph Loscalzo, Richard L. Maas, Calum A. MacRae, J. Carl Pallais, Lance H. Rodan, Edwin K. Silverman, Joan M. Stoler, David A. Sweetser, Melissa Walker, Chris A. Walsh, Cecilia Esteves, Emily Glanton, Ingrid A. Holm, Isaac S. Kohane, Alexa T. McCray, Matthew Might, Kimberly LeBlanc, David P. Bick, Camille L. Birch, Braden E. Boone, Donna M. Brown, Daniel C. Dorset, Angela L. Jones, Jozef Lazar, Shawn E. Levy, Thomas May, J. Scott Newberry, Elizabeth A. Worthey, Gabriel F. Batzli, Heather A. Colley, Jyoti G. Dayal, David J. Eckstein, Sarah E. Gould, Ellen M. Howerton, Donna M. Krasnewich, Laura A. Mamounas, Teri A. Manolio, John J. Mulvihill, Tiina K. Urv, Anastasia L. Wise, Matthew Brush, Jean-Philippe F. Gourdine, Melissa Haendel, David M. Koeller, Jennifer E. Kyle, Thomas O. Metz, Katrina M. Waters, Bobbie-Jo M. Webb-Robertson, Euan A. Ashley, Jonathan A. Bernstein, Devon Bonner, Terra R. Coakley, Jean M. Davidson, Annika M. Dries, Gregory M. Enns, Liliana Fernandez, Paul G. Fisher, Noah D. Friedman, Jason Hom, Yong Huang, Jennefer N. Kohler, Marta M. Majcherska, Shruti Marwaha, Colleen E. McCormack, Jason D. Merker, Chloe M. Reuter, Jacinda B. Sampson, Kevin S. Smith, Daryl M. Waggott, Matthew T. Wheeler, Diane B. Zastrow, Chunli Zhao, Patrick Allard, Hayk Barseghyan, Manish J. Butte, Esteban C. Dell'Angelica, Katrina M. Dipple, Naghmeh Dorrani, Emilie D. Douine, Ascia Eskin, Brent L. Fogel, Hane Lee, Sandra K. Loo, Martin G. Martin, Julian A. Martínez-Agosto, Stan F. Nelson, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, Rebecca Signer, Janet S. Sinsheimer, Eric Vilain, Jijun Wan, Amanda J. Yoon, Allison Zheng, Babak Behnam, Elizabeth A. Burke, Precilla D'Souza, Mariska Davids, David D. Draper, Tyra Estwick, Carlos Ferreira, Rena A. Godfrey, Catherine A. Groden, Jean M. Johnston, C. Christopher Lau, Ellen F. Macnamara, Valerie V. Maduro, Thomas C. Markello, Marie Morimoto, Jennifer L. Murphy, Michele E. Nehrebecky, Donna Novacic, Barbara N. Pusey, Prashant Sharma, CamiloToro, Colleen E. Wahl, Guoyun Yu, Andrea L. Gropman, Eva Baker, David R. Adams, William A. Gahl, May Christine V. Malicdan, Cynthia J. Tifft, Lynne A. Wolfe, John Yang, John H. Postlethwait, Monte Westerfield, Anna Bican, Elly Brokamp, Laura Duncan, Rizwan Hamid, Mary Kozuira, John H. Newman, John A. Phillips, III, Lynette Rives, Amy K. Robertson, Lisa Shakachite, and Joy D. Cogan
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano N, Atsuumi H, Nakamura A, Tanaka Y, Tanaka I, Yao M. 2014. Direct interaction between EFL1 and SBDS is mediated by an intrinsically disordered insertion domain. Biochem Biophys Res Commun 443: 1251–1256. [DOI] [PubMed] [Google Scholar]
- Bodian M, Sheldon W, Lightwood R. 1964. Congenital hypoplasia of the exocrine pancreas. Acta Paediatr 53: 282–293. [DOI] [PubMed] [Google Scholar]
- Burke V, Colebatch JH, Anderson CM, Simons MJ. 1967. Association of pancreatic insufficiency and chronic neutropenia in childhood. Arch Dis Child 42: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapito R, Konantz M, Paillard C, Miao Z, Pichot A, Leduc MS, Yang Y, Bergstrom KL, Mahoney DH, Shardy DL, et al. 2017. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman–Diamond-like features. J Clin Invest 127: 4090–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanraj S, Matveev A, Li H, Lauhasurayotin S, Jardine L, Cada M, Zlateska B, Tailor CS, Zhou J, Mendoza-Londono R, et al. 2017. Biallelic mutations in DNAJC21 cause Shwachman–Diamond syndrome. Blood 129: 1557–1562. [DOI] [PubMed] [Google Scholar]
- Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, Kerr E, Zeidler C, Shimamura A, Shah N, et al. 2011. Draft consensus guidelines for diagnosis and treatment of Shwachman–Diamond syndrome. Ann N Y Acad Sci 1242: 40–55. [DOI] [PubMed] [Google Scholar]
- Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, González Fernández A, Simpson P, D'Santos CS, Arends MJ, et al. 2011. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman–Diamond syndrome. Genes Dev 25: 917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Márquez A, Gijsbers A, de la Mora E, Sánchez-Puig N. 2015. Defective guanine nucleotide exchange in the elongation factor-like 1 (EFL1) GTPase by mutations in the Shwachman–Diamond syndrome protein. J Biol Chem 290: 17669–17678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginzberg H, Shin J, Ellis L, Morrison J, Ip W, Dror Y, Freedman M, Heitlinger LA, Belt MA, Corey M, et al. 1999. Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pediatr 135: 81–88. [DOI] [PubMed] [Google Scholar]
- Kerr EN, Ellis L, Dupuis A, Rommens JM, Durie PR. 2010. The behavioral phenotype of school-age children with Shwachman Diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman–Bodian–Diamond syndrome gene function. J Pediatr 156: 433–438. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin TL, Makitie O, Berdon WE, Lachman RS. 2015. Shwachman–Bodian–Diamond syndrome: metaphyseal chondrodysplasia in children with pancreatic insufficiency and neutropenia. Pediatr Radiol 45: 1066–1071. [DOI] [PubMed] [Google Scholar]
- Menne TF, Goyenechea B, Sánchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, Brost RL, Costanzo M, Boone C, Warren AJ. 2007. The Shwachman–Bodian–Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet 39: 486–495. [DOI] [PubMed] [Google Scholar]
- Meyer AE, Hoover LA, Craig EA. 2010. The cytosolic J-protein, Jjj1, and Rei1 function in the removal of the pre-60 S subunit factor Arx1. J Biol Chem 285: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KC, Bolyard AA, Otto B, Wong TE, Jones AT, Harris RE, Davies SM, Dale DC, Shimamura A. 2014. Variable clinical presentation of Shwachman–Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr 164: 866–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. 2014. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord 7: 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576. [DOI] [PubMed] [Google Scholar]
- Shwachman H, Diamond LK, Oski FA, Khaw KT. 1964. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr 65: 645–663. [DOI] [PubMed] [Google Scholar]
- Stepensky P, Chacón-Flores M, Kim KH, Abuzaitoun O, Bautista-Santos A, Simanovsky N, Siliqi D, Altamura D, Méndez-Godoy A, Gijsbers A, et al. 2017. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in a Shwachman–Diamond like syndrome. J Med Genet 54: 558–566. [DOI] [PubMed] [Google Scholar]
- Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, et al. 2017. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource. Am J Hum Genet 100: 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiviainen-Salo S, Durie PR, Numminen K, Heikkilä P, Marttinen E, Savilahti E, Makitie O. 2009. The natural history of Shwachman–Diamond syndrome–associated liver disease from childhood to adulthood. J Pediatr 155: 807–811 e802. [DOI] [PubMed] [Google Scholar]
- Tummala H, Walne AJ, Williams M, Bockett N, Collopy L, Cardoso S, Ellison A, Wynn R, Leblanc T, Fitzgibbon J, et al. 2016. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60s ribosome subunit maturation. Am J Hum Genet 99: 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren AJ. 2017. Molecular basis of the human ribosomopathy Shwachman–Diamond syndrome. Adv Biol Regul 67: 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Guturu H, Bernstein JA, Bejerano G. 2017. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med 19: 209–214. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, Vetter IR. 2011. Structure–function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80: 943–971. [DOI] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, et al. 2015. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 17: 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.